Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(14):2510-2517. doi:10.7150/jca.25324 This issue Cite
Review
Effects of the intestinal microbial metabolite butyrate on the development of colorectal cancer
Xinqiang Wu1,*, Yuanbing Wu2,*, Liangmei He3, Longhuo Wu4, Xiangcai Wang3, Zhiping Liu5,6 ![]()
1. Gannan Medical University, Ganzhou, Jiangxi, China.
2. The First People's Hospital of Jiashan County, Jiaxing, Zhejiang, China.
3. The First Affiliated Hospital, Gannan Medical University, Ganzhou, Jiangxi, China.
4. College of Pharmacy, Gannan Medical University, Ganzhou, Jiangxi, China.
5. School of Basic Medicine, Gannan Medical University, Ganzhou, Jiangxi, China.
6. Ganzhou Cancer Precision Engineering Research Center, Ganzhou, Jiangxi, China.
* These authors contributed equally to this work.
Received 2018-2-3; Accepted 2018-5-2; Published 2018-6-15
Abstract
Colorectal cancer (CRC) is one of the major health threats in developed countries. Changes in dietary components, such as more protein and lipid intake, can increase the risk of CRC. Diet affects CRC in many ways. They regulate the composition and function of gut microbiota, which have an amazing metabolic capacity and can produce short chain fatty acids (SCFAs), such as propionate, acetate, and butyrate. Butyrate is a principal energy source for colonic epithelial cells and plays an important role in maintaining the stability of gut microbiota and the integrity of intestinal epithelium. However, there are few studies reviewing the anti-CRC potentials of butyrate. This review summarizes the recent research progresses in the effect of gut microbiota imbalance and the decrease in intestinal microbial metabolite butyrate caused by unbalanced diet on CRC development, and discusses the mechanisms of butyrate-induced anti-CRC activities, which may guide people to prevent CRC by improving diet structures.
Keywords: Butyrate, Colorectal cancer, Gut microbiota, Diet.
Introduction
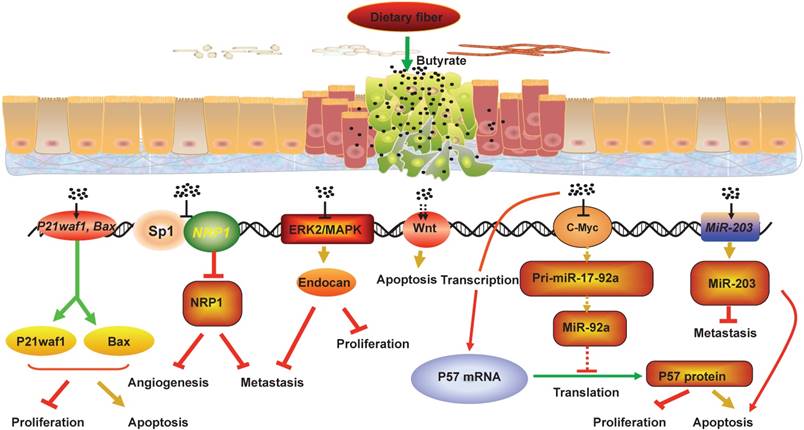
Colorectal cancer (CRC) is the second most common cancer in women and the third in men worldwide[1]. Epidemiological studies have shown that the occurrence and development of CRC is associated with an increase in meat diet and a reduction in dietary fiber, and that changes in food composition can affect CRC risk by affecting intestinal microbial metabolism[2, 3]. Studies in recent decades have proved that gut microbiota, which can be affected by diets, play an important role in the occurrence and development of CRC[4, 5]. After digestion and absorption, food with different structures left residue in the intestinal tract. As food flows through the colon, residues of dietary fiber can be fermented by gut microbiota to produce short chain fatty acids(SCFAs), such as propionate, acetate, and butyrate, which play plurifunctional roles on the gut microbiota and intestinal epithelial cells[6, 7]. Studies have shown that the primary energy source of the intestinal epithelia is butyrate, rather than glucose, which established the basis for the modern theory of mutualism between gut microbiota and intestinal epithelia[8]. Butyrate can inhibit the development of CRC and promote intestinal health through various mechanisms (Figure 1). This review summarizes the recent research progresses in the effect of gut microbiota imbalance and the decrease in the intestinal microbial metabolite butyrate caused by unbalanced diet on CRC development, and discusses the mechanisms of butyrate-induced anti-CRC activity, such as suppression of neuropilin I expression, regulation of miRNA expression, promotion of endocan expression, hyper-activation of Wnt signaling pathway, and upregulation of P21waf1 and bax expression (Figure 2).
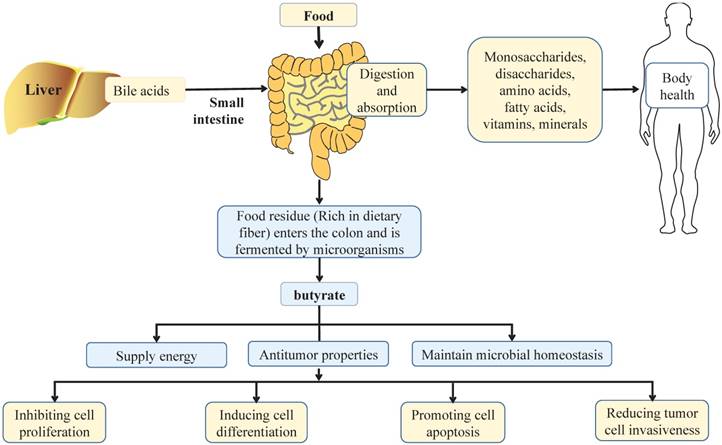
Effect of balanced diet on intestinal health. In a balanced diet, food is mainly digested by digestive enzymes and bile acids from liver in the small intestine, and more than 90% are absorbed in the small intestine. After absorption, nutrients are distributed throughout the body through the circulation of the blood, in which monosaccharides, disaccharides, amino acids, fatty acids, vitamins and minerals play important roles in maintaining the health of the body. When dietary residues flow through the colon, they are broken down by intestinal microbiota, which produce short chain fatty acids, such as butyrate. Butyrate is a principal energy source for the gut microbiota and intestinal epithelial cells, and plays an important role in maintaining the stability of gut microbiota and the integrity of intestinal epithelia. Notably, butyrate can also inhibit cell proliferation, induce cell differentiation, promote cell apoptosis, and reduce tumor cell invasiveness to exert its anti-tumor properties, thus playing an important role in the health of the colon.
