Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(9):1527-1537. doi:10.7150/jca.24488 This issue Cite
Review
Anticancer Properties of Fenofibrate: A Repurposing Use
Xin Lian1,2, Gang Wang1, Honglan Zhou1, Zongyu Zheng1,2, Yaowen Fu1, ![]() , Lu Cai1,2,3,
, Lu Cai1,2,3, ![]()
1. Department of Urology, the First Hospital of Jilin University; 71 Xinmin Street, Changchun 130021, China
2. Pediatric Research Institute, Department of Pediatrics, University of Louisville, Louisville, KY 40202, USA
3. Departments of Radiation Oncology, Pharmacology and Toxicology, University of Louisville, Louisville, KY 40202, USA
Received 2017-12-21; Accepted 2018-2-25; Published 2018-4-6
Citation:
Lian X, Wang G, Zhou H, Zheng Z, Fu Y, Cai L. Anticancer Properties of Fenofibrate: A Repurposing Use. J Cancer 2018; 9(9):1527-1537. doi:10.7150/jca.24488. https://www.jcancer.org/v09p1527.htm
Other stylesAbstract
Cancer is a leading cause of death throughout the world, and cancer therapy remains a big medical challenge in terms of both its therapeutic efficacy and safety. Therefore, to find out a safe anticancer drug has been long goal for oncologist and medical scientists. Among clinically used medicines with no or little toxicity, fenofibrate is a drug of the fibrate class that plays an important role in lowering the levels of serum cholesterol and triglycerides while elevating the levels of high-density lipoproteins. Recently, several studies have implied that fenofibrate may exert anticancer effects via a variety of pathways involved in apoptosis, cell-cycle arrest, invasion, and migration. Given the great potential that fenofibrate may have anticancer effects, this review was to investigate all published works which directly or indirectly support the anticancer activity of fenofibrate. These studies provide evidence that fenofibrate exerted antitumor effects in several human cancer cell lines, such as breast, liver, glioma, prostate, pancreas, and lung cancer cell lines. Among these studies some have further confirmed the possibility and efficacy of fenofibrate anticancer in xenograft mouse models. In the last part of this review, we also discuss the potential mechanisms of action of fenofibrate based on the available information. Overall, we may repurpose fenofibrate as an anticancer drug in cancer treatment, which urgently need further and comprehensively investigated.
Keywords: fenofibrate, lipid-lowering, anticancer drug, repurposing
1. Introduction
Since its clinical introduction as a third-generation fibrate in 1975, fenofibrate has been widely used in the treatment of hypercholesterolemia and hyperlipidemia [1, 2]. The lipid-lowering effect of fenofibrate is believed to be mediated through its stimulation of peroxisome proliferator-activated receptor α (PPARα) [3-5]. In addition to its lipid-lowering function, fenofibrate exerts also pleiotropic effects. For instance, fenofibrate was found to not only slow the progression of diabetic retinopathy and other microvascular complications in patients with type 2 diabetes [6-8], but also protect against retinopathy, nephropathy, and cardiac pathological changes in type 1 diabetes [9-11]. Fenofibrate was established to afford myocardial protecttion through its direct effects on the cardiovascular system [12, 13]. Most recently, PPARα-specific agonists were reported to have anticancer effects in a large number of human cancer types, such as acute myeloid leukemia [14, 15], chronic lymphocytic leukemia [16], and solid tumors, including those of the liver [17], ovary [18], breast, skin, and lungs [19]. Furthermore, fenofibrate inhibited the proliferation of cell lines derived from breast and oral tumors, melanoma, lung carcinoma, glioblastoma, and fibrosarcoma in mouse models [19-21]. Therefore, this review mainly focuses on some recent developments in the anticancer actions of fenofibrate.
Figure 1
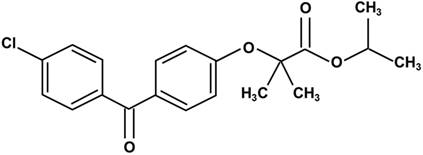
Chemical structure of fenofibrate. Fenofibrate is 2-4[(4-chlorobenzoyl) phenoxy]-2-methylpropanoic acid, 1-methylethyl ester.
Figure 2
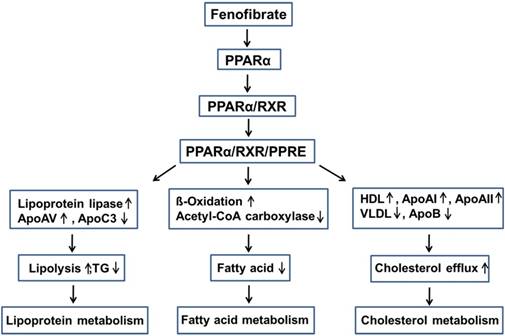
Lowering lipid mechanisms of fenofibrate. Fenifibrate activates PPARα, and forms a heterodimer with RXR, then interacts with PPRE, leading to the activation of target gene transcription of lipid metabolism regulation genes. PPARα: peroxisome proliferator-activated receptor α; RXR: retinoid X receptor; PPRE: peroxisome proliferator response element; Apo: apolipoprotein; HDL: high-density lipoprotein; VLDL: very-low-density lipoprotein; TG: triglyceride.
2. Pharmacology of fenofibrate
Fenofibrate is 2-4[(4-chlorobenzoyl) phenoxy]-2-methylpropanoic acid, 1-methylethyl ester (Fig. 1). Following oral administration, it is rapidly hydrolyzed by esterases in the liver and intestinal wall to its active metabolite, fenofibric acid [22]. The lipid-lowering effects of fenofibrate are mediated by the activation of PPARα [3-5], one of the three isoforms of peroxisome proliferator-activated receptors (PPARs). PPARs belong to a nuclear hormone receptor superfamily, whose functional domains include a DNA-binding domain (DBD) and a ligand-binding domain (LBD) [23, 24]. During the process of binding to the respective ligand, PPARα forms a heterodimer with a retinoid X receptor (RXR), another nuclear receptor and interacts with a peroxisome proliferator response element (PPRE), located in the promoter of the target genes through the DBD. This leads to the activation of target gene transcription, including that of lipid metabolism regulation genes [25, 26].
Fenofibrate can inhibit triglyceride (TG) synthesis through reducing the availability of free fatty acids by promoting β-oxidation [27]. Fenofibrate can decrease the activities of acetyl-CoA carboxylase and fatty acid synthase and thus inhibits the de novo fatty acid synthesis [28]. Moreover, fenofibrate increased the potential for elimination of atherogenic TG-rich lipoproteins from the plasma and lipolysis through the activation of apolipoprotein (Apo) AV and lipoprotein lipase, and the reduced production of the lipoprotein lipase inhibitor ApoCIII [29, 30]. Fenofibrate also stimulates the synthesis of high-density lipoprotein (HDL), ApoAI, and ApoAII, and decreases the production of very-low-density lipoprotein (VLDL) and ApoB, while enhancing the expression of adenosine triphosphate-binding cassette transporter A1 and scavenger receptor-B1, leading to HDL-mediated cholesterol efflux from macrophages [27, 28, 31-33] (Fig. 2).
3. Anticancer activity: in vitro studies
Fenofibrate is widely used in clinical practice as a lipid-lowering agent, but increasing evidence reveals its potential as an anti-cancer drug. Hence, we first provide a summary of the available information predominantly based on in vitro studies, brief overview of which is presented in Table 1.
