Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(11):1915-1924. doi:10.7150/jca.23648 This issue Cite
Research Paper
Mycophenolate Mofetil induces c-Jun-N-terminal kinase expression in 22Rv1 cells: an impact on androgen receptor signaling
Ondrej Zenata, Zdenek Dvorak, Radim Vrzal ![]()
Department of Cell Biology and Genetics, Faculty of Science, Palacky University in Olomouc, Slechtitelu 27, Olomouc, CZ-783 71, Czech Republic
Received 2017-11-3; Accepted 2018-2-17; Published 2018-4-27
Citation:
Zenata O, Dvorak Z, Vrzal R. Mycophenolate Mofetil induces c-Jun-N-terminal kinase expression in 22Rv1 cells: an impact on androgen receptor signaling. J Cancer 2018; 9(11):1915-1924. doi:10.7150/jca.23648. https://www.jcancer.org/v09p1915.htm
Other stylesAbstract
Mycophenolate Mofetil (MYC) is a transplant drug used to prevent rejection in heart and kidneys transplant patients. Inosine monophosphate dehydrogenase (IMPDH), an enzyme involved in de novo synthesis of guanosine nucleotides, was considered as a primary target for MYC. Recently, we described that MYC was activates aryl hydrocarbon receptor and it antagonizes glucocorticoid receptor. Here we describe an androgen receptor (AR) as another off-target for MYC.
We found that MYC increased basal and dihydrotestosterone (DHT)-inducible AR-dependent luciferase activity in AIZ-AR cells. In the same manner it induced or augmented mRNA level of KLK3 (prostate specific antigen; PSA) in 22Rv1 cells. Herein it displayed a hormetic effect on proliferation activity, since it significantly stimulated proliferation in lower concentrations but inhibited in higher (>1 µg/ml) concentrations in the presence of DHT. In contrast, MYC suppressed DHT-inducible KLK3 mRNA expression and cell proliferation in androgen-dependent LNCaP cells. MYC augmented DHT-inducible nuclear translocation of AR and increased the expression of MAPK8/9 (JNK46/54) resulting in the drop of their phosphorylation status. Moreover, MYC sensitized DHT-treated 22Rv1 cells to JNK-IN-8 mediated growth inhibition with the drop of IC50 from 1425 nM to 84 nM within 24 hrs. In conclusion, we suggest that, castrate-resistant prostate cancers progression might be retarded with the combination of MYC and chemical JNK inhibitors, involving AR-dependent mechanism.
Keywords: 22Rv1, LNCaP, JNK, proliferation, Mycophenolate
Introduction
Mycophenolate Mofetil (MYC) (Figure 1A) is an immunosuppressive drug inhibiting the inosine monophosphate dehydrogenase (IMPDH), the rate limiting enzyme in the de novo synthesis of guanosine nucleotides [1]. It is used in the prevention of rejection of heart and kidney transplants due to the higher affinity for IMPDH II isoform, which is present primarily in lymphocytes. Mycophenolate Mofetil is administered orally and it undergoes rapid absorption and hydrolysis to mycophenolic acid (MPA) by liver and plasma esterases. Thereafter, it is almost completely metabolized to glucuronides by UGT1A9 and UGT2B7 [2]. Intravenous or per oral administration of 500 mg MYC resulted in statistically indistinguishable Cmax around 10 µg/ml within approx. 2 hrs [3]. Thus, these or actually lower systemic concentrations can be expected in patients treated with this immunosuppressant.
While IMPDH inhibition is probably the main target of MYC/MPA, compounds itself probably targets other cellular components. Recently, we found that MYC activated aryl hydrocarbon receptor (AhR) in dose-dependent manner and it antagonized AhR as well as glucocorticoid receptor (GR) activation by appropriate ligands in vitro [4]. Moreover, it was described that several factors involved in cell proliferation or cell cycle regulation are strongly down-regulated upon MPA treatment [5]. As the proliferation is undesirable process of cancer cells and many tumors have up-regulated IMPDH [6, 7], off-target use of this compound for cancer treatment offers itself. In the study by Dun et al. it was found that gastric adenocarcinoma (AGS), gastric carcinoma (NCI-N87), colorectal adenocarcinoma (HCT-8), ovarian adenocarcinoma (A2780) and pancreatic adenocarcinoma (BxPC-3) were highly sensitive to MYC treatment [5]. The use of MYC can be expected for the prostate cancer treatment as another inhibitor of IMPDH II, compound AVN944, was found to induce cell cycle arrest and cell death in 4 prostate cancer cell lines, particularly LNCaP, 22Rv1, DU145 and PC-3 [8]. Based on this and our above mentioned observation of the action of MYC towards ligand-activated transcription factors, the nuclear receptors, we decided to investigate the effect of MYC on the activity of androgen receptor (AR) in recently established AR-responsive cell line (AIZ-AR), which was derived from prostate cancer cell line 22Rv1 [9]. In addition, we compared this androgen-independent (22Rv1) and androgen-dependent (LNCaP) cell lines.
Materials and methods
Compounds and reagents
Dimethylsulfoxide (DMSO), dihydrotestosterone (DHT), Mycophenolate Mofetil (MYC), Proliferation assay kit (5-Bromo-2′-deoxy-uridine Labeling and Detection Kit III) and Charcoal-stripped Fetal Bovine Serum (CS-FBS; F6765) were purchased from Sigma-Aldrich (Prague, Czech Republic). Oligonucleotide primers used in RT-PCR reactions were synthesized by Generi Biotech (Hradec Kralove, Czech Republic). LightCycler 480 Probes Master was from Roche Diagnostic Corporation (Intes Bohemia, Czech Republic). All other chemicals were of the highest quality commercially available.
Cell Cultures
Human Caucasian prostate carcinoma (LNCaP; No. 89110211) and human prostate carcinoma epithelial cell line (22Rv1; No. 05092802) were purchased from Public Health England and cultured in RPMI 1640 medium supplemented with 10% of charcoal-stripped fetal bovine serum, 2 mM L-glutamine, 1% non-essential amino acids, and 1 mM sodium pyruvate. Cells were maintained at 37°C and 5% CO2 in a humidified incubator.
