Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(8):1486-1499. doi:10.7150/jca.23277 This issue Cite
Research Paper
Cancer Stem Cells are Regulated by STAT3 Signalling in Wilms Tumour
Yanmei Liu1*, Xuexiang Gao1*, Shuo Wang1, Xuemin Yuan1, Yunqing pang1, Jian Chen2 ![]() , Jing Wang1
, Jing Wang1 ![]()
1. School of Stomatology Lanzhou University, Lanzhou, Gansu Province, PR China
2. Department of Pediatric Surgery, The First Hospital of Lanzhou University, Lanzhou, Gansu Province, PR China
*These authors contributed equally to this work
Received 2017-10-11; Accepted 2018-1-13; Published 2018-4-6
Abstract
The survival rates associated with Wilms tumour (WT) remain dismal despite advancements in detection and treatment strategies. Cancer stem cells (CSCs) are correlated with the initiation, recurrence and metastasis of tumours, but its impact on Wilms cancer stem cell (WCSC) maintenance remains unclear. In this study, CD133+ cells were successfully isolated from a single-cell suspension of the G401 Wilms tumour cell line using magnetic activated cell sorting (MACS). Signal transducers and activators of transcription 3 (STAT3) has been implicated in tumorigenesis, but its contribution to the metastatic progression of WCSCs has not been investigated. Here, we show that STAT3 is overexpressed in WCSCs. Activation of STAT3 in WCSCs initiated a forward feedback loop that was responsible for mediating the aggressive malignant character of Wilms tumour cells in vitro and in vivo. Treatment of CD133+ cells with stattic, a STAT3 inhibitor, also inhibited tumour formation and progression in xenograft animal models in vivo. Collectively, these studies revealed a critical role of STAT3 signalling in WCSC proliferation and motility and a role for CD133 in cancer stem-like cell function, providing evidence for CD133 as a potential therapeutic target in Wilms tumour.
Keywords: Wilms Tumour, Cancer stem cells, STAT3, CD133
Introduction
Wilms tumour has become one of the most common malignant tumours in paediatrics because of its low age of incidence and high malignancy [1]. The National Wilms Tumour Study Group (NWTSG) reported that its incidence throughout the world was approximately 1/1,000,000 over the last three years, accounting for approximately 90% of malignant tumours in children's kidneys [2]. Most nephroblastoma cases are unilateral (one kidney). The bilateral (two kidney) incidence is approximately 10%, and the age of incidence is low and concentrated in childhood. The prevalence rate of adults is very low, only approximately 1% [3]. Presently, the primary treatment of nephroblastoma is surgery combined with chemotherapy, and the 5-year survival rate has been significantly improved. However, the main cause of death in patients with nephroblastoma is metastasis and recurrence of the tumour, and lung metastasis occurs earlier in many cases with a poor prognosis [4-5]. Therefore, how to effectively target treatment of nephroblastoma as well as reduce the metastasis rate and recurrence rate have become the foci of clinical research.
In recent years, the theory of cancer stem cells (CSCs) has laid an important foundation for tumour research as well as clinical diagnosis and treatment and led to a new breakthrough, which has become the forefront and hot spot of current oncology research. Cancer stem cells are just a few of the tumour cell subsets that have the characteristics of stem cells in tumour tissue and have unlimited proliferation potential; CSCs can be updated indefinitely and differentiate into heterogeneous tumour cells. Cancer stem cells offer an important therapeutic target and research foundation for the treatment of multiple malignant tumours, which can kill cancer stem cells in clinical tumour treatment and provide the possibility for the complete cure of tumours [6-7]. In current cancer research, we have successfully detected and isolated cancer stem cells from lung cancer, gastric cancer, colorectal cancer, and breast cancer, among others and further explored the relationship between the presence of cancer stem cells and characteristics of tumour resistance, recurrence, metastasis and adverse prognosis [8-11].
The theory of cancer stem cells provides a new target for the treatment of solid tumours. Biological behaviours, such as the proliferation, invasion and apoptosis of tumour cells, are the basis for the occurrence and development of a tumour [12]. Cancer stem cells have been the most important cell type in basic research of tumour therapy because of their strong ability to proliferate and metastasize and high resistance to radiotherapy and chemotherapy [13].
CD133 is a transmembrane transport protein that was proven to be the surface-specific antigen of human hematopoietic stem cells [14]. Subsequently, CD133 was proven to be a starting factor in epithelial cells, lymphocytes, and angiogenesis. CD133, or CD133 combined with other markers, has been widely used to screen tissue stem cells, such as prostate tissue, colorectal cancer tissue, and glioblastoma tissue [15-17]. At the same time, researchers have used the CD133 polyclonal antibody to separate cancer stem cell groups in tumour tissues, including liver tumours [18] and pancreatic tumours [19-20]. Therefore, as a definite tumour stem cell marker, CD133 provides an important means for identification and sorting of stem cells into various tumours and provides an essential reference for the targeted therapy of tumours.
The signal transduction and transcription activator family (STAT) includes STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b, and STAT6, with STAT3 being an important member [21]. STAT3 is composed of approximately 770 amino acids with three isomers—STAT3α、STAT3β and STAT3γ [22]. Under normal circumstances, STAT3 regulates the transcription of target genes by phosphorescent activation (p-STAT3) and maintains and regulates various biological behaviours in the normal human body through activation of the STAT3 signalling pathway, including cell growth, differentiation, apoptosis, immune and so on [23], and abnormal activation of STAT3 may lead to excessive expression of VEGF, leading to abnormal proliferation of cells and tumorigenesis [24-25]. Studies have demonstrated that phosphorylation of STAT3 in the nucleus activates target gene transcription, promoting tumour angiogenesis and leading to the occurrence, invasion and metastasis of tumours [26]. Rokavec showed that abnormally high expression of STAT3 could inhibit apoptosis of tumour cells by regulating the expression level of the apoptosis-related proteins Cyclin D1 and Bcl-2 to promote colon cancer [27]. Moreover, Schroeder A showed that the elevation of STAT3 expression in prostate cancer was closely related to the adverse prognosis of patients [28].