Table 1
General information for the in vitro studies
| Cancer | Cell line | Findings | PPAR-α dependent or independent | Reference |
|---|---|---|---|---|
| Breast cancer | MDA-MB-231 | Fenofibrate induced apoptosis along with NF-κB pathway activation and induced cell cycle arrest at G0/G1 phase by up-regulating p21, p27/Kip1 and down-regulating Cyclin D1 and Cdk4. | independent | 20 |
| MCF-7 | Fenofibrate inhibited the semaphorin 6B protein expression that is able to prompted tumor invasion and metastasis. | dependent | 34 | |
| Liver cancer | HepG2 | Fenofibrate induced necrotic cell death by increasing ROS and intracellular calcium, decreasing GSH level, and impairing mitochondrial function. | not mentioned | 38 |
| Huh7 | Fenofibrate induced G1 arrest and G2/M arrest through up-regulating CTMP-mediated AKT phosphorylation inhibition. | independent | 39 | |
| Glioma | U87, U343, U251 and T98 | Fenofibrate inhibited cell proliferation and induced apoptosis in all human HGG cell lines through inhibition Akt function. Fenofibrate also induced cell cycle arrest in G0/G1 phase in U87MG cells probably by FoxO1-p27kip signaling pathway. | independent | 46, 47 |
| U87, U251 | Fenofibrate inhibited the transcriptional activity of NF-κB/RelA and disrupted the association of RelA and HIF1α, leading to the decreased PKM2 expression and mitochondrial impairment. | dependent | 50 | |
| glioma stem cells (GSC) | Fenofibrate inhibited GSC invasion by decreasing expression of CD133 and Oct4GSC. | independent | 46 | |
| LN-229 | Fenofibrate induced apoptosis through increasing FoxO3A nuclear accumulation. | dependent | 48 | |
| LN-229 | Fenofibrate induced glioblastoma cytotoxicity by activation of AMPK and inhibition of mTOR activity. | independent | 49 | |
| LN-229, T98 | Fenofibrate inhibited glioma cell motility by ROS accumulating through disturbed mitochondrial potential. | partially dependent | 51 | |
| Prostate cancer | LNCaP | Fenofibrate induced cell cycle arrest and apoptosis, probably by suppressing Akt phosphorylation and enhancing intracellular ROS levels. | not mentioned | 54 |
| DU145 | Fenofibrate inhibited the motility of DU-145 cells by increasing ROS accumulation. | independent | 56 | |
| DU145 | Fenofibrate improved endothelial barrier function by disturbing the endothelial cell adhesion and impairment of motile activity to inhibited metastasis via promoting intracellular ROS accumulation. | partially dependent | 57 | |
| Endometrial cancer | Ishikawa endometrial cancer cells | Fenofibrate inhibited the growth of Ishikawa endometrial cancer cells through downregulating CCND1, along with upregulating p8, p21, GADD45A, GADD45B and TP53. | dependent | 59, 60 |
| Oral cancer | CAL 27 | Fenofibrate inhibited the invasion and migration of CAL 27 cells by NF-κB signaling pathway, which mediated through AMPK signaling. | independent | 61 |
| SAS | Fenofibrate induced cytotoxicity in SAS oral cancer cells through regulation of the Warburg effect and down-regulation of mTOR activity through activation of AMPK signaling. | dependent | 63 | |
| Lung cancer | A549, SKMES-1 | Fenofibrate induced G1 cell cycle arrest of these cancer cells by suppressing NF-κB activity and ERK signaling. | independent | 64 |
| Multiple myeloma (MM) | KMS18, OPM-2, RPMI-8226 and U-266 | Fenofibrate reduced cell viability of MM cell lines by apoptosis. | not mentioned | 67 |
| Mantle cell lymphoma (MCL) | Jeko-1, Mino and SP53 | Fenofibrate induced apoptosis in MCL cells probably through inhibiting TNF-a/NF-κB pathway. | independent | 68 |
| Neuroblastoma | SH-SY5Y, IMR-32 | Fenofibrate inhibited proliferation and migration of NB cells by upregulating TXNIP expression. | independent | 72 |
| Pancreatic cancer | PANC-1, SW1990 | Fenofibrate inhibited pancreatic cancer cells proliferation via upregulating MEG3-mediated p53 function. | not mentioned | 78 |
| Melanoma | B16-F10 | Fenofibrate suppressed melanogenesis through p38 MAPK signaling pathway. | independent | 80 |
| B16-F10, SkMell88 | Fenofibrate decreased metastatic potential of melanoma cell cells by down-regulation of Akt phosphorylation. | dependent | 81 |
3.1 Breast cancer
Breast cancer is the most common malignant cancer in women worldwide. Fenofibrate was found to inhibit the proliferation of breast cancer MDA-MB-231 cell lines by inducing apoptosis and cell-cycle arrest [20]. Fenofibrate increased the expression of Bad, but decreased that of Bcl-xL and Survivin, and activated caspase-3. Fenofibrate also induced cell-cycle arrest at the G0/G1 phase by upregulation of p21, p27/Kip1 and downregulation of cyclin D1 and Cdk4. These effects were not abolished by GW6471 (a PPARα specific inhibitor), suggesting that the induction of breast cancer apoptosis by fenofibrate is independent of PPARα.
In MCF-7 breast adenocarcinoma cells, fenofibrate inhibited the expression of semaphorin 6B protein [34]. Semaphorins are a family of highly conserved transmembrane and secreted proteins that were initially identified as axon guidance and development elements of the nervous system [35]. Recent studies showed that the semaphorin family contains eight classes of members with a wide range of functions, including tumor invasion and metastasis [36]; for example, class 6 semaphorin B (sema 6B) can increase tumor progression capacity [37]. Therefore, fenofibrate may not only induce breast cancer cell death, but also inhibit its progression and metastasis.
3.2 Liver cancer
Fenofibrate has been established to decrease the viability of human Hepatocellular carcinoma HepG2 cells partly by necrotic cell death, but not apoptosis [38]. However, the cell death can be partially prevented by vitamin E, which suggests that the oxidative stress plays an important role in this event. Indeed, in a previous study, fenofibrate increased the levels of reactive oxygen species (ROS) and intracellular calcium, but decreased that of glutathione (GSH, an important cellular antioxidant) and impaired the mitochondrial function in HepG2 cells. The levels of PPARα mRNA were elevated after the treatment with fenofibrate, but whether the increase in the amount applied is related to cell viability inhibition was not elucidated in this investigation.
Besides the induction of necrotic cell death, fenofibrate can also lead to cell-cycle arrest in liver cancer cells. Daisuke et al. revealed that fenofibrate induced G1 and G2/M arrest in Huh7 cells through downregulation of cyclin A, cyclin B, CDK1, and E2F, accompanied with upregulation of p27. Furthermore, fenofibrate activated endogenous PPARα in Huh7, HepG2, and Li7 cell lines, but the antiproliferative activity induced by fenofibrate was not affected by the PPARα inhibitor GW6471 or the knock-down of the expression of PPARα by siRNA. Moreover, LY294002, a PI3K inhibitor, markedly inhibited the growth of Huh7 cells, and fenofibrate suppressed AKT phosphorylation. Fenofibrate also increased the expression of C-terminal modulator protein (CTMP), which binds specifically to AKT, and reduced AKT phosphorylation. Taken together, fenofibrate inhibited cell proliferation in a PPARα-independent manner, through inhibition of AKT phosphorylation by upregulation of CTMP [39].
Some studies identified fenofibrate as a liver tumor promoter in rodents. However, whether fenofibrate increases the risk of liver cancer in humans remains controversial [40]. Therefore, it is important to understand the different mechanisms functioning in different species to properly evaluate the health risk of fenofibrate treatment. First, the gene expression levels of PPARs are different between rodents and humans. PPAR was observed to be much more abundant in the rodent liver than in the human. The levels of PPAR in humans may be sufficient only to lower lipid levels, but not high enough to activate the transcriptional activities of adverse effect-related genes observed in rodents, such as liver enlargement and liver cancer [41]. Second, the same PPARα-target genes have modulation patterns that differ between humans and rodents. For example, the treatment of rats with fenofibrate decreased their ApoAI expression at both the mRNA and protein levels [42], whereas such treatment increased the transcription of ApoAI in humans [43]. Third, most experiments have explored in vitro the biochemical differences among hepatocytes of various species, rather than by studies of the mechanisms of action of the drug in animal models [44].
3.3 Glioma
Glioblastoma is the most frequent type of high-grade glioma (HGG). It is noteworthy that the combined treatment of this cancer by surgery, radiation, and temozolomide therapy can initially improve the survival rates, but tumor recurrence and death may eventually occur afterwards [45]. Therefore, there is an urgent need to discover a new treatment for glioma.