The mechanisms that butyrate suppresses CRC development. Fermentation of fiber in the lumen leads to production of short-chain fatty acids including butyrate. Butyrate can inhibit the development of CRC and promote intestinal health through various mechanisms. For example, butyrate can decrease the NRP-1 expression by inhibiting the transactivation of Sp1 to suppress the angiogenesis, metastasis and survival of CRC cells, and butyrate can promote the apoptosis of CRC cells by hyper-activating Wnt signaling pathway. In addition, butyrate can restrict cell proliferation, colony formation, cell invasion, and induce cell apoptosis in CRC cells through upregulating the expression of miR-203, P21waf1 and bax, and promoting endocan expression. Meanwhile, butyrate can increase p57 mRNA and protein levels by inhibiting c-Myc expression, which reduces miR-17-92a cluster transcription and the level of miR-92a. Collectively, the interactions between butyrate, NRP-1, Wnt, endocan, P21waf1 and bax, miR-203 and miR-92a mediate the anti-proliferation and pro-apoptosis effect of butyrate in CRC cells.
Effects of dietary factors on CRC
The geographical variation in CRC incidence is remarkable, as illustrated by the WHO's data that CRC incidence rate is relatively high in western developed countries while relatively low in developing countries, such as Asia and Africa. A previous study observed a significant change in the CRC incidence after Japanese migrate to Hawaii. Specifically, CRC incidence was low in Japan, and after these people migrated to Hawaii, their CRC incidence became as high as Hawaiian natives within one generation[9]. Further analysis showed that the CRC incidence varies from place to place, mainly due to different dietary habits. The high CRC incidence in western developed countries is induced by imbalanced diet, mainly demonstrated by a lack in fruits, vegetables, cereals, and other high fiber foods. It is well recognized that dietary fiber can reduce the CRC risk, whereas the meat diet can increase the CRC risk[10]. A study showed that up to 30 g/d dietary fiber intake could prevent CRC development[11]. Moreover, based on the characteristics of high fiber diet, gut microbiota is evolved into a model that is suitable for decomposing high fiber food residues. Fiber diet and intestinal microorganisms benefit mutually, and gradually become dependent on each other to form a stable equilibrium. These showed that dietary factors and the gut microbes play an important role in the CRC pathogenesis.
Effects of gut microbes on CRC
Gut harbors a large number of different microorganisms, such as bacteria, viruses, archaea, yeasts, and fungi. The gut microbiota has many important functions for human health[12, 13]. For example, gut microbiota can ferment dietary fiber to produce SCFAs, such as acetate, propionate, and butyrate[14]. Among the gut microbes, there are several types of anaerobic bacteria, such as Faecalibacterium prausnitzii and Roseburia species, which are the main butyrate-producing bacteria[15, 16]. In comparison with healthy controls, CRC patients have significant changes in composition of gut microbiota, such as the increase in Fusobacteria, Porphyromonadaceae, Staphylococcaceae, Akkermansia spp. and Methano-bacteriales, and the decrease in Bifidobacterium, Lactobacillus, Ruminococcus, Faecalibacterium spp., Roseburia, and T-reponema[17-19]. In particular, butyrate-producing Fusobacterium was shown to inhibit CRC development[20, 21]. In addition, the microbial metabolites such as nitrogen components, were reported to be elevated, whereas others metabolites, such as butyrate, decreased in certain CRC patients[22]. Gut microbiota can also induce chronic inflammation, one of the major risk factors for CRC, by directly invading intestinal epithelial barrier or activating immune responses by binding to Toll-like receptors, which leads to the synthesis and secretion of cytokines. The abundance of gut microbes in the lumen is also closely related to the CRC occurrence. For example, the distribution of microorganisms in the distal colon is the densest in all parts of intestine, thus the probability of CRC occurrence in the colon is also the largest[23]. Food residue, intestinal microbiota, and intestinal epithelia, work together to maintain intestinal homeostasis. The structure of the gut microbiota is usually very stable, and the microbes in each part of the digestive tract from the mouth to the rectum are different[24]. The formation of this steady state is also determined by the long-term natural selection in human evolution. Plant food in the lower food chain is relatively readily available than meat food on the top of the food chain. Therefore, the long vegetarian diet of human determines the dependence of intestinal microbiota on fiber diet. When this long-term eating habits change, intestinal microbial disorder tends to occur. Butyrate, the metabolite produced by the fermentation of dietary fiber-rich food residues by the gut microbes, is an important mediator among dietary factors, intestinal microbiota and CRC. In comparison with healthy people, CRC patients have significant reduction in the number of bacteria that produce butyrate and other SCFAs[25]. Therefore, the change of intestinal microbiota caused by the diet change plays an important role in the occurrence and development of CRC.