Currently, treatment using the STAT3 signalling pathway has become the focus of tumour therapy; by inhibiting the STAT3 signalling pathway, the proliferation and invasion of tumour cells are reduced and apoptosis is promoted, achieving the purpose of tumour treatment [29-31]. A Horiguchi used the STAT3 phosphorylation inhibitor WP 1066, which reduces the expression of ALD and CD133, to significantly inhibit the proliferation of tumour cells and enhance apoptosis of tumour cells [32]. Mokoto selectively inhibited the proliferation and invasion of head and neck squamous cell carcinoma cells by inhibiting STAT3 phosphorylation and nuclear transfer, thereby improving the poor prognosis of patients [33]. However, no relevant study has reported on whether inhibition of the STAT3 signalling pathway has an effective killing effect on Wilms tumour stem cells.
Based on the above, we selected CD133 as a marker in the G401 Wilms tumour cell line using magnetic activated cell sorting to select Wilms tumour cells. The properties of stem cells were further identified using the STAT3 inhibitor stattic, which inhibits phosphorylation of STAT3, and by treating WCSCs. The effects of the STAT3 signalling pathway on the characteristics of tumour stem cells and its effect on apoptosis were detected by performing a clone formation experiment, Annexin-v-FITC/PI cell apoptosis experiment, western blot experiment and nude mice tumour test. We preliminary explored the possible mechanism of the STAT3 signalling pathway to promote apoptosis of WCSCs and explored the effect of STAT3 inhibition on apoptosis of tumour stem cells of Wilms tumour, sequentially providing a new therapeutic target and theoretical basis for the clinical treatment of Wilms tumour.
Materials and Methods
Cell lines and Cell culture
G401 cells (human nephroblastoma cell line) were purchased from Peking Union Cell Resource Center and were routinely maintained in McCoy 5A medium supplemented with 10% FBS and penicillin/streptomycin at 37°C in a humidified atmosphere of 5% CO2. The medium was renewed every two days.
Experimental Animals
BALB/c-nu/nu mice (male, aged between 4 and 5 weeks and weighing 16-18 g) were purchased from Beijing Virton Experimental Animal Co., Ltd (Beijing, China). Animals were cared for in the Animal Experiment Center of Gansu University of Traditional Chinese Medicine, were housed under pathogen-free conditions and were fed a standard diet and water. All surgical procedures and care administered to the animals had to conform with the ethics committee of Stomatology of Lanzhou University.
Reagents
McCoy's 5A was obtained from Gibco (Gibco, USA). Dimethyl sulfoxide was purchased from Sigma (Sigma, USA). Jimsa dye solution was purchased from Abcam (Abcam, USA), and Trizol was provided by Takara (Takara, Japan). Anti-CD133, CD133 microbeads, FCR blocking reagent and Labeling Check reagent were all obtained from Miltenyi Biotechnology (Miltenyi Biotec, Germany). The Stattic inhibitor (purity >99%, dissolved in DMSO at a final concentration of 0.1%, and stored at -20°C) was obtained from Selleck (Selleck, USA).
Cell detection
A single-cell suspension of 1×106 /ml Wilms tumour cells was used for cell detection. Cells were incubated with anti-CD133 and anti-CD44 antibodies for 30 min at 4˚C in the dark. After washing in PBS, cells were incubated with secondary antibodies for 30 min at room temperature in the dark. Next, the cells were resuspended in 300 μL of PBS for FCM detection.
Magnetic-activated cell sorting of CD133 cells
G401 cells were digested with 1000 μl of type I collagenase and were passed over 40-μm nylon cell strainers (at a density of 5×107/ml). The cells were resuspended in 300 μL of PBS with 100 µl of FCR blocking regent. Cells were incubated with CD133/l rabbit anti human polyclonal immunomagnetic beads for 30 min at 4˚C. The CD133+ cells obtained from the column were centrifuged and resuspended in a solution of PBS and were analysed using a MACS Multistand (Miltenyi Biotec, Germany). The purity of the CD133+ and CD133- cells was evaluated using standard flow cytometric analysis.
Tumour sphere assay
CD133+ cells, CD133- cells and unsorted G401 cells (at a density of 1×104/well) were plated into 6-well ultra-low attachment plates in McCoy 5A medium supplemented with 2% FBS. Next, spheres (>50 μm) were counted and imaged after 21 days under a microscope, and photos were acquired. Quantification of colony formation was processed using the “Colony area” plugin on ImageJ.
Clone formation experiment
CD133+ cells, CD133- cells and unsorted G401 cells were dissociated into single cells (2000 /well), were plated into culture dishes (diameter, 10 cm) and were allowed to grow for 21 days. The medium was changed every three days until the cells started to grow and formed floating tumour spheres. Next, the media were removed, cell colonies were stained with crystal violet for 30 min, and cells were washed with PBS, air dried at room temperature for 1 h, and washed with water.
Wound healing assay
Cells were cultured in six-well plates until they reached 90% confluence. A horizontal wound was gently created in monolayers using a 1-ml sterile pipette tip. The cells were subsequently washed 3 times with PBS to remove detached cells, the medium was replaced with fresh medium, and three random images were acquired at 50× magnification when the scratch was performed (T0) and after 24 h (T24). The images were captured using an inverted microscope and camera at the designated times to assess the inhibition of wound closure. The migration distance was determined as a reduction in the wound gap using ImageJ software.
RNA extraction and quantitative reverse transcription PCR
Total RNA was isolated from primary sorted CD133+ cells, CD133- cells and parental cells using the RNA Isolation Kit, and cDNA synthesis and quantitative reverse transcription PCR (qRT-PCR) were carried out according to the manufacturer's protocol (Sangon Biotech, Shanghai, China).