In an earlier investigation, fenofibrate significantly inhibited cell proliferation and induced apoptosis in human HGG cell lines, including p53 wild-type (U87 and U343) and p53 mutant (U251 and T98) cells. GW6471 did not reverse the suppression of proliferation or the pro-apoptosis caused by fenofibrate, which is consistent with PPARα-independent anticancer effects discussed earlier. Several signaling pathways were involved in the fenofibrate cytotoxic effect in HGG cells, including a decrease in NF-κB levels and phosphorylation of Akt and cyclin D1. Fenofibrate can also affect apoptotic signaling molecules by upregulation of Bax and downregulation of Bcl-xL. Furthermore, the drug obviously reduced glioma stem cells (GSC) invasion probably through decreasing the expression of CD133 (classical stem-cell markers) and Oct4GSC (associated with a poor prognosis in GSC) [46].
Besides the canonical pathway, fenofibrate can also inhibit cell growth through its impact on Fork-head box (Fox) family. FOX is a family of transcription factors that plays important roles in the regulation of the expression of genes involved in cell growth, proliferation, differentiation, and longevity. Han et al. found that fenofibrate induced cell-cycle arrest in U87MG cells at the G0/G1 phase, which was mediated by the FoxO1-p27kip signaling pathway. Moreover, pretreating U87MG cells with PPARα siRNA significantly rescued fenofibrate-induced cell-cycle arrest, suggesting that G0/G1 arrest by fenofibrate operates via a PPARα-dependent mechanism [47].
FoxO3A is another member of the Fox family that is involved in tumor cell apoptosis in glioblastoma. The treatment with 50 μM fenofibrate induced considerable increase in the levels of apoptosis in the human glioblastoma cell line LN-229, and the apoptotic event was associated with FoxO3A nuclear accumulation. Phosphorylated FoxO3A increased the transcriptional activity and expression of Bim, a FoxO-dependent apoptotic protein. Interestingly, fenofibrate-induced apoptosis was attenuated by siRNA-mediated inhibition of FoxO3A, indicating a direct effect of fenofibrate on glioblastoma cell growth [48].
Energetic metabolism and mitochondrial function also play important roles in anti-tumor effect of fenofibrate. Fenofibrate directly inhibited the mitochondrial respiration in human glioblastoma LN-229 cell lines, which was not reversed by PPARα siRNA and GW6471. This mitochondrial action switched the metabolism from glycolysis to fatty acid β-oxidation. The subsequent decrease of intracellular ATP activated AMPK and inhibited mTOR activity, resulting in fenofibrate-induced glioblastoma cytotoxicity [49]. Han et al. also found that fenofibrate suppressed glycolysis in U87 and U251 glioblastoma cells, and fenofibrate-treated cells in the presence of GW9662 (a synthetic PPARγ inhibitor, also blocks PPARα at higher concentrations) reversed the inhibition of glycolysis, indicating that the effect of fenofibrate on glioblastoma cell glycolysis is PPARα dependent. Furthermore, they proved that fenofibrate inhibited the transcriptional activity of NF-κB/RelA and disrupted the association of RelA and HIF1α, leading to the decreased PKM2 expression and mitochondrial impairment [50]. In addition, there was another study showed that fenofibrate inhibited glioma cell motility by ROS accumulating through disturbed mitochondrial potential, and NAC could rescue the cell motility [51].
3.4 Prostate cancer
Prostate cancer is one of the most widespread cancer types, which is the second leading cause of cancer-related deaths among men in the developed countries [52]. Androgens, such as testosterone and dihydrotestosterone, play an important role in normal prostate development and maintenance, and stimulate the proliferation and progression of prostate cancer cells [53]. Low concentrations of fenofibrate induced cell-cycle arrest and apoptosis in the androgen-dependent prostate cancer cell line LNCaP, but high concentrations were needed for the induction of cell death of the androgen-independent prostate cell line PC-3, suggesting that the androgen receptor (AR) played an important role in fenofibrate-induced apoptosis and G1 arrest [54]. Fenofibrate induced cell-cycle arrest at the G1 phase mediated by downregulation of the protein levels of cyclins D1 and E2F1 in LNCaP cell lines. Meanwhile, fenofibrate induced apoptosis through decreasing Bcl-2 protein expression and increasing that of Bax. Furthermore, fenofibrate reduced the expression levels of AR and AR target genes (prostate-specific antigen and TMPRSS2) by suppression of Akt phosphorylation. Certain amounts of ROS are involved in the maintenance of the intracellular homeostasis, but extra ROS production can induce cell death. Fenofibrate also exerted apoptosis properties by causing oxidative stress. The drug enhanced the intracellular ROS levels accompanied with a decrease in the activities of superoxide dismutase (SOD) and malondialdehyde (MDA), whereas the pretreatment with nacetylcysteine (NAC) significantly attenuated the increase in apoptosis caused by fenofibrate in LNCaP cells.
As is known, cancer cell morphology and motility can be used to evaluate cancer invasion. Fenofibrate has been found to inhibit the invasive potential of prostate cancer cell lines. For example, Wybieralska et al. investigated the effects of fenofibrate on the invasive ability of the DU-145 cell line (androgen-independent prostate cell line) in the context of gap junctional intercellular coupling [55]. They found that the treatment with fenofibrate changed the morphology of DU-145 cells, making them long and thin. This effect was involved in the suppression of 'contact-stimulated' cell motility. Meanwhile, the effect of fenofibrate on DU-145 cell motility was PPARα-independent, because GW9662, which effectively inhibits both PPARγ and PPARα, did not restore the motility of DU-145 cells suppressed by fenofibrate. Furthermore, fenofibrate increased the ROS accumulation in DU-145 cells, whereas NAC partially restored cell motility and gap junctional coupling in fenofibrate-treated DU-145 cell [56].
The research group of Wybieralska further explored the interference of fenofibrate with prostate cancer metastasis by monitoring of the events accompanying the penetration of endothelial continuum by DU-145 cells. They set up a model of DU-145 cells co-cultured with human umbilical vein endothelial cells (HUVEC), which imitates the interface between circulating cancer cells and endothelium. Under normal conditions, DU-145 cells efficiently penetrated the endothelial layer, but the treatment with 25 µM fenofibrate significantly delayed the transendothelial penetration of DU-145 cells through enhancement of the endothelial barrier function, which was correlated with the inhibitory effect on DU-145-induced HUVEC motility. This influence was partly mediated in a PPARα-dependent manner, because GW9662 attenuated fenofibrate-induced cytoskeleton reorganization, but failed to restore the HUVEC motility in the co-cultures with DU-145 cells. Moreover, fenofibrate promoted intracellular ROS accumulation, and NAC attenuated fenofibrate-induced cytoskeletal rearrangements and significantly restored HUVEC motility, suggesting a ROS-dependent manner in the HUVEC responses to fenofibrate. Therefore, fenofibrate improved the endothelial barrier function by disturbing the endothelial cell adhesion and impairment of motile activity, decreased the efficiency of prostate cancer cell diapedesis, and was probably involved in the prostate cancer metastatic cascade [57].
3.5 Other cancer types
Endometrial cancer is the most common gynecologic malignancy globally that accounts for 97% of all uterine cancers [58]. Cathrine et al. showed for the first time that fenofibrate inhibited the growth of Ishikawa endometrial cancer cells in vitro, through downregulation of cyclin D1 (CCND1), which was associated with the upregulation of p8, p21, GADD45A, GADD45B, and TP53 [59] [60].