Figure 1
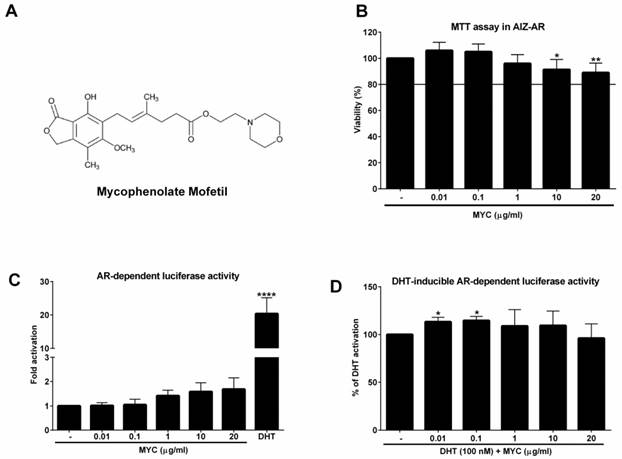
The viability of AIZ-AR cells and the activation of androgen receptor (AR) The structure of Mycophenolate Mofetil (MYC). (B) AIZ-AR cells were treated incubated with Mycophenolate Mofetil (MYC; 0.01-20 µg/ml) and/or DMSO (0.1% v/v) as a vehicle for control for 24 h. Thereafter, MTT assay was performed as described in Materials and Methods section. The data are the mean ± SD from 5 consecutive cell passages and are expressed as % of negative control (DMSO). AIZ-AR cells were incubated with Mycophenolate Mofetil (MYC; 0.01-20 µg/ml) in the absence (C) or presence (D) of DHT (100 nM) and/or DMSO (0.1% v/v) for 24 h. Thereafter, gene reporter assay was performed as described in Materials and Methods section. The data are the mean ± SD from 5 (C) or 7 (D) consecutive cell passages independent experiments and are expressed as fold activation of AR over DMSO-treated cells (C) or as a % of positive control (DHT) (D). *, **, **** - value is significantly different from untreated cells (DMSO) (C) or DHT-treated cells (D) (p < 0.05, 0.01, 0.0001).
Cell viability assay (MTT)
Cell line AIZ-AR was treated with increasing concentrations of MYC (0.01-20 µg/ml) and/or DMSO (0.1%; v/v) for 24 hrs. Thereafter, the medium was replaced by PBS with MTT (MTT = 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) in final concentration of 0.3 mg/ml. The solution was discarded after 30-40 minutes of incubation and replaced by DMSO for dissolution of the formazan crystals. Absorbance was measured at 570 nm with Infinite M200 (TECAN, Austria). Tested concentrations causing decline in viability no greater than 20% were considered as non-toxic for furthers experiments.
Gene reporter assay
For the assessment of AR transcriptional activity, we employed recently established stably transfected gene reporter cell line AIZ-AR (derived from 22Rv1 cells) [9]. Following the plating in medium with CS-FBS, cells were stabilized for 16 h and then treated with MYC (0.01-20 µg/ml) in the absence (Agonist setting) or presence (Antagonist setting) of dihydrotestosterone (DHT; 100 nM) and/or vehicle (DMSO; 0.1% v/v) for 24 hrs in medium with CS-FBS. After the treatments, cells were lysed and luciferase activity was measured with Infinite M200 (TECAN, Austria).
Quantitative reverse transcriptase polymerase chain reaction (qRT-PCR)
The total RNA was isolated using TRI Reagent® (Molecular Research Center, USA). cDNA was synthesized from 1000 ng of total RNA using M-MuLV Reverse Transcriptase (M0253S, New England BioLabs) at 42 °C for 60 min in the presence of random hexamers (S1230, New England BioLabs). qRT-PCR was carried out on Light Cycler 480 II apparatus (Roche Diagnostic Corporation, Prague, Czech Republic). The levels of KLK3 and GAPDH mRNAs were determined using primers and probes from Universal Probes Library (UPL; Roche Diagnostic Corporation, Prague, Czech Republic) as follows: KLK3 (PSA) - UPL 44, forward: GTGCTTGTGGCCTCTCGT, reverse: CAGCAAGATCACGCTTTTGT; GAPDH - UPL 60, forward: CTCTGCTCCTCCTGTTCGAC, reverse: ACGACCAAATCCGTTGACTC. The following program was used for monitoring the expression of both genes: an activation step at 95 °C for 10 min was followed by 45 cycles of PCR (denaturation at 95 °C for 10 s; annealing with elongation at 60 °C for 30 s). The measurements were performed in triplicates. Gene expression was normalized per glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as a housekeeping gene. Data were processed with the delta-delta method. Results are expressed as fold induction over DMSO- treated cells.
Proliferation assay
Proliferation assay was used according to the manufacturer recommendations with minor modifications. Briefly, cells were seeded at the density of 10000 cells per well into 96-well plate (pre-coated with poly-D-lysine for LNCaP) in medium with CS-FBS and stabilized overnight. Next day, the cells were treated with dihydrotestosterone (DHT) alone or together with increasing concentrations of Mycophenolate Mofetil (0.01, 0.1, 1, 10, 20 µg/ml) for 24 hrs. At the end of treatment, solution with 5-Bromo-2′-deoxy-uridine (BrDu) was applied directly into the well for additional 3 hrs. Thereafter, we followed strictly the procedure from manufacturer guide. At the end, the absorbance at 405 nm and reference at 495 nm were measured with Infinite M200Pro (Tecan, Austria). The proliferation capacity was expressed as % of the ratios of absorbances A405/A495 between the sample and DHT, i.e. DHT was set to 100%.
SDS-PAGE and Western blotting
Total protein extracts were prepared from 1 well of 6-well plate dish. Cells were washed once with ice-cold PBS and scraped into 1 ml of PBS. The suspension was centrifuged (2,300x g/5 min/4°C) and the pellet was resuspended in 150 μl of ice-cold lysis buffer (150 mM NaCl; 10 mM Tris pH 7.2; anti-protease and anti-phosphatase cocktails, 1% (v/v) Triton X-100; 1% (v/v) sodium deoxycholate; 5 mM EDTA). The mixture was vortexed and incubated for 10 min on ice and then centrifuged (15,700x g/13 min/4°C). Supernatant was collected and the protein content was determined by the Bradford reagent.