Premier sequences and product sizes
| Gene Name | Sequence(5'-3') | Tm | Amplicon length(bp) |
|---|---|---|---|
| SOX2 | F:CTGAGCGCCCTGCAGTACAA | 64.8 | 82 |
| R:GCGAGTAGGACATGCTGTAGGTG | 63.3 | ||
| NANOG | F:CCTGTGATTTGTGGGCCTGA | 64.4 | 168 |
| R:CTCTGCAGAAGTGGGTTGTTTG | 62.1 | ||
| OCT | F:GAGGAAGCTGACAACAATGAAAATC | 62.4 | 109 |
| R:GGTTGCCTCTCACTCGGTTC | 62.2 | ||
| GADPH | F:GCACCGTCAAGGCTGAGAAC | 63.3 | 138 |
| R:TGGTGAAGACGCCAGTGGA | 64.0 |
Tumorigenicity Experiment In vivo
After one week of acclimation, BALB/ c-nu/nu mice were randomly divided into three groups (n=5 per group) and were then subcutaneously (s.c.; 5×105 / 5×104 / 5×105 in 200 μl per mouse) injected into the right flank with CD133+ cells (5×105), CD133+ cells (5×104) and CD133- cells (5×104). Tumour progression was monitored every 2 days by pulsation, and the tumour size was measured using a digital caliper. The tumour volume was calculated according to the following formula: volume (mm3) = (a*b2) /2, where a and b are the longest and shortest tumour diameters, respectively. Twelve weeks later, the nude mice were sacrificed, and the tumours were obtained.
MTT assays
Exponentially growing CD133+ cells (5×104/ml/well) were plated in a 96-well plate in 100 μl of complete growth medium and were then treated with different concentrations of stattic (0 μM, 0.625 μM, 1.25 μM) on the second day after cell attachment. Incubation was carried out at 37ºC for 24 h. Next, 20 μl of MTT (Sigma-Aldrich) solution was added to each well (5 mg/ml) and was incubated for 4 h. The supernatants were removed from the wells, and the reduced MTT bromide dye was solubilized in 150 μl of DMSO/well. The absorbance at 490 nm was determined on a plate reader.
Annexin V apoptosis assays
CD133+ cells were cultured in McCoy's 5A medium containing 0 μM, 0.625 μM, or 1.25 μM stattic for 24 h, and then, cells were washed twice with serum-containing medium. Next, 300 μl of 1× Binding buffer and 5 μl of Annexin v-FITC binding solution were added, and the samples were incubated for 15 min in the dark. Finally, 5 μl of pi dye liquor was added for flow cytometry detection.
Western blot analysis
Sorted CD133+ cells were treated with McCoy's 5A containing 0 μM, 0.625 μM, or 1.25 μM stattic for 24 h. Thereafter, protein lysates were collected and a protein assay (BioRad) was performed. The concentration of total protein in cells was quantified using the bicinchoninic acid (BCA) protein assay reagent following the manufacturer's instructions. Cells were lysed with MPER (Pierce) supplemented with protease and phosphatase inhibitors for 30 min on ice, and lysates were clarified by centrifugation for 5 min at 12,000 /g. Twenty-five to fifty milligrams of lysates were loaded onto 5% SDS-PAGE gels and were transferred onto PVDF membranes. The membranes were blocked with 5% skim milk for 1 h and were incubated overnight at 4°C with anti-Bcl-2 (ImmunoWay, USA), anti-Bcl-xl (ImmunoWay, USA), anti-P-STAT3 (ImmunoWay, USA), anti- STAT3 (ImmunoWay, USA) antibody, anti-β-actin (ImmunoWay, USA) antibody (1/1000 dilution) was used as the internal loading control. The membranes were washed three times and then were incubated with a secondary antibody (1/1000 dilution) at room temperature for 1 h. Finally, the membranes were washed three times and were exposed in the dark room; ImageJ software was used for image analysis.
Anti-tumour Efficacy in CD133 + Xenografts
Ten male BALB/ c-nu/nu mice at 5 weeks of age were subcutaneously injected into the right flank with 1×105 CD133+ cells, in a total volume of 100 μl of sterile PBS (five mice per condition). Chemotherapy was administered when the tumour diameter reached 4 mm. The mice were intraperitoneally injected (i.p.) with the STAT3 inhibitor stattic (10 mg/kg/day) or vehicle (normal DMSO). Tumour progression was monitored every 2 days by pulsation, and the tumour size was measured using a digital caliper. The tumour volume was calculated according to the following formula: volume (mm3) = (a*b2)/2. The curve of tumour growth was constructed according to the tumour volume and time of implantation. Twelve weeks later, the mice were killed by cervical dislocation, and the tumours were measured. Xenografts were frozen at -80°C for subsequent western blot analysis. Formalin-fixed and paraffin embedded samples were prepared for HE staining. Images were taken with a Nikon camera.
Statistical analysis
The results were analysed using GraphPad Prism 5.0 software. P values less than 0.05 were considered to be statistically significant. The T-test was performed to determine the difference between the control and treated group. All data are shown as the means ± SD.
Results
CD133+ cells are highly expressed in Wilms tumour cells
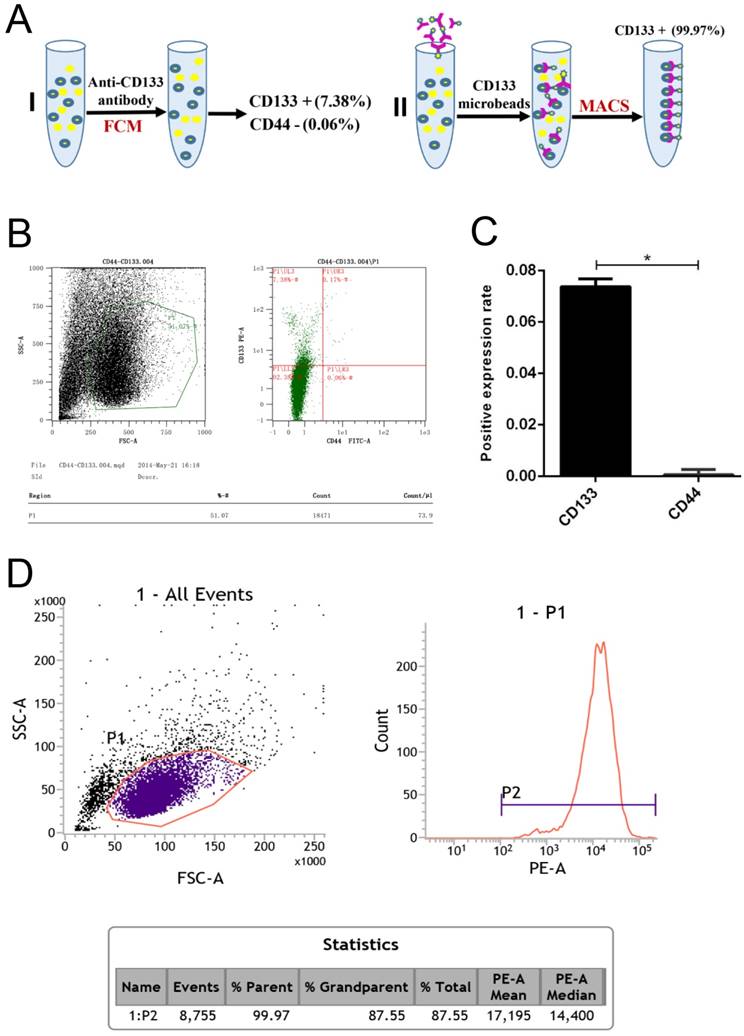
In our experiments, CD133+ cells were detected in primary human Wilms tumour cell samples using flow cytometry (Fig 1.A). The expression rate in CD133+ cells was 7.38%, but that for the CD44+ cells was only 0.06% (Fig 1.B-C). Following immunomagnetic sorting with CD133 beads, three measurements of CD133 expression in the CD133+-sorted population indicated a high mean CD133+ expression rate of 99.97% (Fig 1.D).