Oral squamous cell carcinoma (OSCC) is the most common head-and-neck cancer with poor prognosis because of uncontrolled tumor progression. Fenofibrate inhibits the invasion and migration of human oral cancer CAL 27 cells [61]. In this in vitro study, they found that fenofibrate reduced MMPS protein expression and suppressed the enzyme activities as established by gelatin zymography assay. Fenofibrate downregulated p-IKKa/b, p-IκBa, and nuclear NFκB (p65 and p50), which are involved in the NF-κB pathway, and upregulated AMPK-related proteins, such as LKB1, p-AMPKα, p-AMPKβ1, and AMPKγ1. Furthermore, compound C (an AMPK inhibitor) or a dominant negative AMPK mutant (DN-AMPKα) obviously attenuated the anti-invasive effect of fenofibrate. Meanwhile, fenofibrate did not stimulate the expression of the nuclear fraction of PPARα and significantly altered the trafficking of PPARα into nuclei. These results suggested that the inhibition of fenofibrate in CAL 27 cells was realized by the NF-κB signaling pathway, mediated through AMPK signaling but not PPARα. Furthermore, activation of AMPK has been reported to regulate the mechanistic targeting of the rapamycin complex 1 (mTORC1) pathway through TSC1/TSC2, which may be linked to the induction of a progressive metabolic reprogramming, thus leading to the promotion of cancer cell death [62]. Therefore, these authors continued to explore whether fenofibrate suppresses the development of oral tumors by mediation of metabolic changes through regulation of the Warburg effect and/or downregulation of mTOR activity through activation of AMPK signaling [63]. They found that fenofibrate induced cytotoxicity in SAS oral cancer cells, which was caused by decreasing the mitochondrial respiration rate (OCR/ECAR) and reducing the ATP content. Meanwhile, fenofibrate decreased the protein expression levels of hexokinase II (HK II) and voltage-dependent anion channel (VDAC), and increased those of pyruvate kinase and pyruvate dehydrogenase, which are associated with the Warburg effect. Moreover, fenofibrate suppressed mTOR activity by activating AMPK signaling and inactivating Akt signaling in a TSC1/2-dependent pathway. Additionally, fenofibrate modulated mTOR activity through direct suppression of raptor, a TSC1/2-independent pathway.
Lung cancer is the leading cause of cancer-related deaths in North America. Fenofibrate inhibited cell proliferation in both TP53 wild-type A549 lung cancer cells and TP53-deficient SKMES-1 lung cancer cells. Fenofibrate induced G1 cell-cycle arrest accompanied by upregulation of cyclin D1 and E and downregulation of cyclin A and B1. The suppression of NF-κB activity and ERK signaling may have contributed to the inhibition of cell proliferation by fenofibrate. The treatment with GW6471 failed to reverse the antiproliferative effects exerted by fenofibrate in A549 and SK-MES-1 cells. These results indicated that the inhibition of cell proliferation induced by fenofibrate is independent of the PPARα receptor [64].
Multiple myeloma (MM) is a hematological malignancy and is primarily treated with chemotherapy, but most MM patients suffered relapse [65, 66]. Schmeel et al. found that fenofibrate reduced cell viability of several MM cell lines and other lymphoma cell lines by apoptosis [67].
Mantle cell lymphoma (MCL) is a type of aggressive B-cell non-Hodgkin's lymphoma, which is characterized by continual resistance to conventional chemotherapy. Fenofibrate can suppress the viability of MCL cells by apoptosis through increasing the levels of cleaved caspase 3 and poly (ADP-ribose) polymerase, and decreasing those of BCL2L1 and BCL2A1, which are members of the BCL-2 family associated with impairment of mitochondrial membrane potential. Furthermore, TNF-α and NF-κB were also downregulated by fenofibrate. To conclude, fenofibrate induced apoptosis in MCL cells, and the associated mechanism might be through inhibition of the TNF-α/NF-κB pathway axis, but the fenofibrate-mediated inhibition of cell growth was not significantly affected by GW6471 [68].
Neuroblastoma (NB) is one of the most common extracranial solid tumors in children, with no effective treatment available for metastatic NB [69]. In related examinations, fenofibrate inhibited the proliferation and migration of NB cells, SH-SY5Y and IMR-32; induced cell apoptosis through upregulation of Bax, downregulation of Bcl-2, and activation of caspase-3. Thioredoxin-binding protein (TXNIP) is able to bind to and inhibit the thioredoxin activity of scavenging ROS, and TXNIP was involved in many solid tumors, such as gastric, breast, and bladder cancers [70, 71]. Fenofibrate induced TXNIP expression, resulting in the increase in intracellular ROS; the inhibition of TXNIP expression alleviated the antitumor effects of fenofibrate, but the suppression of PPARα expression did not affect the inhibitory influence on NB cell growth induced by fenofibrate. These results indicated that the antiproliferative and antimigratory effects of fenofibrate on NB cells were dependent on the upregulation of TXNIP, but were independent of PPARα [72].
Hu et al. exposited whether fenofibrate suppressed the proliferation of pancreatic cancer cells, PANC-1 and SW1990 cell lines, and using Long noncoding RNA (LncRNA) microarray analysis, they found that the human maternally expressed gene 3 (MEG3) expression obviously increased after fenofibrate treatment. LncRNAs are classically defined as RNA transcripts with a length greater than 200 nucleotides that have no or limited protein coding ability [73]. Aberrant expression of lncRNAs was reportedly involved in the development of conditions, such as cardiovascular diseases [74], neurodegenerative disorders [75], diabetes, and obesity [76], as well as cancer biology. MEG3 is a member of LncRNA which is expressed in many normal tissues, but its expression is lost in some human tumors [77]. In the study of Hu et al., the knockdown of MEG3 by siRNA attenuated the cytotoxicity induced by fenofibrate and the transfectious overexpression of MEG3 through induced cell death and increased p53 expression. The results indicated that fenofibrate inhibited pancreatic cancer cells proliferation via activation of p53 mediated by upregulation of MEG3 [78].
Melanin is synthesized by melanocytes and acts as a main regulator of pigmentation [79]. In an earlier investigation, fenofibrate inhibited the melanogenesis in B16-F10 melanoma cells through decreasing the intracellular tyrosinase activity, which was confirmed by the DOPA-staining activity; reducing the expression of melanocortin 1 receptor (MC1R), as well as activating the p38 MAPK signaling pathway. Moreover, the p38 MAPK inhibitor SB203580 prevented the repressive effects of fenofibrate on the melanin production, but GW6471 did not alter the melanin levels. These findings suggested that fenofibrate suppressed the melanogenesis through p38 MAPK signaling pathway, independently of PPARα [80]. But Grabacka et al. found that fenofibrate decreased metastatic potential of mouse (B16F10) and human (SkMell88) melanoma cells by down-regulating Akt phosphorylation. These effects were reversed by GW9662, suggesting that the anti-metastasis effects of fenofibrate on melanoma cells were also dependently of PPARα [81].
4. In vivo evidence
In addition to human in vitro studies of the anti-cancer potential of fenofibrate, we also reviewed some investigations in animal models (Table 2).
Table 2
General information for the in vivo studies
| Cancer | Animal model | Treatment | Findings | Reference |
|---|---|---|---|---|
| Breast cancer | Tumor-bearing mouse model was established by implanting MDA-MB-231 cells into six-week-old female BALB/c nude mice. | At the 7 days after implantation, fenofibrate at 200 mg/kg was given daily via intragastric administration for 21 days. | Fenofibrate inhibited tumor growth, showing by reduced tumor sizes, weights, and ratios of tumor weight/body weight. | 20 |
| Oral cancer | Cancers were induced in 6-week-old male C57BL/6JNarl mice with drinking water containing 200 μg/ml 4-NQO and 500 μg/ml arecoline hydrobromide for 8 weeks. | Mice received fenofibrate orally either at a low dose of 0.1% diet/mouse/day (about 200 mg/kg body weight/day) or at a high dose of 0.3% diet/mouse/day (about 500 mg/kg body weight/day) for the following 12 or 20 weeks after 4-NQO administration. | High-dose of fenofibrate suppressed the tumor incidence and reduced the multiplicity of tongue lesions after continuing administration for 20 weeks. However, low-dose of fenofibrate lacked anti-tumor activity. In addition, high dose of fenofibrate also decreased EGFR expression and immunoreactivity of COX2 in squamous cell carcinoma (SCC). | 21 |
| 2 groups of Mice were pre-treated with fenofibrate for 2 weeks in order to explore the preventive potential of fenofibrate on tumorigenesis. In addition, administration of fenofibrate from weeks 9, 13, 17, 21, or 25 to week 28 was performed to evaluate the therapeutic efficacy of fenofibrate on tumor progression. | Fenofibrate had preventive efficacy on tumorigenesis and prevented tumor progression, particularly at an early stage of tumor. The underlying mechanisms might include the suppression of mTOR activity through activation of AMPK and inactivation of Akt, mediated by TSC1/2-dependent and independent signaling pathways. | 63 | ||
| Melanoma | Tumor-bearing mice with B16-F10 cells were made in SCID mice. | Once tumors reached 100 mm3, fenofibrate at 200 mg/kg were administered daily by gavage for 20 days. | Fenofibrate inhibited B16-F10 tumor growth by 61%. | 19 |
| Lewis lung carcinoma | Tumor-bearing mice with LLC cells were made in SCID mice. | Once tumors reached 100 mm3, fenofibrate at 200 mg/kg were administered daily by gavage for 20 days. | Fenofibrate inhibited LLC tumor growth by 58%. | 19 |
| Glioblastoma | Tumor-bearing mice with U87 cells were made in SCID mice. | Once tumors reached 100 mm3, fenofibrate at 200 mg/kg were administered daily by gavage for 24 days. | Fenofibrate inhibited U87 tumor growth by 72%. | 19 |
| Fibrosarcoma | Tumor-bearing mice withHT1080 cells were made in SCID mice. | Once tumors reached 100 mm3, fenofibrate at 200 mg/kg were administered daily by gavage for 28 days. | Fenofibrate inhibited HT1080 tumor growth by 66%. | 19 |
4.1 Breast cancer
Li et al. injected MDA-MB-231 cells (2 × 106) into six-week-old female BALB/c nude mice to establish a tumor-bearing mouse model. Seven days after the injection, the researchers administered intragastrically 200 mg/kg of fenofibrate daily to the mice in treatment group or an equal volume of a vehicle (5% sodium carboxymethylcellulose) in the control group [20]. Fenofibrate considerably inhibited the growth of the tumors as evidenced by their sizes and weights, and the ratio of tumor weight/mice body weight. Meanwhile, fenofibrate induced approximately two-fold increase apoptotic cells, as detected by TUNEL assay. Furthermore, they also evaluated the safety of fenofibrate and found no statistical differences between the control and treatment groups in body weight, white blood cell count, and the levels of hemoglobin, platelets, alanine transaminase, aspartate aminotransferase, and blood urea nitrogen. These results confirmed that fenofibrate suppressed tumor growth and induced apoptosis in a xenograft mouse model with a good safety profile [20].