Isolation of cytosolic and nuclear extract was performed with the use of Nuclear extract kit (Active Motif; 40010) according to the manufacturer's protocol.
SDS-PAGE gels (10%) were run on a BioRad apparatus according to the general procedure followed by the protein transfer onto PVDF membrane. The membrane was saturated with 5% non-fat dried milk for 1 h at room temperature in 1x TBS-Tween20 (0.5%). Blots were probed with primary antibodies against AR (mouse monoclonal, sc-7305, 441, diluted 1:500, lot H3112) and actin (mouse monoclonal, sc-47778, C4, lot K1414), both purchased from Santa Cruz Biotechnology and with p-JNK (rabbit polyclonal,9251, lot 3, diluted 1:1000), JNK (rabbit polyclonal; 9252, lot 9, diluted 1:1000), both purchased from Cell Signaling Technology, overnight at 4°C. Chemiluminescence detection was performed using horseradish peroxidase-conjugated secondary antibodies (Santa Cruz Biotechnology) and Western Sure Premium Chemiluminescent Substrate (C60429, LiCor). Densitometric analyses were performed with the C-DiGit Chemiluminescent Western Blot Scanner and Image Studio Digits software, both from LiCor.
Statistical Analysis
Differences between the groups were compared using the Student's unpaired two-tailed t-test. Two-way analysis of variance with a Dunnett's post hoc test was applied to the data if more than two groups were analysed. All of the statistical analyses were performed using GRAPH-PAD PRISM 6 software (GraphPad Software Inc., San Diego, CA, USA). A P-value of <0.05 was considered to be statistically significant.
Results
Mycophenolate Mofetil activates androgen receptor
As a first step, we investigated if MYC can activate androgen receptor in recently developed AR-responsive cell line [9]. To this purpose, we had to determine the non-toxic concentrations of MYC in this cell line. We chose the range which corresponds to plasma concentrations [3] with the maximal concentration set to 20 µg/ml (approx. 62 µM). Since the viability of cells was not decreased by more than 10% at highest concentrations (Figure 1B) we could use full range of concentrations for further experiments. In the Agonist setting, the positive control DHT (100 nM) induced the androgen receptor (AR)-dependent luciferase activity (18-fold) which served as a proof of the functionality of the system. MYC induced the activity as well (1.4-1.7 fold), however insignificantly (Figure 1C). In the Antagonist setting, i.e. in the presence of DHT, we observed significant approx. 13% increase of luciferase activity above DHT alone for 0.01 and 0.1 µg/ml concentrations (Figure 1D). All other concentrations were without significant effect.
Mycophenolate Mofetil modulates KLK3 expression and proliferation of prostate cells
The finding of increased AR-dependent luciferase activity led us to focus on the expression of one of the most known AR target genes, KLK3 (prostate specific antigen, PSA). First, we monitored the expression of KLK3 mRNA in 22Rv1, which the AIZ-AR cell line was derived from. The positive control (DHT) induced KLK3 mRNA significantly (Figure 2A) but the average fold induction did not exceed 2-fold. Mycophenolate mofetil induced significantly KLK3 mRNA to approx. 1.5-fold for 3 highest concentrations (Figure 2A). This induction corresponds to almost 65% of induction caused by DHT. An interesting and consistent observation with gene reporter assay (Figure 1D) we made when we co-treated 22Rv1 cells with MYC and DHT. The presence of MYC significantly stimulated the KLK3 mRNA induction over DHT alone by almost 25 % for MYC concentration of 1 µg/ml (Figure 2B). Since the cell cycle of prostate cancer cells is modulated by AR, we further focused on monitoring the proliferative activity of 22Rv1 cells. We observed that concentrations 0.01 and 0.1 µg/ml significantly stimulated proliferation of cells but higher concentrations had the inhibitory effect (Figure 2C). While two lowest concentrations reflected the pattern observed in DHT-inducible KLK3 mRNA expression, the other three (starting from 1 µg/ml) are probably high enough to inhibit the primary target of MYC, inosine monophosphate dehydrogenase (IMPDH) with consequent impact on short-lived proteins involved in cell cycle, i.e. the proliferation. Since two lowest concentrations stimulated proliferation above control, we further monitored this effect for longer period of time, 48 hrs. However, no effect was observed for these two concentrations (Figure 2C).
It is known that 22Rv1 cells have two forms of AR, one being full length (AR-FL) and second one truncated missing ligand-binding domain (AR-v7). Therefore, we decided to compare AR-independent cells (22Rv1) with AR-dependent cells (LNCaP), which lack AR-v7 isoform. Positive control induced KLK3 mRNA in average 15-fold (Figure 2D), while MYC slightly but insignificantly induced KLK3 1.4-fold over control cells. In the presence of DHT, we observed opposite effect on inducible KLK3 mRNA in contrast to 22Rv1 cells, i.e. the decrease in mRNA level (Figure 2E). Monitoring the proliferation of LNCaP cells revealed a pattern similar with the inducible KLK3 mRNA. The comparison of these two cell lines revealed lower anti-proliferative activity of MYC in LNCaP cells (IC50 > 20 µg/ml) than in 22Rv1 cells (IC50 ~ 1.0 µg/ml) (Figure 2F, C).
Mycophenolate mofetil stimulates DHT-dependent AR translocation in 22Rv1 cells
Since MYC stimulated DHT-mediated AR-dependent luciferase activity (Figure 1D) as well as KLK3 mRNA induction (Figure 2B), we decided to further investigate this effect. We monitored AR translocation in the presence of MYC. We used MYC at concentration of 1 µg/ml as this was the lowest threshold concentration causing any effect in our previous assays. We found that the presence of MYC stimulated DHT-induced translocation of full length version of AR (AR-FL) into the nucleus after 60 minutes (Figure 3A, B). Moreover, MYC also reduced nuclear level of truncated form of AR (AR-v7) as well (Figure 3A, C). Since AR-FL/AR-v7 isoforms were slightly increased in cytosolic fraction upon MYC treatment (Figure 3A, D) with simultaneous decrease in nucleus (Figure 3A, C), we considered an idea of MYC-stimulated degradation of AR isoforms by proteasome. However, co-treatment of MYC with proteasome inhibitor bortezomib had no effect on protein level of any AR isoforms, suggesting no involvement of proteasome in this process (Figures 3E, F).