Flow cytometry analysis of Wilms tumour stem cell markers in G401 cells. A, Schematic for the isolation of CD133+ cells by magnetic-activated cell sorting (MACS). B, Flow cytometry analysis of CD133 and CD44 expression in G401 cells. C, Quantitative histogram of the cell number, *p<0.05. D, Flow cytometry revealed a high CD133+ expression rate in the CD133-sorted population. The percentage of CD133+-expressing cells in Wilms tumour is shown. Each experiment was repeated three times.
CD133 + cells have functional features of CSCs
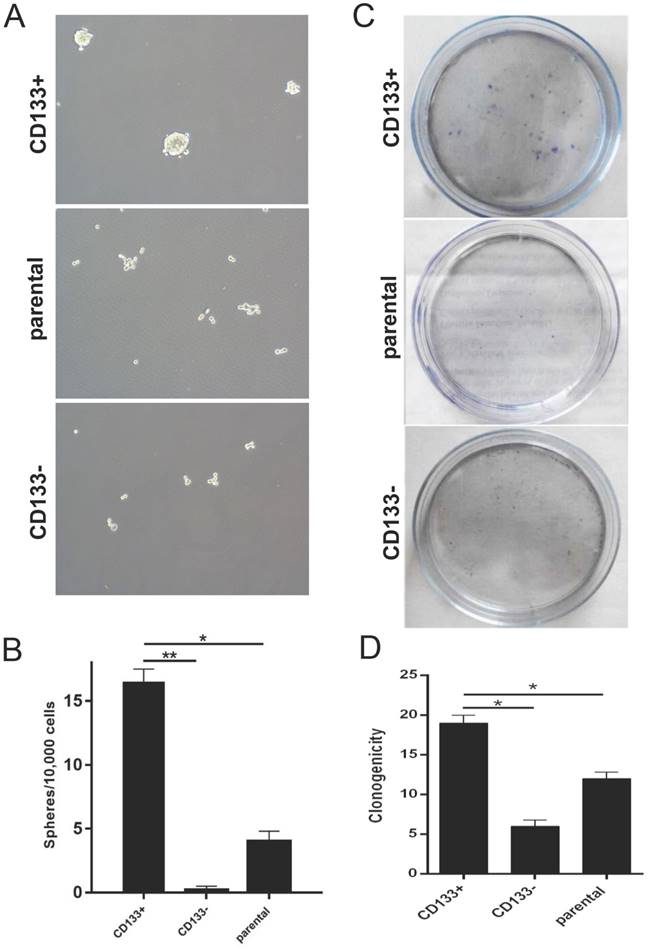
We next examined the self-renewal property of CD133+ cells using the in vitro sphere formation assay, colony assay and wound healing test. We found that CD133+ cells exhibited a significantly higher oncosphere efficiency, colony-forming efficiency and migration capabilities than CD133- cells and primary Wilms tumour cells (Fig 2.A-D). Thus, we defined CD133+ cells as WCSCs.
In vitro identification of Wilms tumour cancer stem cells (WCSCs). A-B, Mammospheres were formed and differentiated to mature mammospheres after 21 days. A, View of representative photos; B, quantitation of sphere formation. Cell were imaged using light microscopy (magnification, 10×) and are shown as the means ±SD. C-D, Colony formation of CD133+, CD133- and parental cells. C, View of representative wells; D, quantitation of colony formation; the data represent the average of at least three samples. One of 3 experiments is shown. The numbers of spheres are expressed as the means ± SD, *p < 0.05, **p< 0.01.
WCSCs originated from G401 cells expressing the stem cell markers
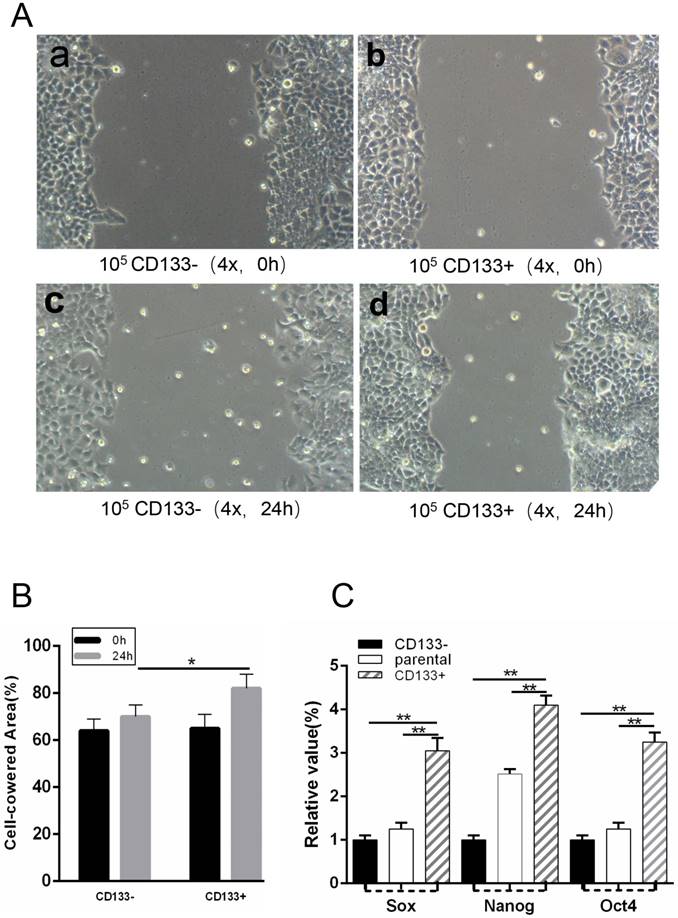
CD133+ cells expressed more cancer stem cells markers, such as Sox2, Nanog and Oct4, than CD133- cells. To isolate WCSCs from G401 cells, we detected the mRNA levels of Sox2, Nanog and Oct4 in CD133+ cells, CD133- cells and unsorted cells by RT-PCR. We found that CD133+ cells exhibited significantly higher Sox2, Nanog and Oct4 expression levels than CD133- and primary Wilms tumour cells (Fig 3.C).