4.2 Oral cancer
Chang et al. established an oral cancer mouse model induced by a 4-nitroquinoline 1-oxide (4-NQO)/arecoline administration for eight weeks, followed by treatment with fenofibrate (0.1% or 0.3%) for 12 or 20 weeks. The scientists found that the treatment with 0.3% fenofibrate suppressed the tumor incidence and tumor formation after continuing administration for 20 weeks. Moreover, the multiplicity of tongue lesions was reduced at the 28-week time point. However, in this study, the application of 0.1% fenofibrate did not show antitumor activity. Furthermore, the authors found that the incidence rate of tongue lesions in the pre-treated with fenofibrate mice was significantly lower than that in the 4-NQO/arecoline-only mice, and the incidence rate of tongue lesions was significantly lower after treatment of the mice with fenofibrate at an early stage of tumor development (week 9). Considering that the epidermal growth factor receptor (EGFR) was found to be the most frequently amplified and highly expressed gene in both human and mouse oral tumors [82], and EGFR contributed to the elevated levels of cyclooxygenase-2 (COX-2) [83], they examined the expression levels of EGFR and COX2 after the fenofibrate treatment. They found that the administration of 0.3% fenofibrate increased the EGFR expression levels in hyperplastic and dysplastic lesions, but decreased the expression of EGFR in squamous cell carcinoma (SCC). Meanwhile, the immunoreactivity of COX2 was elevated in the dysplasia lesions but was reduced in SCC lesions [21]. There were also other molecular mechanisms, including the suppression of mTOR activity through activation of AMPK and inactivation of Akt, which were mediated by TSC1/2-dependent signaling or through direct suppression of raptor, which is mediated by a TSC1/2-independent pathway [63]. Therefore, fenofibrate exhibited a high potential to prevent and inhibit the formation of SCC.
4.3 Other cancers
Tumor-bearing mice with B16-F10 melanoma, Lewis lung carcinoma (LLC), glioblastoma (U87), and fibrosarcoma (HT1080) tumor cells were made using C57BL/6 or SCID mice. Once the tumors reached 100 mm3, fenofibrate, combined with other PPARα ligands (bezafibrate and gemfibrozil) was administered by daily gavage for 20-28 days. In this examination, fenofibrate (200 mg/kg per day) was found to inhibit B16-F10, LLC, U87, and HT1080 tumor growth by 61%, 58%, 72%, and 66%, respectively, and had more potent anticancer effects than other fibrates, such as bezafibrate and gemfibrozil [19].
5. Clinical case reports
Inflammatory hepatocellular adenomas (IHCA) account from 40% to 50% of all hepatic adenomas, and large HCA (more than 5 cm in diameter) are prone to bleeding and development of hepatocellular carcinoma. The risk factors for IHCA include prolonged use of oral contraceptives (OC), high alcohol consumption, overweight, and insulin resistance. The cardinal feature of IHCAs is the activation of the IL6/JAK/STAT pathway.
Poupon et al. reported a case of a 52-year-old woman presenting with severe multiple typical IHCA established by biochemistry and liver MRI, and confirmed by histology and immunohistochemistry of liver biopsy specimens taken from the tumor and non-tumoral tissue. She had been taking OC for about ten years until the discovery of the liver nodules in 2011. There were no major changes of the liver lesions in the following year (2012). She was exceedingly reluctant to invasive treatment (liver surgery) to prevent complications, so she received fenofibrate (400 mg/day). Six months later, the biochemistry parameters were quite normalized, and liver MRI showed a 50% reduction of the area of the three major lesions. Further evaluations in 2014 and 2015 confirmed the normalization of biochemistry as well as the major regression of the nodules. No side-effects related to fenofibrate treatment were observed. This is the first case report, but also the only one available so far, suggesting that fenofibrate might suppress the inflammation associated with IHCA and induce tumor regression in humans [84].
Figure 3
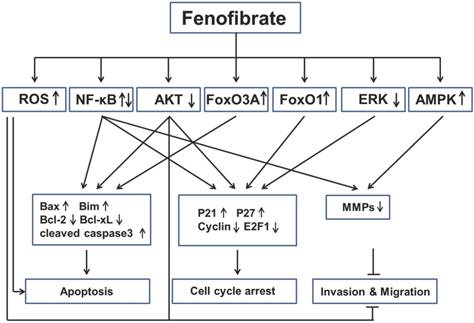
Key signaling mechanisms involved in anticancer activities of fenofibrate. Fenofibrate inhibited multiple cancers by regulation of apoptosis, cell cycle arrest, invasion and migration through different pathways. ROS: reactive oxygen species; NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells; FoxO3A: Fork-head box O3A; FoxO1: Fork-head box O1; ERK: extracellular-signal-regulated kinase; AMPK: 5' adenosine monophosphate-activated protein kinase; Bax: Bcl-2-associated X protein; Bim: Bcl-2-like protein 11; Bcl-2: B-cell lymphoma 2; bcl-xL: B-cell lymphoma-extra large; MMPs: Matrix metalloproteinases.
6. Possible mechanisms by which fenofibrate inhibits cancer
Based on the findings described above, we have learned that fenofibrate inhibited multiple cancers through induction of apoptosis, induction of cell-cycle arrest and inhibition of tumor invasion and migration (Fig. 3). However, we are also aware that the mechanisms of anti-cancer property of fenofibrate are very complex via several pathways (see below).