Mycophenolate Mofetil induces JNK expression
We based our further investigation on our recent publication [4]. Here we found that MYC activated and inhibited AhR activity in the presence of dioxin. Moreover, activity of GR was inhibited concentration-dependently by MYC in the presence of glucocorticoid receptor ligand, dexamethasone. Since the likelihood of one compound being a ligand of three different receptors with limited overlapping spectra of ligands is low in general, we focused on the idea of ligand-independent co-stimulation of AR activity by kinase event. We focused on c-Jun N-terminal kinase (JNK) as similar observation for MYC and JNK was already published [10, 11]. Treatment of 22Rv1 cells with MYC for 30 minutes revealed mild reproducible decrease of phospho status of JNKs while total JNKs were elevated (Figure 4A). Sorbitol, a JNK activator significantly induced the phosphorylation while had no effect on total JNK protein level. This finding suggested that MYC suppressed basal JNK activity by increasing total level of JNK. In order to investigate the origin of this effect, we focused on determination of either stabilization or induction of JNKs. An experiment with proteosynthesis inhibitor, cycloheximide (CHX), revealed that CHX significantly suppressed MYC-induced increase of JNKs. This suggested transcriptional event. Monitoring the expression at mRNA level revealed mild but significant induction of expression of JNK54 (MAPK9) and JNK46 (MAPK9) (Figure 4C). To further confirm that either activation or inhibition of JNK affects transcription activity of AR, we treated AIZ-AR cells with JNK activator sorbitol or JNK inhibitors (SP600125 and JNK-IN-8). Sorbitol dose-dependently increased while both tested inhibitors decreased the AR-dependent luciferase activity (Figure 4D). It is worth to notice that lower concentrations of JNK inhibitors had mild co-stimulating effect on AR-mediated luciferase activity similarly like MYC (Figure 1D).
Figure 2
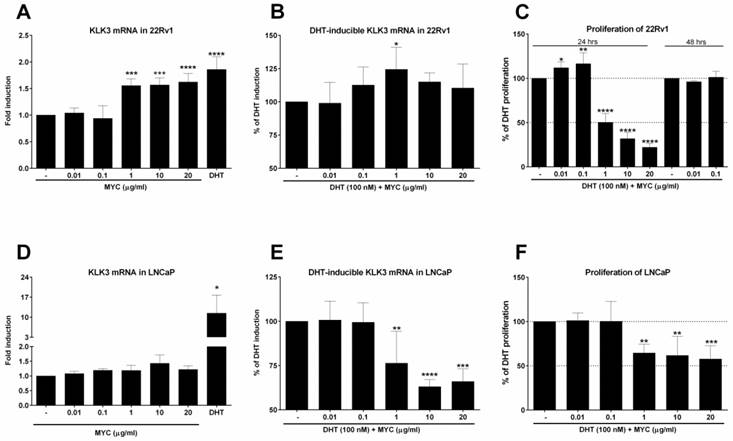
The effect of Mycophenolate Mofetil on KLK3 mRNA and cellular proliferation 22Rv1 or LNCaP cells were treated for 24 h with Mycophenolate Mofetil (MYC; 0.01-20 µg/ml) in the absence (A, D) or presence (B, E) of DHT (100 nM) and/or DMSO as a vehicle for negative control. Thereafter KLK3 mRNA was determined as described in Materials and Methods section. Results are expressed as fold induction over DMSO-treated cells ± SD (A) or as % of DHT induction ± SD (B). The data are mean from 5 consecutive cell passages and were normalized per GAPDH mRNA levels. *, **, ***, **** - value is significantly different from DMSO-treated cells (A, D) or DHT-treated cells (B, E) (p < 0.05, 0.01, 0.001, 0.0001). (C) 22Rv1 cells were treated in the presence of DHT (100 nM) for 24 hrs with MYC (0.01-20 µg/ml) or for 48 hrs with MYC (0.01, 0.1 µg/ml). (F) LNCaP cells were treated in the presence of DHT (100 nM) with MYC (0.01-20 µg/ml) for 24 hrs. Thereafter, detection of proliferation was measured as described in Materials and Methods section. Results are expressed as % of DHT proliferation from 4 (C) and 5 (F) consecutive passages. *, **, ***, **** - value is significantly different from DHT-treated cells (p < 0.05, 0.01, 0.001, 0.0001).