(T0) A wound was made in the monolayer, and cells were allowed to migrate for 24 h (T24). A, Representative images at T0 and T24 are shown. Scale bars, 100 μm. B, Quantification of wound closure. The bars represent the average of three independent experiments. C, Expression levels of Sox, Nanog and Oct4 in CD133+ cells, CD133- cells and parental cells by RT-PCR. *p < 0.05, **p< 0.01.
CD133 + cells possess more powerful tumorigenicity than CD133 - cells
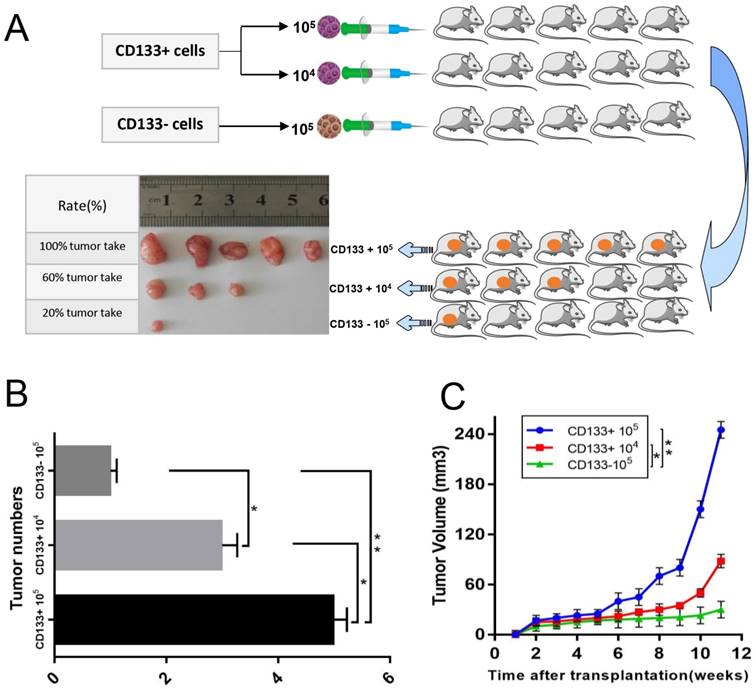
The above results suggested that CD133+ could be an important determinant of therapeutic resistance in Wilms tumour cells. We next assessed whether CD133+ cells were more tumorigenic than CD133- cells or unsorted cells. We found that the tumour formation rate of nude mice injected with CD133+ cells (80%) was higher than that of nude mice injected with CD133- cells (20%), and the growth rate was much faster (Fig 4.A-C).
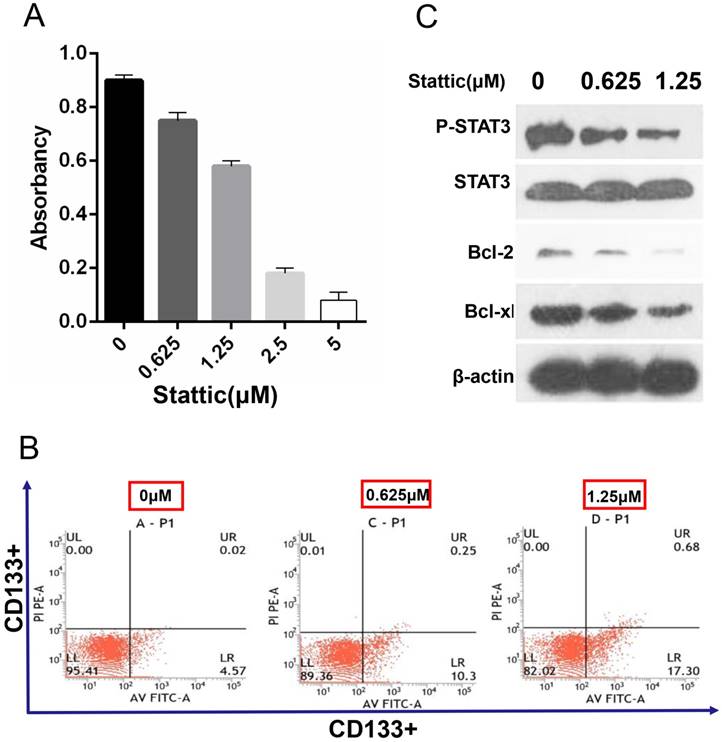
The effective inhibition concentration of stattic is detected by the MTT assay
The results showed that the survival rate of CD133+ cells was decreased with the increase in the stattic drug concentration (0-5 μM). Under concentrations of 0.625 μM and 1.25 μM stattic, the inhibition rates of stattic were 25% and 46%, respectively (Fig 5.A). Therefore, we chose 0.625 μM and 1.25 μM as the effective concentrations of stattic in CD133+ cells of Wilms tumours.
Annexin-v-FITC/PI detection shows that stattic promotes the apoptosis of CD133 + cells
Annexin-v-FITC/PI double staining is a sensitive index to detect early apoptosis. The results showed that the apoptosis level of CD133+ cells was increased significantly with the concentration of stattic and that the number of apoptotic CD133+ cells was increased with increased stattic concentration (Fig 5.B).