Effects of the gut microbiota metabolites butyrate on CRC
In a balanced diet, gut microbes ferment fiber-rich food residue in colon and produce SCFA metabolites including butyrate. Butyrate can provide energy for intestinal epithelial cells[6, 26], and has anti-inflammatory and anti-tumor properties[27-29]. A recently proposed mechanism about the effect of butyrate on CRC progression is based on the down-regulation of butyrate transporter protein in CRC tissue, which will reduce the transport of butyrate in the intestine and reduce the metabolism of butyrate[30]. The atrophy of butyrate metabolism is associated with the pathogenesis of various colonic diseases[31]. It is well known that the pathogenesis of CRC is closely related to the dysfunction of intestinal epithelial barrier, which activates tumor-associated macrophages, leads to the production of inflammatory cytokines, and ultimately leads to tumor growth and progression[32]. Butyrate can promote the assembly of tight junction by activating the Amp-activated protein kinase (AMPK), and can induce the expression of MUC2 in human colon cancer cell line LS174T cells, to maintain the function of intestinal epithelial barrier[33-35]. The movement of the gastrointestinal tract decreased and the subsequent intestinal transport slowed down in the long time, which also affected the incidence of CRC. Studies show that butyrate can interfere with the internal nervous system and regulate the excitability of neurons, causing the increase of colonic motility and the extracorporeal contraction reaction[36-38]. Therefore, butyrate is critical for intestinal microbial balance and colon health. In addition, a number of studies have shown that butyrate can inhibit the occurrence and development of CRC through various mechanisms (Table 1).
Effects of butyrate on the development of colorectal cancer
| Signaling pathway | Key molecules | Experimental systems | Main effects on colorectal cancer | References |
|---|---|---|---|---|
| NRP-1/VEGF | NRP-1 | HCT116 HT29 Caco-2 | Butyrate can decrease NRP-1 expression by inhibiting the transactivation of Sp1 to suppress the angiogenesis, metastasis and survival of CRC cells | [55, 56,57] |
| ERK2/MAPK | Endocan | RKO | Butyrate can inhibit CRC cell proliferation, clone formation and migration by promoting endocan expression through ERK2/MAPK signaling pathway | [61, 62] |
| Wnt | β-catenin | DLD-1 HCT116 | Butyrate can promote the apoptosis of CRC cells by hyper-activating Wnt signaling pathway | [67-71] |
| p53 | P21wafl bax | HT-29, SW48, SW1116 | Butyrate can enhance the expression of P21waf1 and bax to inhibit the proliferation and induce apoptosis of CRC cells and block the cell cycle in G1 phase | [72, 73] |
| microRNA | miR-203 miR-92a | SW837 HT-29 Caco-2 HCT116 | Butyrate can up-regulate miR-203 expression and decrease miR-92a expression to restrict cell proliferation, colony formation, cell invasion, and induce cell apoptosis in CRC cells | [89, 91] |
As a fatty acid, butyrate can be converted to acetyl-coA by beta-oxidation, and then completely oxidized through three carboxylic acid cycle. The metabolization of butyrate to acetyl-CoA is essential for stimulating the activity of histone acetyltransferase (HAT). Due to the Warburg effect, butyrate does not metabolize normally in cancer cells, but can be accumulated as a histone deacetylase inhibitor (HDACi) in the nucleus, thereby regulating the expression of downstream target genes. Histone deacetylase (HDAC) is very important for gene expression, and the expression of these enzymes in tumor cells increase and vary by tumor types [39]. For example, HDAC1 is highly expressed in prostate cancer, gastric cancer, lung cancer, esophageal cancer and breast cancer, and HDAC2 is highly expressed in colorectal cancer, cervical cancer and gastric cancer. In addition, HDAC3 is highly expressed in breast cancer and colorectal cancer, while HDAC6 is highly expressed in nerve cell carcinoma[40, 41]. In most of tumors, acetylation will result in expression changes of some genes, which may be involved in signaling pathways such as ERK, Wnt, and further affect the protease system as well as the activities of some kinases (such as protein kinase C) [42]. On the structure, HDACi can induce cell death through different processes, depending on the target cells, including changes in gene expression, histone modifications, and epigenetic changes[43]. Inhibition of histone acetylation leads to chromatin lysis and reduction in concentration, making DNA more readily accessible. The decrease in the activity of histone deacetylase was associated with inhibition of tumor cell growth[44]. As an HDACi, butyrate has complicated roles in histone acetylation. Butyrate can influence the development of tumor by inducing cell cycle stagnation and apoptosis.
Butyrate reduces the expression of neuropilin by inhibiting the transactivation of Sp1(specificity protein 1) to suppress the angiogenesis, metastasis and survival of CRC
Tumor growth and metastasis are highly dependent on angiogenesis, which is the initial stage of new blood vessel formation[45]. Vascular endothelial growth factor (VEGF) is the core regulator of angiogenesis[46]. The expression of VEGF is up-regulated in many solid tumors, including primary and metastatic CRC[47]. Multiple studies have found that butyrate, as an HDACi, can down-regulate the expression of VEGF to inhibit the tumor angiogenesis[48, 49]. Neuropilin (NRP) is a receptor for VEGF, which is expressed in normal epithelial cells and is highly expressed in tumor cells[50, 51]. The binding of VEGF to Neuropilin-1 (NRP-1) promotes the migration and survival of CRC cells[52]. Clinical studies indicate that CRC with high expression of NRP-1 has a greater incidence of metastases, increased proliferation index and reduced numbers of apoptotic cancer cells than CRCs with low expression of NRP-1, suggesting that NRP-1 can protect CRC cells from apoptosis[53]. Silence of NRP-1 expression by siRNA can induce apoptosis of tumor cells, suggesting that the decrease in NRP-1 expression can be used to increase sensitivity of tumor cells to chemotherapy[54]. Previous studies revealed that NRP-1 is down-regulated by butyrate in CRC patients[55]. Meanwhile, in vitro experiment also showed that butyrate can down-regulated NRP-1 and VEGF at the mRNA and protein level in CRC cell lines through inhibiting Sp1 transactivation [56, 57]. HCT116, HT29 and Caco-2 lines represent a range of CRC cell types. When different concentrations of butyrate were used to treat these three CRC cell lines, the expression of NRP-1 were suppressed, and the binding of VEGF to NRP-1 also decreased, thus inhibiting angiogenesis [49].