Figure 3
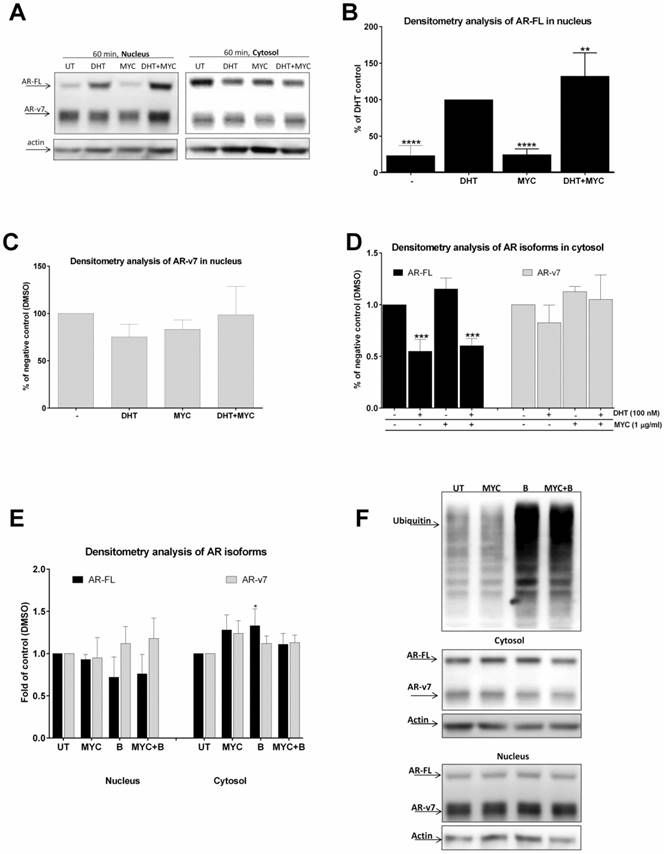
The effect of MYC on translocation of AR (A-D) 22Rv1 cells were treated incubated with Mycophenolate Mofetil (MYC; 1 µg/ml) and/or DMSO (0.1% v/v) and/or DHT (100 nM) for 60 minutes. Thereafter, cytosolic and nuclear fractions were isolated as described in Materials and Methods section; semi-quantitation was performed with LiCOR scanner. (A) One representative western blot of 5 consecutive passages is shown. (E) 22Rv1 cells were treated incubated with Mycophenolate Mofetil (MYC; 1 µg/ml) and/or DMSO (0.1% v/v) and/or Bortezomib (B; 100 nM) for 60 minutes. Thereafter, cytosolic and nuclear fractions were isolated as described in Materials and Methods section; semi-quantitation was performed with LiCOR scanner. (F) One representative western blot of 3 independent experiments is shown. *, **, ***, **** - value is significantly different from DHT-treated cells (B) or DMSO-treated cells (D, E) (p < 0.05, 0.01, 0.001, 0.0001).
Mycophenolate Mofetil has synergistic antiproliferative effect with JNK inhibitor
Since MYC induced JNK protein, we focused on idea of MYC-induced sensitization of 22Rv1 cells with consequent higher impact on viability of these cells when co-treated with JNK inhibitors. By employing MTT assay, we observed nothing but additive effect of tested concentrations of MYC in the presence of JNK inhibitor, JNK-IN-8 (Figure 5A). Similar data we obtained with reversible JNK inhibitor SP600125 (data not shown). However, we have noticed that presence of DHT had protective effect as increased viability was observed in DHT-plus rather than DHT-free medium (data not shown). However, this protective effect of DHT was observed for JNK activator, sorbitol, as well (data not shown). This suggests that no matter of JNK activation or inhibition, DHT has protective effect on viability of 22Rv1 cells.
In the next step, we monitored the proliferation of 22Rv1 cells co-treated with MYC and JNK inhibitor, JNK-IN-8 [12]. Proliferation of 22Rv1 cells was concentration-dependently inhibited by JNK-IN-8 with observed IC50 approx. 1425 nM (Figure 5B). When we combined JNK-IN-8 with MYC, the presence of two lowest concentrations (0.01 and 0.1 µg/ml) had no effect on the proliferation (data not shown) similarly like on the viability (Figure 5A). However, the presence of MYC at 1 µg/ml shifted the proliferation curve to lower values with the drop of IC50 to approx. 84 nM within 24 hrs (Figure 5B). That represents a 17-fold lower IC50 than for JNK inhibitor alone.
Figure 4
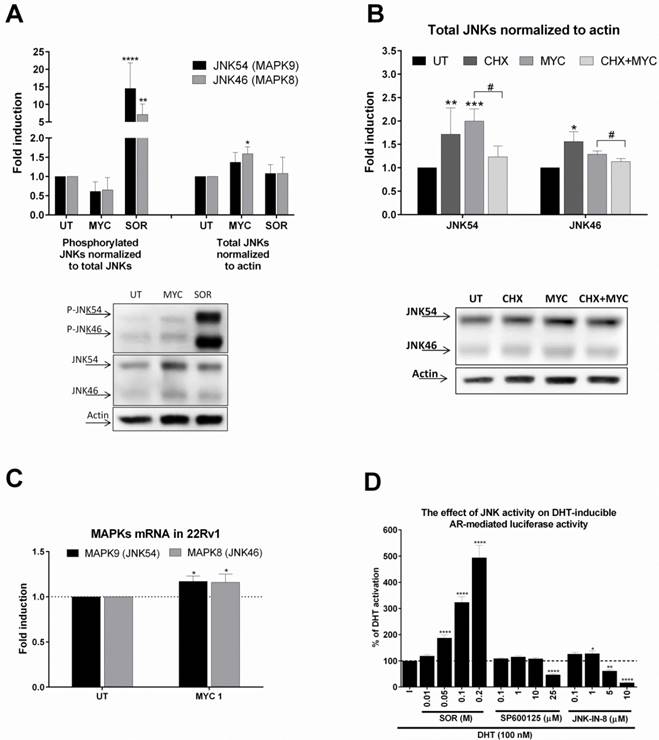
The effect of MYC on JNKs status (A) 22Rv1 cells were incubated with Mycophenolate Mofetil (MYC; 1 µg/ml) and/or DMSO (0.1% v/v) or Sorbitol (SOR; 0.4 M) for 30 minutes. Thereafter, total protein lysate was isolated as described in Materials and Methods section with subsequent protein detection and semiquantitation performed with LiCOR scanner. Phosphorylated JNKs normalized to total JNKs protein (left side of A) and total JNKs normalized to actin (right side of A). One representative western blot of 6 independent experiments is shown in lower part of A. (B) 22Rv1 cells were incubated with Mycophenolate Mofetil (MYC; 1 µg/ml), cycloheximide (CHX; 20 µg/ml) and/or DMSO (0.1% v/v) or combination of both for 30 minutes. Thereafter, total protein lysate was isolated as described in Materials and Methods section with subsequent protein detection and semi-quantitation performed with LiCOR scanner. Total JNKs were normalized to actin and data are expressed as fold induction over negative control (DMSO). One representative western blot of 4 independent experiments is shown in lower part of B. (D) AIZ-AR cell line was treated with DHT (100 nM) in the presence or absence of Sorbitol (0.01-0.2 M), SP600125 (0.1-25 µM), JNK-IN-8 (0.1-10 µM) for 24 hrs. Thereafter luciferase activity was monitored as described in Materials and Methods section. *, **, ***, **** - value is significantly different from DMSO-treated cells (A,B,C) or DHT-treated cells (D) (p < 0.05, 0.01, 0.001, 0.0001). # - significant difference between two groups (p<0.05)
Figure 5
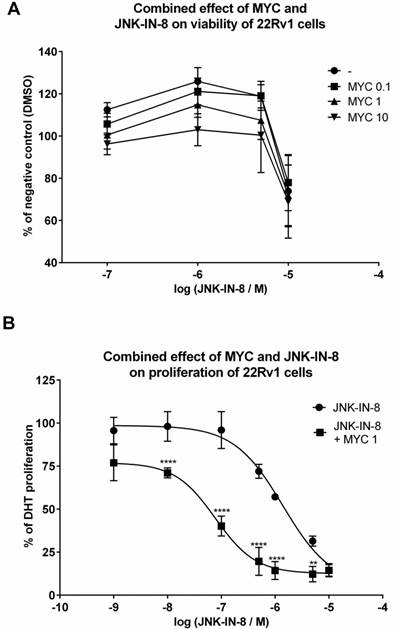
The effect of MYC and JNK inhibitor co-treatment on viability and proliferation 22Rv1 cells were treated JNK-IN-8 (0.001-10 µM) and/or with Mycophenolate Mofetil (MYC; 1 µg/ml) and/or DMSO (0.1% v/v) in the presence of DHT for 24 hrs. Thereafter, viability (A) or proliferation (B) were performed as described in Materials and Methods section. Results are expressed as % of viability for DMSO (A) or % of DHT proliferation (B) from 3 consecutive passages. **, **** - value is significantly different from JNK-IN-8-treated cells only (p < 0.01, 0.0001).