In vivo xenograft study. A, Sorted CD133+ cells and CD133- cells were subcutaneously injected into the right flank of athymic nude mice. Representative image of a xenograft tumour is shown. B, Quantification of the tumour numbers. C, Tumour volume of mice. The data were pooled from 3 independent experiments and are presented as the means ± SD, *p < 0.05, **p< 0.01.
Effect STAT3 inhibition on drug sensitivity. A, The effective concentration of stattic was measured by the MTT assay. B, CD133+ cells were treated with different concentrations (0 μM, 0.625 μM, and 1.25 μM) of the STAT3 inhibitor stattic for 24 hours. After staining with Annexin V-FITC and PI, apoptotic cells were analysed by flow cytometer. The numbers in each plot indicate the percentage of apoptotic cells. C, The levels of phosphorylated STAT3, STAT3 and apoptosis markers (Bcl-2 and Bcl-xl) were compared by Western blot analysis. β-Actin protein expression served as the loading control. Representative blots are shown. Saline was used as a control in stattic treatment experiments.
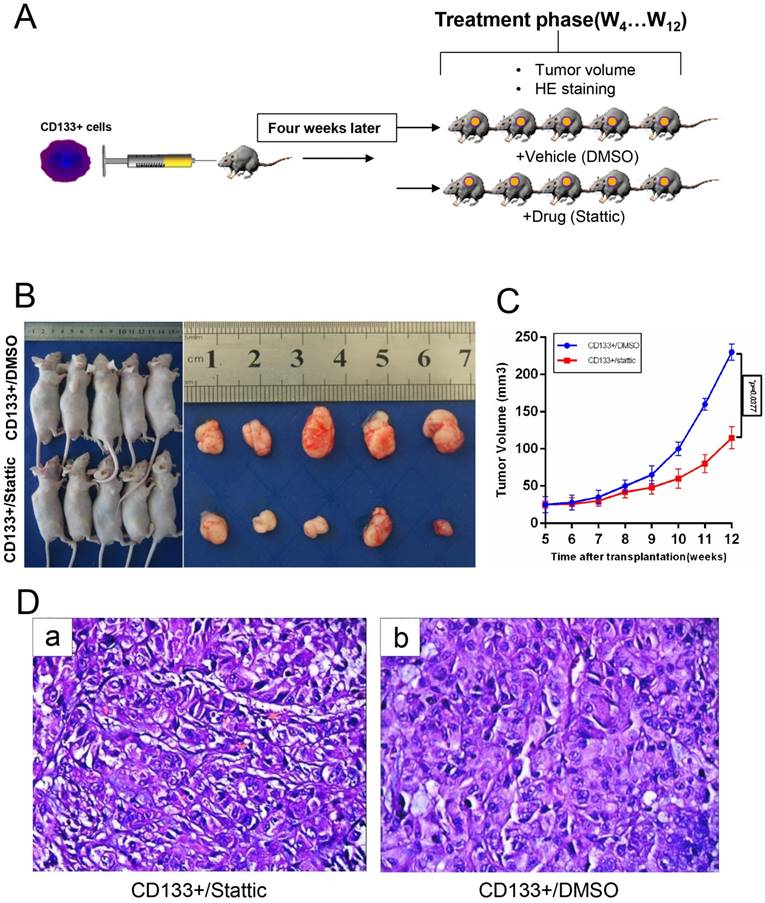
Anti-cancer Efficacy of Stattic on the CD133+ Xenograft Tumour Model In vivo
To confirm that stattic could inhibit tumour growth in vivo, a model was generated in which CD133+ cells were administered by subcutaneous injection into male nude mice (Fig 6.A). Wilms tumours in nude mice were induced by administering stattic or DMSO. At 7 weeks of stattic exposure, CD133+/stattic displayed tiny Wilms tumours. By contrast, CD133+/DMSO mice showed Wilms tumours of larger sizes (Fig 6.B-C). Tumours in stattic- or DMSO-treated mice were confirmed by histology staining (Fig 6.D), which showed cellular heterogeneity in the control group compared with the positive group, including chromatin condensation, nuclear condensation and disappearance of cell structure. By contrast, the nuclei of the control group were deeply stained and mitotic figures were increased, indicating that the cells were still in a more active proliferative state. Overall, stattic administration effectively suppressed Wilms tumour growth compared with DMSO treatment. Altogether, STAT3 is implicated in the tumorigenesis of Wilms tumour. Consequently, stattic-silenced CD133+ cells formed a smaller tumour burden and displayed much lower xenograft tumour formation rates than DMSO-treated cells. Altogether, these data suggest that CD133+ contributes to the activation of STAT3 signalling and tumorigenesis of Wilms tumour.
Western blot analysis
Expression of p-STAT3 and STAT3 was decreased significantly after stattic treatment, which showed that stattic could effectively inhibit phosphorylation of STAT3. At the same time, after stattic treatment, expression of Bcl-2, Bcl-xl decreased, which showed that stattic inhibited expression of Bcl-2 and Bcl-xl protein, thus promoting apoptosis of CD133+ cells of Wilms tumour (Fig 5.C).
In vivo validation of the STAT3 pathway for drug sensitivity. A, CD133+ cells were subcutaneously injected into the right flank of 5-week-old BALB/c nude mice (n=5), and then the mice were treated with DMSO or stattic (10 mg/kg/day). B-C, Transplanted tumour size and volume of the mice, *p < 0.05. D, Haematoxylin and eosin staining of the tumour tissues isolated from each group.
Discussion
In terms of cancer stem cell markers in Wilms tumours, Shukrun and colleagues reported that the tumors' heterogeneous cell population contains a small fraction of cancer stem cells (CSC) identified by two markers: (NCAM1) expression and Aldehyde dehydrogenase 1 (ALDH1) enzymatic activity, confirmed that NCAM1+ALDH1+ cells have been identified as the CSC fraction in WT. The capability of the tumor to grow and propagate is maintained solely by these cells [34]. Pode-Shakked et al. demonstrates a new approach for the treatment of the largest and clinically most difficult class of Wilms' tumours. The identification of the NCAMþALDH1þWT CSC is an essential step towards a further increase in (long-term) survival of the patients [35].