First, fenofibrate, as a PPARα agonist, induced transcriptional activation of fatty acid β-oxidation machinery and switched energy metabolism from the glucose to fatty acid utilization, both resulting in decreased ATP levels and activated AMPK [85]. Fenofibrate inhibits the invasion and migration of human oral cancer CAL 27 cells through AMPK signaling [61]. Second, fenofibrate induced ROS accumulation, which might be also due to the elevated peroxisomal β-oxidation [38]. Fenofibrate induced apoptosis in neuroblastoma and inhibited prostate cancer metastasis by increasing intracellular ROS [56, 57, 72]. Third, fenofibrate was able to increase plasma membrane rigidity in a manner similar to elevated cholesterol content in cell membranes [86], so we can speculate that fenofibrate may alter the membrane-bound growth factor receptors such as type 1 insulin-like growth factor receptor (IGF-IR) or EGFR, which has a close relationship with Akt and ERK. Fenofibrate induced apoptosis in liver cancer cells [39], induced prostate cancer cells apoptosis and cell cycle arrest [54], and inhibited metastasis in melanoma cells [81], all which were mediated by decreasing Akt activity. In addition, fenofibrate reduced phosphorylation of ERK protein in lung cancer cells [64]. Fourth, FoxO family proteins could be regulated by many upstream signals, such as Akt, Erk and AMPK [87], but the authors, who reported FOXO family proteins involved in the anti-proliferation effect of fenofibrate on glioblastoma cells [47, 48], did not explored the underlying mechanisms. Last, fenofibrate had bidirectional modulatory effects on NF-κB activity in different cancers. For instance, fenofibrate activated NF-κB pathway in breast cancer by increasing the DNA binding of NF-κB through down-regulation of p-IκBα, which sequestered NF-κB protein in the cytoplasm [20]. But fenofibrate inhibited NF-κB pathway in lung cancer by decreasing the DNA binding of NF-κB, without increasing of IκBa protein. All these possibilities might be the result of direct interaction between fenofibrate and NF-kB [64]. The detailed mechanisms should be uncovered in the future.
7. Conclusion
Fenofibrate is an effective lipid-lowering agent, exerting its beneficial effects by increasing HDL levels and decreasing the levels of triglycerides and LDL cholesterol, that has been widely applied in clinic for the treatment of hyperlipidemia [21, 88-90]. Nevertheless, fenofibrate has many other therapeutic properties, including protection against retinopathy and nephropathy in diabetes, as well as myocardial protection, even in type 1 diabetes. Recently, a large number of studies have explored the anticancer activities of fenofibrate and the related pathways involved in apoptosis, cell-cycle arrest, invasion, and migration. The in vivo experimental results discussed herein confirm that fenofibrate exert positive effects against various tumor types, although only its application in high doses (200 mg/kg or 0.3%) inhibited the tumor growth. Therefore, we may regard fenofibrate as an adjuvant drug in cancer treatment, which can be used in combination with chemotherapy or targeted molecular drugs in future research.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Staels B, Dallongeville J, Auwerx J, Schoonjans K, Leitersdorf E, Fruchart JC. Mechanism of action of fibrates on lipid and lipoprotein metabolism. Circulation. 1998;98:2088-93
2. Soska V. [Present status of fibrates in the treatment of hyperlipoproteinemias]. Vnitrni lekarstvi. 1999;45:438-40
3. Staels B, Auwerx J. Regulation of apo A-I gene expression by fibrates. Atherosclerosis. 1998;137(Suppl):S19-23
4. Srivastava RA. Fenofibrate ameliorates diabetic and dyslipidemic profiles in KKAy mice partly via down-regulation of 11beta-HSD1, PEPCK and DGAT2. Comparison of PPARalpha, PPARgamma, and liver x receptor agonists. European journal of pharmacology. 2009;607:258-63
5. Pettersen JC, Pruimboom-Brees I, Francone OL, Amacher DE, Boldt SE, Kerlin RL. et al. The PPARalpha agonists fenofibrate and CP-778875 cause increased beta-oxidation, leading to oxidative injury in skeletal and cardiac muscle in the rat. Toxicologic pathology. 2012;40:435-47
6. Noonan JE, Jenkins AJ, Ma JX, Keech AC, Wang JJ, Lamoureux EL. An update on the molecular actions of fenofibrate and its clinical effects on diabetic retinopathy and other microvascular end points in patients with diabetes. Diabetes. 2013;62:3968-75
7. Czupryniak L, Joshi SR, Gogtay JA, Lopez M. Effect of micronized fenofibrate on microvascular complications of type 2 diabetes: a systematic review. Expert opinion on pharmacotherapy. 2016;17:1463-73
8. Keating GM. Fenofibrate: a review of its lipid-modifying effects in dyslipidemia and its vascular effects in type 2 diabetes mellitus. American journal of cardiovascular drugs: drugs, devices, and other interventions. 2011;11:227-47
9. Chen Y, Hu Y, Lin M, Jenkins AJ, Keech AC, Mott R. et al. Therapeutic effects of PPARalpha agonists on diabetic retinopathy in type 1 diabetes models. Diabetes. 2013;62:261-72
10. Cheng Y, Zhang J, Guo W, Li F, Sun W, Chen J. et al. Up-regulation of Nrf2 is involved in FGF21-mediated fenofibrate protection against type 1 diabetic nephropathy. Free radical biology & medicine. 2016;93:94-109
11. Zhang J, Cheng Y, Gu J, Wang S, Zhou S, Wang Y. et al. Fenofibrate increases cardiac autophagy via FGF21/SIRT1 and prevents fibrosis and inflammation in the hearts of Type 1 diabetic mice. Clinical science. 2016;130:625-41
12. Balakumar P, Rohilla A, Mahadevan N. Pleiotropic actions of fenofibrate on the heart. Pharmacological research. 2011;63:8-12
13. Tabernero A, Schoonjans K, Jesel L, Carpusca I, Auwerx J, Andriantsitohaina R. Activation of the peroxisome proliferator-activated receptor alpha protects against myocardial ischaemic injury and improves endothelial vasodilatation. BMC pharmacology. 2002;2:10
14. Laurora S, Pizzimenti S, Briatore F, Fraioli A, Maggio M, Reffo P. et al. Peroxisome proliferator-activated receptor ligands affect growth-related gene expression in human leukemic cells. The Journal of pharmacology and experimental therapeutics. 2003;305:932-42
15. Scatena R, Nocca G, Sole PD, Rumi C, Puggioni P, Remiddi F. et al. Bezafibrate as differentiating factor of human myeloid leukemia cells. Cell death and differentiation. 1999;6:781-7
16. Liu H, Zang C, Fenner MH, Liu D, Possinger K, Koeffler HP. et al. Growth inhibition and apoptosis in human Philadelphia chromosome-positive lymphoblastic leukemia cell lines by treatment with the dual PPARalpha/gamma ligand TZD18. Blood. 2006;107:3683-92
17. Muzio G, Maggiora M, Oraldi M, Trombetta A, Canuto RA. PPARalpha and PP2A are involved in the proapoptotic effect of conjugated linoleic acid on human hepatoma cell line SK-HEP-1. International journal of cancer. 2007;121:2395-401
18. Shigeto T, Yokoyama Y, Xin B, Mizunuma H. Peroxisome proliferator-activated receptor alpha and gamma ligands inhibit the growth of human ovarian cancer. Oncology reports. 2007;18:833-40
19. Panigrahy D, Kaipainen A, Huang S, Butterfield CE, Barnes CM, Fannon M. et al. PPARalpha agonist fenofibrate suppresses tumor growth through direct and indirect angiogenesis inhibition. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:985-90
20. Li T, Zhang Q, Zhang J, Yang G, Shao Z, Luo J. et al. Fenofibrate induces apoptosis of triple-negative breast cancer cells via activation of NF-kappaB pathway. BMC cancer. 2014;14:96