Butyrate inhibits CRC progression by up-regulating the endocan gene expression via ERK2/MAPK signaling pathway
Endocan is a sulfate proteoglycan and plays an important role in inflammation, angiogenesis, and metastasis of tumor cells[58-60]. The expression of Endocan in CRC tissues was lower than control tissues, and the Endocan expression was inversely correlated with the degree of malignancy[61]. A study showed that butyrate increased endocan expression at both transcription and translation level in the RKO colon cancer cells. Moreover, over-expression of endocan can significantly inhibit the proliferation, colony formation, and migration of RKO cells[62]. In addition, knocking-down of the endogenous endocan level reversed the inhibitory effect of butyrate on tumor proliferation and metastasis. This study also showed that butyrate increased the endocan expression through inhibiting ERK2/MAPK signaling pathway, thus suppressing the proliferation, migration, and colony formation of CRC cells[62].
Butyrate regulates Wnt activity levels and apoptosis in CRC cells
Wnt signaling pathway is deregulated in most cases of CRC[63]. The core protein in this pathway is β-catenin, which is bound by the adenomatous polyposis coli (APC) protein. When Wnt signaling is not activated, β-catenin is phosphorylated by glycogen synthase kinase 3β(GSK-3β) and targeted for degradation[64]. When Wnt signaling pathway is activated, the activity of GSK-3 kinase is suppressed, and β-catenin accumulates in the nucleus and binds to the Tcf/Lef (T-cell factor/lymphocyte enhancer factor) family to activate the transcription of target genes. However, in CRC, the mutation in the components of the pathway (e.g., β-catenin and / or APC) leads to constitutive transcriptional activation of the Wnt target gene, which ultimately leads to unlimited cell growth[65, 66].
Previous studies showed that butyrate induced higher Wnt activity and apoptosis in CRC cells, and Wnt levels were correlated with apoptosis levels. However, the underlying mechanism by which butyrate induced CRC cell apoptosis in hyper-activation of Wnt signaling pathway is not well understood[67-69]. The correlation between high Wnt activity and apoptosis induced by butyrate was also observed in DLD-1 and HCT116 cells with dominant negative (dn) Tcf4, in which the decrease of Wnt activity was associated with the decrease of apoptosis[70]. In addition, the CRC cells with higher Wnt activity had higher levels of apoptosis, while the samples with low or no Wnt activity had lower levels of apoptosis after the treatment of butyrate[70]. Meanwhile, another study confirms that butyrate can induce changes in expression of Wnt-signaling-specific genes in CRC cells[71]. Thus, the hyper-activated Wnt activity is causatively related to the induction of apoptosis in CRC cells treated with butyrate.
Butyrate enhances p21wafl and bax expression to inhibit proliferation and induce apoptosis of CRC cells
p53 is a tumor suppressor gene that has a high correlation with human tumor regression. p53 mutation can be detected in more than 50% of human cancers. Some chemicals or food ingredients can induce the expression of p53 target genes (such as p21wafl, bax) and potentially suppress tumor development. A study showed that butyrate induced p21wafl and bax expression both at mRNA and protein level in HT-29 cells[72]. In addition, butyrate with the concentration above 2.5 mmol/L can inhibit the growth of HT-29 cells. Furthermore, butyrate treatment increased the percentage of G1 phase cells. According to the results of Annexin-V/PI staining, the apoptosis rates of butyrate-treated group were significantly higher than those of the control group[72]. Studies using other CRC cell lines (SW48, SW1116, and SW837) also showed that butyrate could suppress cell proliferation and enhance cell apoptosis[73].
Butyrate upregulates miR-203 expression and downregulates miR-92a expression to restrict cell proliferation, colony formation, cell invasion, and induces cell apoptosis in CRC cells
The ability of butyrate to inhibit CRC cells is also associated with its regulation on microRNA (miRNA), such as the miR-106b family and the miR-17-92 family[74, 75]. miRNAs are small and non-coding RNAs that regulate cell cycle arrest, cell apoptosis, proliferation and differentiation[76, 77]. More than 1000 miRNAs have been identified, and many of them have hundreds of target genes[78]. Genetic or epigenetic modifications affect the expression of miRNAs in various human tumors[79]. Moreover, the disorder of miRNA expression is involved in the process of tumorigenesis through regulating the expression of proto-oncogenes and tumor suppressor genes[80]. In CRC cells, a variety of miRNAs have been found to affect cell growth, migration and invasion[81-84]. MiR-203, as a tumor suppressor miRNA, has lower expression levels in CRC tissues than in non-tumor tissues[85]. Studies reveal that miR-203 could inhibit cell proliferation, invasion and metastasis, and promote cell apoptosis[86, 87]. A previous study of skin tumors found that butyrate was able to decrease the expression of miR-203 to inhibit tumor development[88]. Another study investigated the effect of butyrate on miR-203 expression in CRC cell lines, HT-29 and Caco-2. The results showed that butyrate could increase miR-203 to inhibit cell proliferation, clone formation, cell invasion, and induce apoptosis in CRC cells. A further study indicated that NEDD9, a target gene of miR-203, could partially reverse the effect of miR-203 on the colony formation and invasion of CRC cells[89].