Discussion
In the current study, we investigated the effect of immunosuppressive drug, Mycophenolate Mofetil (MYC) on the activity of androgen receptor (AR) in prostate cancer cell lines. We found that MYC slightly activated androgen receptor in androgen-independent cells 22Rv1 and induced AR-target gene expression (KLK3 mRNA) herein while this was not observed in AR-dependent cells (LNCaP). The combination of MYC and DHT synergistically stimulated KLK3 mRNA induction in 22Rv1 cells while the opposite, i.e. the suppression was observed in LNCaP cells. Mycophenolate had anti-proliferative effect in both cell lines with IC50s more than 20-times lower for androgen-independent (22Rv1) than dependent (LNCaP) cells. This finding shows androgen-independent (but AR positive) cells as more sensitive to anti-proliferative effect of Mycophenolate Mofetil. Moreover, we found that MYC stimulated DHT-inducible translocation of AR-FL to nucleus and induced MAPK8/9 (JNK46/54) expression, which resulted in the decrease of JNKs' phosphorylation status. The decline of phosphorylation is consistent with the study performed in proximal tubule cell line (HK-2) [10]. In that report, the inhibition of hydrogen peroxide-induced phosphorylation of JNK by MYC was described in the concentration range between 50-500µM (approx. 16-160 µg/mL).
The primary molecular target of MYC was always considered IMPDH, particularly isoform II for which mycophenolic acid (active form of MYC) is 5-fold more potent inhibitor than for isoform I [1]. However, our recent study suggested that this compound can affect other molecular targets like ligand-activated transcription factors (AhR, GR) [4]. In stably transfected HeLa cells responsive for glucocorticoid receptor, we observed strong antagonistic behavior of MYC in the presence of ligand dexamethasone. In addition, we observed dose-dependent activation of AhR and synergistic action with AhR ligand dioxin in hepatocellular carcinoma HepG2 cells. However, probably due to IMPDH inhibition at higher concentrations, the dose-response curves had reversed-U shape profile [4]. More or less the same as for AhR, we can now see for DHT-stimulated AR activation as well as for KLK3 mRNA expression in 22Rv1 cells, an in vitro model of castrate-resistant prostate cancer (CRPC). The reason why KLK3 mRNA goes down at higher concentrations of MYC is likely due to the insufficient number of nucleotides as IMPDH inhibition takes place.
There may be many reasons for different behavior of LNCaP and 22Rv1 cells upon MYC treatment. One among many would be the different expression level of IMPDH II. Different profiles of inducible KLK3 mRNA expression level might suggest such a scenario. If there was higher level of IMPDH II in 22Rv1 than in LNCaP cells, then the decline in KLK3 mRNA would be faster for LNCaP cells than 22Rv1 cells. However, this scenario is out of question as equal levels of IMPDH II were detected in LNCaP as well as 22Rv1 cells [8].
Among other plausible mechanisms of MYC action belongs the ability to change the expression of adhesion molecules. There are several studies which investigated this phenomenon. One of the first studies revealed MYC-triggered downregulation of E-selectin and P-selectin expression on endothelial cell membranes [13]. It also significantly inhibited lipopolysaccharide/interferon-gamma-stimulated expression of intercellular adhesion molecule (ICAM-1) and MHC-II on monocytes [14]. Another important adhesion molecule NCAM (neural cell adhesion molecule) was decreased in the presence of MYC in neuroblastoma cells [15]. This suggested an increased risk of tumor metastasis since several studies revealed an inverse correlation between NCAM expression and tumor invasiveness [16-18]. Surprisingly, treatment of human colon adenocarcinoma cell line WiDr with MYC prevented the attachment to E-selection and blocked binding to endothelium [19]. In DU-145 prostate cancer cell line, MYC blocked dose-dependently (0.1-1µM ~0.04-0.4 µg/ml) adhesion of these cells in monolayer adhesion assay probably via increased expression of beta1 integrins like CD49a, CD49b, CD49e [20]. This study indicated that MYC possessed anti-tumor activity particularly to prostate cells. The disturbance of other cell machinery components was already described in prostate cancer cells after incubation with another IMPDH inhibitor, AVN944 [8]. Here, more pronounced induction of pro-apoptotic Bok, cyclin E and decrease of cyclin A proteins, was observed after 2 days at 5 µM of AVN944 in 22Rv1 cells than in LNCaP cells. While this may be partially addressed to rapid decline in the expression of some key molecules with short half-life in the cells due to the IMPDH inhibition, other proteins are increased as a reaction on the presence of MYC in order to deal with the IMPDH inhibition.