21. Chang NW, Tsai MH, Lin C, Hsu HT, Chu PY, Yeh CM. et al. Fenofibrate exhibits a high potential to suppress the formation of squamous cell carcinoma in an oral-specific 4-nitroquinoline 1-oxide/arecoline mouse model. Biochimica et biophysica acta. 2011;1812:558-64
22. Adkins JC, Faulds D. Micronised fenofibrate: a review of its pharmacodynamic properties and clinical efficacy in the management of dyslipidaemia. Drugs. 1997;54:615-33
23. Willson TM, Brown PJ, Sternbach DD, Henke BR. The PPARs: from orphan receptors to drug discovery. Journal of medicinal chemistry. 2000;43:527-50
24. Elijah IE, Borsheim E, Maybauer DM, Finnerty CC, Herndon DN, Maybauer MO. Role of the PPAR-alpha agonist fenofibrate in severe pediatric burn. Burns: journal of the International Society for Burn Injuries. 2012;38:481-6
25. Berger J, Moller DE. The mechanisms of action of PPARs. Annual review of medicine. 2002;53:409-35
26. Issemann I, Green S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature. 1990;347:645-50
27. Fruchart JC, Duriez P. Mode of action of fibrates in the regulation of triglyceride and HDL-cholesterol metabolism. Drugs of today. 2006;42:39-64
28. Schoonjans K, Staels B, Auwerx J. Role of the peroxisome proliferator-activated receptor (PPAR) in mediating the effects of fibrates and fatty acids on gene expression. Journal of lipid research. 1996;37:907-25
29. Ito MK. Long-chain omega-3 fatty acids, fibrates and niacin as therapeutic options in the treatment of hypertriglyceridemia: a review of the literature. Atherosclerosis. 2015;242:647-56
30. Schoonjans K, Peinado-Onsurbe J, Lefebvre AM, Heyman RA, Briggs M, Deeb S. et al. PPARalpha and PPARgamma activators direct a distinct tissue-specific transcriptional response via a PPRE in the lipoprotein lipase gene. The EMBO journal. 1996;15:5336-48
31. Berthou L, Duverger N, Emmanuel F, Langouet S, Auwerx J, Guillouzo A. et al. Opposite regulation of human versus mouse apolipoprotein A-I by fibrates in human apolipoprotein A-I transgenic mice. The Journal of clinical investigation. 1996;97:2408-16
32. Arakawa R, Tamehiro N, Nishimaki-Mogami T, Ueda K, Yokoyama S. Fenofibric acid, an active form of fenofibrate, increases apolipoprotein A-I-mediated high-density lipoprotein biogenesis by enhancing transcription of ATP-binding cassette transporter A1 gene in a liver X receptor-dependent manner. Arteriosclerosis, thrombosis, and vascular biology. 2005;25:1193-7
33. Forcheron F, Cachefo A, Thevenon S, Pinteur C, Beylot M. Mechanisms of the triglyceride- and cholesterol-lowering effect of fenofibrate in hyperlipidemic type 2 diabetic patients. Diabetes. 2002;51:3486-91
34. Murad H, Collet P, Huin-Schohn C, Al-Makdissy N, Kerjan G, Chedotal A. et al. Effects of PPAR and RXR ligands in semaphorin 6B gene expression of human MCF-7 breast cancer cells. International journal of oncology. 2006;28:977-84
35. Kolodkin AL, Matthes DJ, Goodman CS. The semaphorin genes encode a family of transmembrane and secreted growth cone guidance molecules. Cell. 1993;75:1389-99
36. Christensen CR, Klingelhofer J, Tarabykina S, Hulgaard EF, Kramerov D, Lukanidin E. Transcription of a novel mouse semaphorin gene, M-semaH, correlates with the metastatic ability of mouse tumor cell lines. Cancer research. 1998;58:1238-44
37. Collet P, Domenjoud L, Devignes MD, Murad H, Schohn H, Dauca M. The human semaphorin 6B gene is down regulated by PPARs. Genomics. 2004;83:1141-50
38. Jiao HL, Zhao BL. Cytotoxic effect of peroxisome proliferator fenofibrate on human HepG2 hepatoma cell line and relevant mechanisms. Toxicology and applied pharmacology. 2002;185:172-9
39. Yamasaki D, Kawabe N, Nakamura H, Tachibana K, Ishimoto K, Tanaka T. et al. Fenofibrate suppresses growth of the human hepatocellular carcinoma cell via PPARalpha-independent mechanisms. European journal of cell biology. 2011;90:657-64
40. Cornu-Chagnon MC, Dupont H, Edgar A. Fenofibrate: metabolism and species differences for peroxisome proliferation in cultured hepatocytes. Fundamental and applied toxicology: official journal of the Society of Toxicology. 1995;26:63-74
41. Holden PR, Tugwood JD. Peroxisome proliferator-activated receptor alpha: role in rodent liver cancer and species differences. Journal of molecular endocrinology. 1999;22:1-8
42. Staels B, van Tol A, Andreu T, Auwerx J. Fibrates influence the expression of genes involved in lipoprotein metabolism in a tissue-selective manner in the rat. Arteriosclerosis and thrombosis: a journal of vascular biology. 1992;12:286-94
43. Hahn SE, Goldberg DM. Modulation of lipoprotein production in Hep G2 cells by fenofibrate and clofibrate. Biochemical pharmacology. 1992;43:625-33
44. Bentley P, Calder I, Elcombe C, Grasso P, Stringer D, Wiegand HJ. Hepatic peroxisome proliferation in rodents and its significance for humans. Food and chemical toxicology: an international journal published for the British Industrial Biological Research Association. 1993;31:857-907
45. Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC. et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. The Lancet Oncology. 2009;10:459-66
46. Binello E, Mormone E, Emdad L, Kothari H, Germano IM. Characterization of fenofibrate-mediated anti-proliferative pro-apoptotic effects on high-grade gliomas and anti-invasive effects on glioma stem cells. Journal of neuro-oncology. 2014;117:225-34
47. Han DF, Zhang JX, Wei WJ, Tao T, Hu Q, Wang YY. et al. Fenofibrate induces G0/G1 phase arrest by modulating the PPARalpha/FoxO1/p27 kip pathway in human glioblastoma cells. Tumour biology: the journal of the International Society for Oncodevelopmental Biology and Medicine. 2015;36:3823-9
48. Wilk A, Urbanska K, Grabacka M, Mullinax J, Marcinkiewicz C, Impastato D. et al. Fenofibrate-induced nuclear translocation of FoxO3A triggers Bim-mediated apoptosis in glioblastoma cells in vitro. Cell cycle. 2012;11:2660-71
49. Wilk A, Wyczechowska D, Zapata A, Dean M, Mullinax J, Marrero L. et al. Molecular mechanisms of fenofibrate-induced metabolic catastrophe and glioblastoma cell death. Molecular and cellular biology. 2015;35:182-98
50. Han D, Wei W, Chen X, Zhang Y, Wang Y, Zhang J. et al. NF-kappaB/RelA-PKM2 mediates inhibition of glycolysis by fenofibrate in glioblastoma cells. Oncotarget. 2015;6:26119-28
51. Drukala J, Urbanska K, Wilk A, Grabacka M, Wybieralska E, Del Valle L. et al. ROS accumulation and IGF-IR inhibition contribute to fenofibrate/PPARalpha -mediated inhibition of glioma cell motility in vitro. Molecular cancer. 2010;9:159
52. Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA: a cancer journal for clinicians. 2009;59:225-49
53. Kaarbo M, Klokk TI, Saatcioglu F. Androgen signaling and its interactions with other signaling pathways in prostate cancer. BioEssays: news and reviews in molecular, cellular and developmental biology. 2007;29:1227-38
54. Zhao H, Zhu C, Qin C, Tao T, Li J, Cheng G. et al. Fenofibrate down-regulates the expressions of androgen receptor (AR) and AR target genes and induces oxidative stress in the prostate cancer cell line LNCaP. Biochemical and biophysical research communications. 2013;432:320-5
55. Friedl P, Wolf K. Plasticity of cell migration: a multiscale tuning model. The Journal of cell biology. 2010;188:11-9
56. Wybieralska E, Szpak K, Gorecki A, Bonarek P, Miekus K, Drukala J. et al. Fenofibrate attenuates contact-stimulated cell motility and gap junctional coupling in DU-145 human prostate cancer cell populations. Oncology reports. 2011;26:447-53
57. Piwowarczyk K, Wybieralska E, Baran J, Borowczyk J, Rybak P, Kosinska M. et al. Fenofibrate enhances barrier function of endothelial continuum within the metastatic niche of prostate cancer cells. Expert opinion on therapeutic targets. 2015;19:163-76