In addition, another study showed that butyrate increased the expression of other miRNA families and clusters, including the oncogenic miR-17-92a cluster, also known as oncomiR-1 and C13orf25 in human sporadic colon cancer[90-92]. MiR-92a is overexpressed in CRC patients and associated with tumor metastasis and poor prognosis[93]. Further studies found that microbe-derived butyrate inhibits the oncogenic miR-92a by regulating c-Myc expression in CRC. Butyrate treatment on HCT116 and HT29 human CRC cells led to the significant decrease in miR-92a level. Other HDACi, such as hydroxamic acid (SAHA) and valproic acid, also have similar effects upon butyrate treatment. In addition, butyrate treatment also reduces the level of other members of the miR-17-92a family, including miR-17, miR-18a, miR-19a/b, and miR-20a. Exogenous supplement of miR-92a can reverse the expression of the tumor suppressor gene of p57, and growth inhibition and apoptosis induced by butyrate. Collectively, these findings reveal a new mechanism by which butyrate suppress CRC development through the interaction among butyrate, c-Myc, miR-92a, and p57, leading to the decrease in cell proliferation and increase in cell apoptosis[91].
Conclusions and future prospects
The geographical distribution of CRC is mainly due to the difference of dietary habits. Gut microbes rely on high fiber foods residues to maintain its normal structure and metabolism, wherein metabolite butyrate plays an important role in maintaining intestinal homeostasis and the health of intestinal epithelia. Butyrate is the basic energy source of the intestinal epithelium, and can inhibit intestinal inflammation. Above all, as an HDACi and a product of fiber fermentation, it can mediate the protective effect of dietary fiber against CRC by a variety of mechanisms. In recent years, studies on the interaction among dietary factors, gut microbes and metabolites in the development of CRC are very active. However, many unknown and the unanswered questions remain. Better understanding on the molecular mechanisms and signaling pathways induced by butyrate treatment will facilitate the development of new therapeutic means to cure CRC.
Acknowledgements
This work was supported by funds from Talents' Start-up Fund of Gannan Medical University (QD201404), Natural Science Foundation of Jiangxi Province (20151BAB205061 and 20171ACB20024), National Natural Science Foundation of China (31560260), and The Key Project from Department of Education of Jiangxi Province (Gjj150937) (All to Zhiping Liu).
Abbreviations
CRC, colorectal cancer; SCFAs, short chain fatty acids; HAT, histone acetyltransferase; HDAC, histone deacetylase; HDACi, histone deacetylase inhibitor; VEGF, vascular endothelial growth factor; NRP-1, neuropilin-1; Sp1, specificity protein 1; APC, adenomatous polyposis coli; GSK-3β, glycogen synthase kinase 3β; Tcf/Lef, T-cell factor/lymphocyte enhancer factor.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Cancer incidence and mortality worldwide: IARC CancerBase No. International Journal of Cancer. 2010;136:E359-E86
2. Aune D, Chan DS, Lau R, Vieira R, Greenwood DC, Kampman E. et al. Dietary fibre, whole grains, and risk of colorectal cancer: systematic review and dose-response meta-analysis of prospective studies. BMJ (Clinical research ed). 2011;343:d6617
3. Tuan J, Chen YX. Dietary and Lifestyle Factors Associated with Colorectal Cancer Risk and Interactions with Microbiota: Fiber, Red or Processed Meat and Alcoholic Drinks. Gastrointestinal Tumors. 2016;3:17-24
4. Cho M, Carter J, Harari S, Pei Z. The Interrelationships of the Gut Microbiome and Inflammation in Colorectal Carcinogenesis. Clinics in Laboratory Medicine. 2014;34:699-710
5. Arthur JC, Jobin C. The Struggle Within: Microbial Influences on Colorectal Cancer. Inflammatory Bowel Diseases. 2011;17:396-409
6. Kuwahara A. Contributions of colonic short-chain Fatty Acid receptors in energy homeostasis. Frontiers in Endocrinology. 2014;5:144
7. Macfarlane GT, Englyst HN. Starch utilization by the human large intestinal microflora. Journal of Applied Bacteriology. 1986;60:195-201
8. Roediger WE. Utilization of nutrients by isolated epithelial cells of the rat colon. Gastroenterology. 1982;83:424-9
9. Le Marchand L, Kolonel LN. [Cancer in Japanese migrants to Hawaii: interaction between genes and environment]. Revue d'epidemiologie et de sante publique. 1992;40:425-30
10. Deng Y. Rectal Cancer in Asian vs. Western Countries: Why the Variation in Incidence? Current Treatment Options in Oncology. 2017;18:64
11. Ferguson LR, Harris PJ. The dietary fibre debate: more food for thought. Lancet. 2003;361:1487-8
12. von Martels JZH, Sadaghian Sadabad M, Bourgonje AR, Blokzijl T, Dijkstra G, Faber KN. et al. The role of gut microbiota in health and disease: In vitro modeling of host-microbe interactions at the aerobe-anaerobe interphase of the human gut. Anaerobe. 2017;44:3-12