Furthermore, more recently, Wilms Tumor Protein 1 (WT1) is also shown to be a transcription factor which is highly expressed by MPM [36] and is supposed to be a key regulator of EMT [37]. Plönes and colleagues' research found that knockdown of WT1 suppresses chemoresistance in both human MPM cell lines (MSTO and H2052) compared with control (scrambled siRNA). WT1 plays a key role in several features with relation to MPM treatment. WT1 may be an interesting target in Human Mesothelioma, which withstands most oncological treatment [38]. Brigitte Royer-Pokora1 et al. describe the establishment and characterization of long-term cell cultures derived from five individual WTs with WT1 mutations. Gene expression profiling revealed that the WT cell lines are highly similar to human mesenchymal stem cells (MSCs)[39]. Inoue K et al 's study implied that WT1 gene plays a critical role in leukemogenesis and performs an oncogenic function in hematopoietic progenitor cells [40].
Cancer stem cells (CSC), also called tumour-initiating cells, are considered to be the origin of human malignant tumour cells. The existence of cancer stem cells affects the invasion ability of malignant tumours, leading to resistance to radiotherapy and chemotherapy and a poor prognosis [41]. Therefore, targeted therapy of cancer stem cells is the focus of current tumour research. Targeted therapy of cancer stem cells involves specific surface markers and intrinsic signalling pathways of different tumours. The signalling pathway of tumour stem cells plays a critical role in the development of tumours, such as malignant glioma [42], squamous cell carcinoma [43] and lung cancer [44]. The tumour suppressor signalling pathway is closely related to the occurrence, metastasis and recurrence of cancer stem cells. Therefore, targeted killing of cancer stem cells can inhibit the self-renewal and differentiation potential of cancer stem cells, thus providing more effective treatment of tumours.
The surface of the cancer stem cell membrane has specific protein molecules that are closely linked to the clinical identification and diagnosis of the tumour. In recent years, various specific cancer stem cell surface markers, such as pancreatic cancer stem cell markers CD44 and CD24 [45], breast cancer stem cell markers CD44 and ALDH [46], liver cancer stem cell markers CD133 and CD90 [47] among others, have been found. These markers can accurately screen and isolate cancer stem cells, thus providing important help for tumour treatment [48]. Among them, CD133+ cells, an eminent cancer stem cell subpopulation, whose proportion among tumour cells is approximately 1-10%, are often used in to screen and identify multiple tumour stem cells [49]. Singh et al. used CD133 as a cancer stem cell marker to obtain brain cancer stem cells for the first time [50]. Subsequently, CD133 was used for the sorting and purification of various cancer stem cell surface markers, as shown by Silva and Dah-Ching Ding, who used CD133 to obtain ovarian cancer stem cells and endometrial cancer stem cells [51-52]. In this study, the G401 cell line was selected as the study object, and the typical cancer stem cell markers CD133 and CD44 were chosen as the candidate markers. We found that CD44 showed almost no expression in the G401 cell line by flow detection, while the expression level of CD133 was approximately 7.38%. Therefore, we chose CD133 as the marker of the G401 cell line. High purity (98.9%) of CD133+ cells was obtained from the G401 cell line by immunomagnetic bead sorting, and CD133+ cells exhibited significantly higher Sox2, Nanog and Oct4 expression levels.In the current study, CD133+ cells have the characteristics of cancer stem cells in G401 cell lines, and the proliferation and metastasis of CD133+ cells are more potent than those of CD133- cells and parental cells in vitro and in vivo.
Although CD133 plays a very important role in current research on cancer stem cells, there are still many questions and uncertainties about CD133. Shmelkov et al. report that CD133-colon cancer cells also have the characteristics of cancer stem cells [53]. Meng et al. detected A549 and H445 lung cancer cells found that CD133 negative cells and CD133 positive cells have the same tumorigenicity, self-renewal, proliferation, differentiation, invasion and resistance to chemotherapy, and that A549 and H446 lung cancer cells in the CD133- cells and CD133+ cells contain lung cancer stem cells [54]. On the other hand, the level of oxygen concentration affects the expression of CD133. It has been found [55]that decreasing the oxygen supply (20% down to 2%-3%) in the culture of medulloblastoma and glioma cells increases the expression of CD133 mRNA by AC133 and AC141 and hypoxia has been shown to promote CD 133 Positive pancreatic cancer cells are more aggressive and proliferative [56]. Therefore, further experiments are needed to determine whether oxygen concentrations in different environments are an important experimental variable in cancer stem cell experiments. Moreover, the biological function of CD133 is not clear. Whether CD133 or its glycosylated epitopes play a direct role in regulating the phenotype of tumor stem cells is unclear yet, and little is known about the biological function of CD133 [57]. Besides, Bidlingmaier et al. proposed that analysis of CD133-targeted knockdown in CD133 + glioma cells followed by functional studies and tumorigenesis will help clarify whether CD133 is involved in the functional regulation of cancer stem cells [58]. Therefore, whether CD133 is a specific marker molecule shared by cancer stem cells requires a large amount of further experimental studies to confirm.