Based on recent study by Dun et al., it is clear that MYC affects many cellular pathways either by the change in expression or phosphorylation status [5]. A dramatic reduction of phosphorylated PI3K, AKT and mTOR were observed in AGS (human Caucasian gastric adenocarcinoma) cell line in vitro. Moreover, many cellular regulators like CDK4, BUB1 or Aurora A were reduced as well. Consistently, mice treated with MYC and bearing AGS xenografts had significantly smaller tumors [5]. These data provide a proof of beneficial effect of MYC as anticancer drug apparently without adverse immunosuppressive effect, which would be expected for immunosuppressant. This also suggests that monitoring the IMPDH expression level in tumors might be used as a prognostic marker for justification the repurposing use of Mycophenolate Mofetil for treatment of cancer patients, particularly those with prostate cancer. Since there is a great knowledge about this compound, no new clinical trials must be performed in order to assess the safety of this drug. Moreover, the retrospective studies which monitored the treatment of patients with MYC found usually no association with increased risk of post-transplant malignancies in registry or multi-center studies [21, 22]. In addition, there was a trend to lower incidence/risk of malignancies.
In our study, we show that MYC has anti-proliferative effect on prostate cancer cells 22Rv1 and LNCaP. However, the second one seems to be more resistant than the first one. In accordance with criteria by Dun et al., we may consider LNCaP as very resistant (IC50 > 20 µg/ml) and 22Rv1 as intermediately sensitive (0.5 µg/ml < IC50 < 20 µg/ml). This is quite surprising, as cell line (22Rv1) regularly used as in vitro model of CRPC should display quite opposite behavior, i.e. the resistance, as pure logic suggests. Since DHT-inducible KLK3 mRNA profile was similar with that of proliferation, we may suggest that AR-v7 variant is an important factor standing beyond our observations in 22Rv1 cells. This variant was demonstrated to be present commonly in CRPC samples [23, 24] and it is considered as emerging mechanism by which prostate cancer escapes. However, this truncated variant might be also the Achilles heel similarly like it seems to be the greatest advantage of CRPC. One thing which is relatively intensively studied towards AR-v7 is the regulation of its expression. Recent paper revealed that multiple kinases or phosphatases may affect the expression level of AR-v7 [25]. This is extremely important as new therapeutic strategies against CRPC may arise. Moreover, this also emphasizes that inhibition of AR-v7 activity rather than AR-FL may be more important for defeating CRCP. We contribute with this work to this general phenomenon, where alternative pathways should be inhibited in order to slow down cancer progression. It is clear that in order to defeat CRCP, one compound won't be enough. However, right combination of some promising drugs, like galeterone [26] or INK-128 [27] with Mycophenolate Mofetil might represent a viable strategy against CRCP.
In conclusion, we demonstrate that well-known immunosuppressant Mycophenolate Mofetil affects androgen receptor-mediated signaling in vitro and this differs between two types of commonly used prostate cancer cell models. Together with many other studies revealing general change in expression of adhesion molecules or components of cell cycle machinery by MYC, we suggest to consider Mycophenolate Mofetil as a good candidate that may be repurposed for anticancer therapy, particularly for prostate cancer patients.
Acknowledgements
This work was supported by the grant from the Czech Science Foundation P303/12/G163.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Carr SF, Papp E, Wu JC, Natsumeda Y. Characterization of human type I and type II IMP dehydrogenases. The Journal of biological chemistry. 1993;268:27286-90
2. Picard N, Ratanasavanh D, Premaud A, Le Meur Y, Marquet P. Identification of the UDP-glucuronosyltransferase isoforms involved in mycophenolic acid phase II metabolism. Drug metabolism and disposition: the biological fate of chemicals. 2005;33:139-46
3. Pescovitz MD, Conti D, Dunn J, Gonwa T, Halloran P, Sollinger H. et al. Intravenous mycophenolate mofetil: safety, tolerability, and pharmacokinetics. Clinical transplantation. 2000;14:179-88
4. Vrzal R, Zenata O, Bachleda P, Dvorak Z. The effects of drugs with immunosuppressive or immunomodulatory activities on xenobiotics-metabolizing enzymes expression in primary human hepatocytes. Toxicology in vitro: an international journal published in association with BIBRA. 2015;29:1088-99
5. Dun B, Xu H, Sharma A, Liu H, Yu H, Yi B. et al. Delineation of biological and molecular mechanisms underlying the diverse anticancer activities of mycophenolic acid. International journal of clinical and experimental pathology. 2013;6:2880-6
6. Fellenberg J, Bernd L, Delling G, Witte D, Zahlten-Hinguranage A. Prognostic significance of drug-regulated genes in high-grade osteosarcoma. Modern pathology: an official journal of the United States and Canadian Academy of Pathology, Inc. 2007;20:1085-94
7. Fellenberg J, Kunz P, Sahr H, Depeweg D. Overexpression of inosine 5'-monophosphate dehydrogenase type II mediates chemoresistance to human osteosarcoma cells. PloS one. 2010;5:e12179
8. Floryk D, Thompson TC. Antiproliferative effects of AVN944, a novel inosine 5-monophosphate dehydrogenase inhibitor, in prostate cancer cells. International journal of cancer Journal international du cancer. 2008;123:2294-302
9. Bartonkova I, Novotna A, Dvorak Z. Novel stably transfected human reporter cell line AIZ-AR as a tool for an assessment of human androgen receptor transcriptional activity. PloS one. 2015;10:e0121316