13. Shen TD. Diet and Gut Microbiota in Health and Disease. Nestle Nutrition Institute workshop series. 2017;88:117-26
14. Holscher HD. Dietary Fiber and Prebiotics and the Gastrointestinal Microbiota. Gut Microbes. 2017;8:172-84
15. Miquel S, Martín R, Rossi O, Bermúdez-Humarán LG, Chatel JM, Sokol H. et al. Faecalibacterium prausnitzii and human intestinal health. Current Opinion in Microbiology. 2013;16:255-61
16. Duncan SH, Barcenilla A, Stewart CS, Pryde SE, Flint HJ. Acetate utilization and butyryl coenzyme A (CoA):acetate-CoA transferase in butyrate-producing bacteria from the human large intestine. Applied and Environmental Microbiology. 2002;68:5186-90
17. Zhu Q, Jin Z, Wu W, Gao R, Guo B, Gao Z. et al. Analysis of the intestinal lumen microbiota in an animal model of colorectal cancer. PloS one. 2014;9:e90849
18. Wu N, Yang X, Zhang R, Li J, Xiao X, Hu Y. et al. Dysbiosis Signature of Fecal Microbiota in Colorectal Cancer Patients. Microbial Ecology. 2013;66:462-70
19. Mira-Pascual L, Cabrera-Rubio R, Ocon S, Costales P, Parra A, Suarez A. et al. Microbial mucosal colonic shifts associated with the development of colorectal cancer reveal the presence of different bacterial and archaeal biomarkers. Journal of Gastroenterology. 2015;50:167-79
20. Moore WE, Moore LH. Intestinal floras of populations that have a high risk of colon cancer. Applied and Environmental Microbiology. 1995;61:3202-7
21. Kostic A, Chun E, Robertson L, Glickman J, Gallini CA, Michaud M. et al. Fusobacterium nucleatum Potentiates Intestinal Tumorigenesis and Modulates the Tumor-Immune Microenvironment. Cell Host & Microbe. 2013;14:207-15
22. Gonçalves P, Martel F. Butyrate and colorectal cancer: the role of butyrate transport. Current Drug Metabolism. 2013;14:994-1008
23. Uronis JM, Muhlbauer M, Herfarth HH, Rubinas TC, Jones GS, Jobin C. Modulation of the intestinal microbiota alters colitis-associated colorectal cancer susceptibility. PloS one. 2009;4:e6026
24. Neish AS. Microbes in Gastrointestinal Health and Disease. Gastroenterology. 2008;136:65-80
25. Wu GD, Compher C, Chen EZ, Smith SA, Shah RD, Bittinger K. et al. Comparative metabolomics in vegans and omnivores reveal constraints on diet-dependent gut microbiota metabolite production. Gut. 2016;65:63-72
26. Daly K, Shirazi-Beechey SP. Microarray analysis of butyrate regulated genes in colonic epithelial cells. DNA and Cell Biology. 2006;25:49-62
27. Mathewson ND, Jenq R, Mathew AV, Koenigsknecht M, Hanash A, Toubai T. et al. Gut microbiome derived metabolites modulate intestinal epithelial cell damage and mitigate Graft-versus-Host Disease. Nature Immunology. 2016;17:505
28. Macfarlane GT, Gibson GR, Cummings JH. Comparison of fermentation reactions in different regions of the human colon. Journal of Applied Bacteriology. 1992;72:57-64
29. Cummings JH, Beatty ER, Kingman SM, Bingham SA, Englyst HN. Digestion and physiological properties of resistant starch in the human large bowel. British Journal of Nutrition. 1996;75:733-47
30. Goncalves P, Martel F. Butyrate and colorectal cancer: the role of butyrate transport. Current Drug Metabolism. 2013;14:994-1008
31. Leonel AJ, Alvarez-Leite JI. Butyrate: implications for intestinal function. Current Opinion in Clinical Nutrition Metabolic Care. 2012;15:474-9
32. Wang K, Karin M. Common flora and intestine: A carcinogenic marriage. Cell Logist. 2013;3:e24975
33. Renaud F, Vincent A, Mariette C, Crepin M, Stechly L, Truant S. et al. MUC5AC hypomethylation is a predictor of microsatellite instability independently of clinical factors associated with colorectal cancer. International Journal of Cancer. 2015;136:2811-21
34. Peng L, Li ZR, Green RS, Holzman IR, Lin J. Butyrate enhances the intestinal barrier by facilitating tight junction assembly via activation of AMP-activated protein kinase in Caco-2 cell monolayers. The Journal of Nutrition. 2009;139:1619-25
35. Hatayama H, Iwashita J, Kuwajima A, Abe T. The short chain fatty acid, butyrate, stimulates MUC2 mucin production in the human colon cancer cell line, LS174T. Biochemical and Biophysical Research Communications. 2007;356:599-603
36. Soret R, Chevalier J, De Coppet P, Poupeau G, Derkinderen P, Segain JP. et al. Short-chain fatty acids regulate the enteric neurons and control gastrointestinal motility in rats. Gastroenterology. 2010;138:1772-82
37. Hurst NR, Kendig DM, Murthy KS, Grider JR. The short chain fatty acids, butyrate and propionate, have differential effects on the motility of the guinea pig colon. Neurogastroenterology Motility. 2014;26:1586-96
38. Canani RB, Costanzo MD, Leone L, Pedata M, Meli R, Calignano A. Potential beneficial effects of butyrate in intestinal and extraintestinal diseases. World Journal of Gastroenterology. 2011;17:1519-28