58. Rose PG. Endometrial carcinoma. The New England journal of medicine. 1996;335:640-9
59. Holland CM, Saidi SA, Evans AL, Sharkey AM, Latimer JA, Crawford RAF. et al. Transcriptome analysis of endometrial cancer identifies peroxisome proliferator-activated receptors as potential therapeutic targets. Mol Cancer Ther. 2004;3:993-1001
60. Saidi SA, Holland CM, Charnock-Jones DS, Smith SK. In vitro and in vivo effects of the PPAR-alpha agonists fenofibrate and retinoic acid in endometrial cancer. Molecular cancer. 2006;5:13
61. Tsai SC, Tsai MH, Chiu CF, Lu CC, Kuo SC, Chang NW. et al. AMPK-dependent signaling modulates the suppression of invasion and migration by fenofibrate in CAL 27 oral cancer cells through NF-kappaB pathway. Environmental toxicology. 2016;31:866-76
62. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274-93
63. Jan CI, Tsai MH, Chiu CF, Huang YP, Liu CJ, Chang NW. Fenofibrate Suppresses Oral Tumorigenesis via Reprogramming Metabolic Processes: Potential Drug Repurposing for Oral Cancer. International journal of biological sciences. 2016;12:786-98
64. Liang H, Kowalczyk P, Junco JJ, Klug-De Santiago HL, Malik G, Wei SJ. et al. Differential effects on lung cancer cell proliferation by agonists of glucocorticoid and PPARalpha receptors. Molecular carcinogenesis. 2014;53:753-63
65. Kuehl WM, Bergsagel PL. Multiple myeloma: evolving genetic events and host interactions. Nature reviews Cancer. 2002;2:175-87
66. Hideshima T, Mitsiades C, Tonon G, Richardson PG, Anderson KC. Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. Nature reviews Cancer. 2007;7:585-98
67. Schmeel LC, Schmeel FC, Schmidt-Wolf IGH. In Vitro Apoptosis Induction by Fenofibrate in Lymphoma and Multiple Myeloma. Anticancer research. 2017;37:3513-20
68. Zak Z, Gelebart P, Lai R. Fenofibrate induces effective apoptosis in mantle cell lymphoma by inhibiting the TNFalpha/NF-kappaB signaling axis. Leukemia. 2010;24:1476-86
69. Bottino C, Dondero A, Bellora F, Moretta L, Locatelli F, Pistoia V. et al. Natural killer cells and neuroblastoma: tumor recognition, escape mechanisms, and possible novel immunotherapeutic approaches. Frontiers in immunology. 2014;5:56
70. Woolston CM, Madhusudan S, Soomro IN, Lobo DN, Reece-Smith AM, Parsons SL. et al. Thioredoxin interacting protein and its association with clinical outcome in gastro-oesophageal adenocarcinoma. Redox biology. 2013;1:285-91
71. Woolston CM, Zhang L, Storr SJ, Al-Attar A, Shehata M, Ellis IO. et al. The prognostic and predictive power of redox protein expression for anthracycline-based chemotherapy response in locally advanced breast cancer. Modern pathology: an official journal of the United States and Canadian Academy of Pathology, Inc. 2012;25:1106-16
72. Su C, Shi A, Cao G, Tao T, Chen R, Hu Z. et al. Fenofibrate suppressed proliferation and migration of human neuroblastoma cells via oxidative stress dependent of TXNIP upregulation. Biochemical and biophysical research communications. 2015;460:983-8
73. Silva A, Bullock M, Calin G. The Clinical Relevance of Long Non-Coding RNAs in Cancer. Cancers. 2015;7:2169-82
74. Schonrock N, Harvey RP, Mattick JS. Long noncoding RNAs in cardiac development and pathophysiology. Circulation research. 2012;111:1349-62
75. Pastori C, Wahlestedt C. Involvement of long noncoding RNAs in diseases affecting the central nervous system. RNA biology. 2012;9:860-70
76. Kim J, Kim KM, Noh JH, Yoon JH, Abdelmohsen K, Gorospe M. Long noncoding RNAs in diseases of aging. Biochimica et biophysica acta. 2016;1859:209-21
77. Zhou Y, Zhang X, Klibanski A. MEG3 noncoding RNA: a tumor suppressor. Journal of molecular endocrinology. 2012;48:R45-53
78. Hu D, Su C, Jiang M, Shen Y, Shi A, Zhao F. et al. Fenofibrate inhibited pancreatic cancer cells proliferation via activation of p53 mediated by upregulation of LncRNA MEG3. Biochemical and biophysical research communications. 2016;471:290-5
79. Lin JY, Fisher DE. Melanocyte biology and skin pigmentation. Nature. 2007;445:843-50
80. Huang YC, Liu KC, Chiou YL, Yang CH, Chen TH, Li TT. et al. Fenofibrate suppresses melanogenesis in B16-F10 melanoma cells via activation of the p38 mitogen-activated protein kinase pathway. Chemico-biological interactions. 2013;205:157-64
81. Grabacka M, Plonka PM, Urbanska K, Reiss K. Peroxisome proliferator-activated receptor alpha activation decreases metastatic potential of melanoma cells in vitro via down-regulation of Akt. Clinical cancer research: an official journal of the American Association for Cancer Research. 2006;12:3028-36
82. Sheu JJ, Hua CH, Wan L, Lin YJ, Lai MT, Tseng HC. et al. Functional genomic analysis identified epidermal growth factor receptor activation as the most common genetic event in oral squamous cell carcinoma. Cancer research. 2009;69:2568-76
83. Moraitis D, Du B, De Lorenzo MS, Boyle JO, Weksler BB, Cohen EG. et al. Levels of cyclooxygenase-2 are increased in the oral mucosa of smokers: evidence for the role of epidermal growth factor receptor and its ligands. Cancer research. 2005;65:664-70
84. Poupon R, Cazals-Hatem D, Arrive L. Fenofibrate-induced massive regression of mutiple inflammatory hepatocellular adenoma. Clinics and research in hepatology and gastroenterology. 2016;40:e1-3
85. Grabacka M, Reiss K. Anticancer Properties of PPARalpha-Effects on Cellular Metabolism and Inflammation. PPAR research. 2008;2008:930705
86. Gamerdinger M, Clement AB, Behl C. Cholesterol-like effects of selective cyclooxygenase inhibitors and fibrates on cellular membranes and amyloid-beta production. Molecular pharmacology. 2007;72:141-51
87. Dansen TB, Burgering BM. Unravelling the tumor-suppressive functions of FOXO proteins. Trends in cell biology. 2008;18:421-9
88. Niemeijer-Kanters SD, Banga JD, Erkelens DW. Lipid-lowering therapy in diabetes mellitus. The Netherlands journal of medicine. 2001;58:214-22
89. Valensi P, Picard S. Lipids, lipid-lowering therapy and diabetes complications. Diabetes & metabolism. 2011;37:15-24
90. Saurav A, Kaushik M, Mohiuddin SM. Fenofibric acid for hyperlipidemia. Expert opinion on pharmacotherapy. 2012;13:717-22
Author contact
![]() Corresponding authors: Yaowen Fu, Department of Urology, the First Hospital of Jilin University; 71 Xinmin Street, Changchun 130021, China. Tel: +86-13756661667; Email: fuyaowencom.cn; Lu Cai, Department of Pediatrics, University of Louisville, Louisville, KY 40202, USA. Tel: +1-5028522214; Fax: +1-5028525634; Email: l0cai001edu
Corresponding authors: Yaowen Fu, Department of Urology, the First Hospital of Jilin University; 71 Xinmin Street, Changchun 130021, China. Tel: +86-13756661667; Email: fuyaowencom.cn; Lu Cai, Department of Pediatrics, University of Louisville, Louisville, KY 40202, USA. Tel: +1-5028522214; Fax: +1-5028525634; Email: l0cai001edu
Citation styles
APA
Lian, X., Wang, G., Zhou, H., Zheng, Z., Fu, Y., Cai, L. (2018). Anticancer Properties of Fenofibrate: A Repurposing Use. Journal of Cancer, 9(9), 1527-1537. https://doi.org/10.7150/jca.24488.
ACS
Lian, X.; Wang, G.; Zhou, H.; Zheng, Z.; Fu, Y.; Cai, L. Anticancer Properties of Fenofibrate: A Repurposing Use. J. Cancer 2018, 9 (9), 1527-1537. DOI: 10.7150/jca.24488.
NLM
Lian X, Wang G, Zhou H, Zheng Z, Fu Y, Cai L. Anticancer Properties of Fenofibrate: A Repurposing Use. J Cancer 2018; 9(9):1527-1537. doi:10.7150/jca.24488. https://www.jcancer.org/v09p1527.htm
CSE
Lian X, Wang G, Zhou H, Zheng Z, Fu Y, Cai L. 2018. Anticancer Properties of Fenofibrate: A Repurposing Use. J Cancer. 9(9):1527-1537.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.