10. Andreucci M, Faga T, Lucisano G, Uccello F, Pisani A, Memoli B. et al. Mycophenolic acid inhibits the phosphorylation of NF-kappaB and JNKs and causes a decrease in IL-8 release in H2O2-treated human renal proximal tubular cells. Chemico-biological interactions. 2010;185:253-62
11. Olejarz W, Bryk D, Zapolska-Downar D, Malecki M, Stachurska A, Sitkiewicz D. Mycophenolic acid attenuates the tumour necrosis factor-alpha-mediated proinflammatory response in endothelial cells by blocking the MAPK/NF-kappaB and ROS pathways. European journal of clinical investigation. 2014;44:54-64
12. Zhang T, Inesta-Vaquera F, Niepel M, Zhang J, Ficarro SB, Machleidt T. et al. Discovery of potent and selective covalent inhibitors of JNK. Chemistry & biology. 2012;19:140-54
13. Blaheta RA, Leckel K, Wittig B, Zenker D, Oppermann E, Harder S. et al. Inhibition of endothelial receptor expression and of T-cell ligand activity by mycophenolate mofetil. Transplant immunology. 1998;6:251-9
14. Glomsda BA, Blaheta RA, Hailer NP. Inhibition of monocyte/endothelial cell interactions and monocyte adhesion molecule expression by the immunosuppressant mycophenolate mofetil. Spinal cord. 2003;41:610-9
15. Blaheta RA, Bogossian H, Beecken WD, Jonas D, Hasenberg C, Makarevic J. et al. Mycophenolate mofetil increases adhesion capacity of tumor cells in vitro. Transplantation. 2003;76:1735-41
16. Owens GC, Orr EA, DeMasters BK, Muschel RJ, Berens ME, Kruse CA. Overexpression of a transmembrane isoform of neural cell adhesion molecule alters the invasiveness of rat CNS-1 glioma. Cancer Res. 1998;58:2020-8
17. Edvardsen K, Pedersen PH, Bjerkvig R, Hermann GG, Zeuthen J, Laerum OD. et al. Transfection of glioma cells with the neural-cell adhesion molecule NCAM: effect on glioma-cell invasion and growth in vivo. International journal of cancer Journal international du cancer. 1994;58:116-22
18. Blaheta RA, Hundemer M, Mayer G, Vogel JU, Kornhuber B, Cinatl J. et al. Expression level of neural cell adhesion molecule (NCAM) inversely correlates with the ability of neuroblastoma cells to adhere to endothelium in vitro. Cell communication & adhesion. 2002;9:131-47
19. Leckel K, Beecken WD, Jonas D, Oppermann E, Coman MC, Beck KF. et al. The immunosuppressive drug mycophenolate mofetil impairs the adhesion capacity of gastrointestinal tumour cells. Clinical and experimental immunology. 2003;134:238-45
20. Engl T, Makarevic J, Relja B, Natsheh I, Muller I, Beecken WD. et al. Mycophenolate mofetil modulates adhesion receptors of the beta1 integrin family on tumor cells: impact on tumor recurrence and malignancy. BMC cancer. 2005;5:4
21. David KM, Morris JA, Steffen BJ, Chi-Burris KS, Gotz VP, Gordon RD. Mycophenolate mofetil vs. azathioprine is associated with decreased acute rejection, late acute rejection, and risk for cardiovascular death in renal transplant recipients with pre-transplant diabetes. Clinical transplantation. 2005;19:279-85
22. Robson R, Cecka JM, Opelz G, Budde M, Sacks S. Prospective registry-based observational cohort study of the long-term risk of malignancies in renal transplant patients treated with mycophenolate mofetil. American journal of transplantation: official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2005;5:2954-60
23. Hu R, Dunn TA, Wei S, Isharwal S, Veltri RW, Humphreys E. et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 2009;69:16-22
24. Guo Z, Yang X, Sun F, Jiang R, Linn DE, Chen H. et al. A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res. 2009;69:2305-13
25. Li Y, Xie N, Gleave ME, Rennie PS, Dong X. AR-v7 protein expression is regulated by protein kinase and phosphatase. Oncotarget. 2015;6:33743-54
26. Kwegyir-Afful AK, Ramalingam S, Purushottamachar P, Ramamurthy VP, Njar VC. Galeterone and VNPT55 induce proteasomal degradation of AR/AR-V7, induce significant apoptosis via cytochrome c release and suppress growth of castration resistant prostate cancer xenografts in vivo. Oncotarget. 2015;6:27440-60
27. Jiang SJ, Wang S. Dual targeting of mTORC1 and mTORC2 by INK-128 potently inhibits human prostate cancer cell growth in vitro and in vivo. Tumour biology: the journal of the International Society for Oncodevelopmental Biology and Medicine. 2015;36:8177-84
Author contact
![]() Corresponding author: Email: radim.vrzalcz (RV). Department of Cell Biology and Genetic, Faculty of Science, Palacky University Olomouc, Slechtitelu 27, 783 71 Olomouc, Czech Republic. Tel.: +420-58-5634904; Fax.: +420-58-5634901
Corresponding author: Email: radim.vrzalcz (RV). Department of Cell Biology and Genetic, Faculty of Science, Palacky University Olomouc, Slechtitelu 27, 783 71 Olomouc, Czech Republic. Tel.: +420-58-5634904; Fax.: +420-58-5634901
Citation styles
APA
Zenata, O., Dvorak, Z., Vrzal, R. (2018). Mycophenolate Mofetil induces c-Jun-N-terminal kinase expression in 22Rv1 cells: an impact on androgen receptor signaling. Journal of Cancer, 9(11), 1915-1924. https://doi.org/10.7150/jca.23648.
ACS
Zenata, O.; Dvorak, Z.; Vrzal, R. Mycophenolate Mofetil induces c-Jun-N-terminal kinase expression in 22Rv1 cells: an impact on androgen receptor signaling. J. Cancer 2018, 9 (11), 1915-1924. DOI: 10.7150/jca.23648.
NLM
Zenata O, Dvorak Z, Vrzal R. Mycophenolate Mofetil induces c-Jun-N-terminal kinase expression in 22Rv1 cells: an impact on androgen receptor signaling. J Cancer 2018; 9(11):1915-1924. doi:10.7150/jca.23648. https://www.jcancer.org/v09p1915.htm
CSE
Zenata O, Dvorak Z, Vrzal R. 2018. Mycophenolate Mofetil induces c-Jun-N-terminal kinase expression in 22Rv1 cells: an impact on androgen receptor signaling. J Cancer. 9(11):1915-1924.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.