39. Donohoe DR, Collins LB, Wali A, Bigler R, Sun W, Bultman SJ. The Warburg effect dictates the mechanism of butyrate-mediated histone acetylation and cell proliferation. Molecular Cell. 2012;48:612-26
40. Li Y, Zhang X, Polakiewicz RD, Yao TP, Comb MJ. HDAC6 is required for epidermal growth factor-induced beta-catenin nuclear localization. The Journal of Biological Chemistry. 2008;283:12686-90
41. Godman CA, Joshi R, Tierney BR, Greenspan E, Rasmussen TP, Wang HW. et al. HDAC3 impacts multiple oncogenic pathways in colon cancer cells with effects on Wnt and vitamin D signaling. Cancer Biology & Therapy. 2008;7:1570-80
42. Bordonaro M, Lazarova DL, Sartorelli AC. The activation of beta-catenin by Wnt signaling mediates the effects of histone deacetylase inhibitors. Experimental Cell Research. 2007;313:1652-66
43. Federico M, Bagella L. Histone deacetylase inhibitors in the treatment of hematological malignancies and solid tumors. Journal of Biomedicine & Biotechnology. 2011:475641
44. Mottamal M, Zheng S, Huang TL, Wang G. Histone deacetylase inhibitors in clinical studies as templates for new anticancer agents. Molecules. 2015;20:3898-941
45. Flores-Perez A, Rincon DG, Ruiz-Garcia E, Echavarria R, Marchat LA, Alvarez-Sanchez E. et al. Angiogenesis Analysis by In Vitro Coculture Assays in Transwell Chambers in Ovarian Cancer. Methods in Molecular Biology (Clifton, NJ). 2018;1699:179-86
46. Mahecha AM, Wang H. The influence of vascular endothelial growth factor-A and matrix metalloproteinase-2 and -9 in angiogenesis, metastasis, and prognosis of endometrial cancer. OncoTargets and Therapy. 2017;10:4617-24
47. Lan J, Li H, Luo X, Hu J, Wang G. BRG1 promotes VEGF-A expression and angiogenesis in human colorectal cancer cells. Experimental Cell Research. 2017;360:236-42
48. Pellizzaro C, Coradini D, Daidone MG. Modulation of angiogenesis-related proteins synthesis by sodium butyrate in colon cancer cell line HT29. Carcinogenesis. 2002;23:735-40
49. Yu DC, Waby JS, Chirakkal H, Staton CA, Corfe BM. Butyrate suppresses expression of neuropilin I in colorectal cell lines through inhibition of Sp1 transactivation. Molecular Cancer. 2010;9:276
50. Chaudhary B, Khaled YS, Ammori BJ, Elkord E. Neuropilin 1: function and therapeutic potential in cancer. Cancer Immunology, Immunotherapy: CII. 2014;63:81-99
51. Guttmann-Raviv N, Kessler O, Shraga-Heled N, Lange T, Herzog Y, Neufeld G. The neuropilins and their role in tumorigenesis and tumor progression. Cancer letters. 2006;231:1-11
52. Staton CA, Koay I, Wu JM, Hoh L, Reed MW, Brown NJ. Neuropilin-1 and neuropilin-2 expression in the adenoma-carcinoma sequence of colorectal cancer. Histopathology. 2013;62:908-15
53. Ochiumi T, Kitadai Y, Tanaka S, Akagi M, Yoshihara M, Chayama K. Neuropilin-1 is involved in regulation of apoptosis and migration of human colon cancer. International Journal of Oncology. 2006;29:105
54. Wey JS, Gray MJ, Fan F, Belcheva A, Mccarty MF, Stoeltzing O. et al. Overexpression of neuropilin-1 promotes constitutive MAPK signalling and chemoresistance in pancreatic cancer cells. British Journal of Cancer. 2005;93:233-41
55. Yu DC, Bury JP, Tiernan J, Waby JS, Staton CA, Corfe BM. Short-chain fatty acid level and field cancerization show opposing associations with enteroendocrine cell number and neuropilin expression in patients with colorectal adenoma. Molecular Cancer. 2011;10:27
56. Yu ChenWei. The effect of butyrate on VEGF and Neuropilin-1 expression in colon cancer. University of Sheffield. 2012
57. Yu DC, Waby JS, Chirakkal H, Staton CA, Corfe BM. Butyrate suppresses expression of neuropilin I in colorectal cell lines through inhibition of Sp1 transactivation. Molecular Cancer. 2010;9:276
58. Rennel E, Mellberg S, Dimberg A, Petersson L, Botling J, Ameur A. et al. Endocan is a VEGF-A and PI3K regulated gene with increased expression in human renal cancer. Experimental Cell Research. 2007;313:1285
59. Leroy X, Aubert S, Zini L, Franquet H, Kervoaze G, Villers A. et al. Vascular endocan (ESM-1) is markedly overexpressed in clear cell renal cell carcinoma. Histopathology. 2010;56:180-7
60. Chen LY, Liu X, Wang SL, Qin CY. Over-expression of the Endocan gene in endothelial cells from hepatocellular carcinoma is associated with angiogenesis and tumour invasion. Journal of International Medical Research. 2010;38:498-510
61. Su-Mei Zhang, Ruo-Lei Hua-Qing, Qing Zhou. et al. Correlation between expression and differentiation of endocan in colorectal cancer. World Journal of Gastroenterology. 2008;14:4562-8
62. Zuo L, Lu M, Zhou Q, Wei W, Wang Y. Butyrate suppresses proliferation and migration of RKO colon cancer cells though regulating endocan expression by MAPK signaling pathway. Food & Chemical Toxicology. 2013;62:892-900
63. Bienz M, Clevers H. Linking colorectal cancer to Wnt signaling. Cell. 2000;103:311-20
64. Wodarz A, Nusse R. Mechanisms of Wnt signaling in development. Annual Review of Cell & Developmental Biology. 2003;14:59
65. Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87:159-70
66. Bienz M, Clevers H. Linking colorectal cancer to Wnt signaling. Cell. 2000;103:311
67. Bordonaro M, Lazarova DL. Butyrate, Wnt Signaling And Colorectal Cancer. 2014.
68. Bordonaro M, Lazarova DL, Augenlicht LH, Sartorelli AC. Cell type- and promoter-dependent modulation of the Wnt signaling pathway by sodium butyrate. International Journal of Cancer. 2002;97:42-51
69. Bordonaro M, Lazarova DL, Sartorelli AC. Butyrate and Wnt signaling: a possible solution to the puzzle of dietary fiber and colon cancer risk? Cell Cycle. 2008;7:1178-83
70. Lazarova DL, Bordonaro M, Carbone R, † ACS. Linear relationship between Wnt activity levels and apoptosis in colorectal carcinoma cells exposed to butyrate. International Journal of Cancer. 2004;110:523-31
71. Lazarova DL, Chiaro C, Bordonaro M. Butyrate induced changes in Wnt-signaling specific gene expression in colorectal cancer cells. BMC Research Notes. 2014;7:226
72. Liu CX, Zhang SZ, Zhang XW, Huang LH, Tie-Jun LI, Zhang J. et al. Sodium butyrate induces apoptosis and regulates p53 target genes in HT-29 colorectal cancer cells. Chinese Journal of Cancer Biotherapy. 2006;13:185-90
73. Abaza MSI. Augmentation of the anticancer effects of proteasome inhibitors by combination with sodium butyrate in human colorectal cancer cells. Experimental & Therapeutic Medicine. 2010;1:675-93
74. Humphreys KJ, Cobiac L, Le Leu RK, Van der Hoek MB, Michael MZ. Histone deacetylase inhibition in colorectal cancer cells reveals competing roles for members of the oncogenic miR-17-92 cluster. Molecular Carcinogenesis. 2013;52:459-74
75. Hu S, Dong TS, Dalal SR, Wu F, Bissonnette M, Kwon JH. et al. The microbe-derived short chain fatty acid butyrate targets miRNA-dependent p21 gene expression in human colon cancer. PloS one. 2011;6:e16221
76. Schickel R, Boyerinas B, Park SM, Peter ME. MicroRNAs: key players in the immune system, differentiation, tumorigenesis and cell death. Oncogene. 2008;27:5959-74
77. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281-97
78. Slaby O, Svoboda M, Michalek J, Vyzula R. MicroRNAs in colorectal cancer: translation of molecular biology into clinical application. Molecular Cancer. 2009;8:102
79. Ventura A, Jacks T. MicroRNAs and cancer: short RNAs go a long way. Cell. 2009;136:586-91
80. Garzon R, Calin GA, Croce CM. MicroRNAs in Cancer. Annual Review of Medicine. 2009;60:167-79
81. Zhang W, Zou C, Pan L, Xu Y, Qi W, Ma G. et al. MicroRNA-140-5p inhibits the progression of colorectal cancer by targeting VEGFA. Cell Physiology and Biochemistry. 2015;37:1123-33
82. Xu K, Liu X, Mao X, Xue L, Wang R, Chen L. et al. MicroRNA-149 suppresses colorectal cancer cell migration and invasion by directly targeting forkhead box transcription factor FOXM1. Cell Physiology and Biochemistry. 2015;35:499-515
83. Li HT, Zhang H, Chen Y, Liu XF, Qian J. MiR-423-3p enhances cell growth through inhibition of p21Cip1/Waf1 in colorectal cancer. Cell Physiology and Biochemistry. 2015;37:1044-54
84. Fang Y, Sun B, Xiang J, Chen Z. MiR-301a promotes colorectal cancer cell growth and invasion by directly targeting SOCS6. Cell Physiology and Biochemistry. 2015;35:227-36
85. Fu Q, Zhang J, Xu X, Qian F, Feng K, Ma J. miR-203 is a predictive biomarker for colorectal cancer and its expression is associated with BIRC5. Tumor Biology. 2016;37:1-7
86. Zhang F, Yang Z, Cao M, Xu Y, Li J, Chen X. et al. MiR-203 suppresses tumor growth and invasion and down-regulates MiR-21 expression through repressing Ran in esophageal cancer. Cancer letters. 2014;342:121-9
87. Abella V, Valladares M, Rodriguez T, Haz M, Blanco M, Tarrio N. et al. miR-203 regulates cell proliferation through its influence on Hakai expression. PloS one. 2012;7:e52568
88. Tiwari P, Gupta KP. Modulation of miR-203 and its regulators as a function of time during the development of 7, 12 dimethylbenz [a] anthracene induced mouse skin tumors in presence or absence of the antitumor agents. Toxicology and Applied Pharmacology. 2014;278:148-58
89. Han R, Sun Q, Wu J, Zheng P, Zhao G. Sodium Butyrate Upregulates miR-203 Expression to Exert Anti-Proliferation Effect on Colorectal Cancer Cells. Cell Physiology and Biochemistry. 2016;39:1919-29
90. He L, Thomson JM, Hemann MT, Hernandomonge E, Mu D, Goodson S. et al. A microRNA polycistron as a potential human oncogene. Nature. 2005;435:828-33
91. Hu S, Liu L, Chang EB, Wang JY, Raufman JP. Butyrate inhibits pro-proliferative miR-92a by diminishing c-Myc-induced miR-17-92a cluster transcription in human colon cancer cells. Molecular Cancer. 2015;14:180
92. Ota A, Tagawa H, Karnan S, Tsuzuki S, Karpas A, Kira S. et al. Identification and characterization of a novel gene, C13orf25, as a target for 13q31-q32 amplification in malignant lymphoma. Cancer Research. 2004;64:3087-95
93. Zhou T, Zhang G, Liu Z, Xia S, Tian H. Overexpression of miR-92a correlates with tumor metastasis and poor prognosis in patients with colorectal cancer. International Journal of Colorectal Disease. 2013;28:19-24
Author contact
![]() Corresponding author: Zhiping Liu, School of Basic Medicine, Gannan Medical University, Ganzhou, Jiangxi, 341000, China. Tel: (86) 13879793709; Email: Zhiping.Liuedu.cn or Xiangcai Wang, The First-Affiliated Hospital, Gannan Medical University, Ganzhou, Jiangxi, 341000, China. Tel: (86) 13576694980; Email: wangxiangcaiorg.cn
Corresponding author: Zhiping Liu, School of Basic Medicine, Gannan Medical University, Ganzhou, Jiangxi, 341000, China. Tel: (86) 13879793709; Email: Zhiping.Liuedu.cn or Xiangcai Wang, The First-Affiliated Hospital, Gannan Medical University, Ganzhou, Jiangxi, 341000, China. Tel: (86) 13576694980; Email: wangxiangcaiorg.cn