STATs, which play a key role in the signalling pathway mediated by cytokines and interferon, are activated by phosphorylation, with consequent formation of dimers, entry into the cell nucleus, activation of specific promoter sequences, and induction of a series of physiological phenomena, including cell growth, differentiation, proliferation, and apoptosis [59-60]. STAT3 is an important component of the signal transduction and transcription activator family. Activated STAT3 dimerizes for nuclear translocation and to regulate tumour cell growth, invasion, inhibition of apoptosis and a series of biological reactions in various human malignant tumours[61-62]. Research suggests that the occurrence and development of the tumour and resistance of chemotherapeutic drugs are mainly due to anti-apoptosis of tumour cells, and the anti-apoptotic protein Bcl-2 family includes the key transduction factors in the tumour cell apoptosis pathway [63]. This family includes the bh3 proteins, such as bid, BIM, bad, noxa, puma, and anti-apoptosis survival proteins, such as Bcl-2, Bcl-2-like 1 (Bcl -xl), and myeloid leukemia-1 (MCL-1). Bcl-2-associated x protein (Bax) and bak are additional apoptotic proteins that induce apoptosis [64]. The Bcl-2 gene was first detected in follicular b lymphoma, its location is between chromosomes 18 and 14 and activation is controlled by the immunoglobulin heavy chain promoter. Bcl-xl is the protein product of the Bcl-xl gene and can inhibit apoptosis [65]. Bcl-2 and Bcl-xl have been proven to enhance the apoptosis resistance of tumour cells [66], enhancing cell viability and drug resistance in various solid tumours. Much literature has reported that the expression level of vascular endothelial growth factor (VEGF) is up-regulated after activation of STAT3 and the expression of cyclin D1, Bcl-2 and Bcl-xl in downstream apoptotic proteins is down-regulated, thus enhancing the resistance to characteristics of tumour cells [67-68]. In renal cell tumours, the effect of STAT3 inhibition on downstream apoptosis-related genes is not yet known. Therefore, this study investigated the effect of STAT3 inhibition on apoptosis of renal cell tumour cells after STAT3 suppression, preliminarily selected Bcl-2 and Bcl-xl as research objects, and discussed the possible mechanism of apoptosis the possible downstream mechanism of apoptosis.
In this study, phosphorylation of STAT3 was blocked, proliferation of tumour cells was decreased significantly, and expression of Bcl-2 and Bcl-xl was decreased after WCSCs were inhibited by stattic. Meanwhile, the experimental results in nude mice showed that the growth volume of the stattic treatment group was significantly lower than that of the control group and that the growth rate was decreased significantly. Collectively, our data indicate that we can effectively inhibit the growth of Wilms tumour by interfering with the STAT3 signalling pathway, thus providing an important and effective scientific basis for the treatment of Wilms tumour.
Abbreviations
WT: wilms tumour; CSC: cancer stem cells; WCSC: wilms cancer stem cells; MACS: magnetic activated cell sorting; STAT3: signal transducers and activators of transcription 3; NWTSG: national wilms tumor study group; Bcl-2: b-cell lymphoma-2; Bcl-xl: b-cell lymphoma-xl.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
The authors received a grant from the National Natural Science Foundation of China (No.81773942) and the International Scientific and Technological Cooperation Project of Gansu Province (No. 17YF1WA165) and the Natural Science foundation of Gansu province (No.1208RJZA217).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Green D M. The evolution of treatment for Wilms tumor[J]. Journal of pediatric surgery. 2013;48(1):14-19
2. Green D M, Breslow N E, D'Angio G J. et al. Outcome of patients with stage II/favorable histology Wilms tumor with and without local tumor spill: a report from the National Wilms Tumor Study Group[J]. Pediatric blood & cancer. 2014;61(1):134-139
3. Kalapurakal J A, Perlman E J, Seibel N L. et al. Outcomes of patients with revised stage I clear cell sarcoma of kidney treated in National Wilms Tumor Studies 1-5[J]. International Journal of Radiation Oncology* Biology* Physics. 2013;85(2):428-431
4. Metzger M L, Dome J S. Current therapy for Wilms' tumor[J]. The Oncologist. 2005;10(10):815-826
5. Chan K W, Lee K H, Mou J W. et al. Surgery for Wilms Tumor in Children in a Tertiary Centre in Hong Kong: A 15-year Retrospective Review[J]. Hong Kong Journal of Paediatrics (New Series). 2012;17(2):103-108
6. Kreso A, Dick J E. Evolution of the cancer stem cell model[J]. Cell stem cell. 2014;14(3):275-291
7. Al-Hajj M, Clarke M F. Self-renewal and solid tumor stem cells[J]. Oncogene. 2004;23(43):7274-7282
8. Sarvi S, Mackinnon A C, Avlonitis N. et al. CD133 + cancer stem-like cells in small cell lung cancer are highly tumorigenic and chemoresistant but sensitive to a novel neuropeptide antagonist[J]. Cancer research. 2014;74(5):1554-1565
9. Wakamatsu Y, Sakamoto N, Oo H Z. et al. Expression of cancer stem cell markers ALDH1, CD44 and CD133 in primary tumor and lymph node metastasis of gastric cancer[J]. Pathology international. 2012;62(2):112-119
10. Todaro M, Gaggianesi M, Catalano V. et al. CD44v6 is a marker of constitutive and reprogrammed cancer stem cells driving colon cancer metastasis[J]. Cell stem cell. 2014;14(3):342-356
11. Lawson D A, Bhakta N R, Kessenbrock K. et al. Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells[J]. Nature. 2015;526(7571):131-135
12. Malanchi I, Peinado H, Kassen D. et al. Cutaneous cancer stem cell maintenance is dependent on &bgr;-catenin signalling[J]. Nature. 2008;452(7187):650-653
13. Donnenberg V S, Donnenberg A D. Multiple drug resistance in cancer revisited: the cancer stem cell hypothesis[J]. The Journal of Clinical Pharmacology. 2005;45(8):872-877
14. Suetsugu A, Nagaki M, Aoki H. et al. Characterization of CD133 + hepatocellular carcinoma cells as cancer stem/progenitor cells[J]. Biochemical and biophysical research communications. 2006;351(4):820-824
15. Miki J, Furusato B, Li H. et al. Identification of Putative Stem Cell Markers, CD133 and CXCR4, in hTERT-Immortalized Primary Nonmalignant and Malignant Tumor-Derived Human Prostate Epithelial Cell Lines and in Prostate Cancer Specimens[J]. Cancer research. 2007;67(7):3153-3161
16. Prabhu V V, Allen J E, Dicker D T. et al. Small-Molecule ONC201/TIC10 Targets Chemotherapy-Resistant Colorectal Cancer Stem-like Cells in an Akt/Foxo3a/TRAIL-Dependent Manner[J]. Cancer Research. 2015;75(7):1423
17. Emlet D R, Gupta P, Holgadomadruga M. et al. Targeting a glioblastoma cancer stem-cell population defined by EGF receptor variant III.[J]. Cancer Research. 2014;74(4):1238-49
18. Ma S, Chan K W, Lee T K W. et al. Aldehyde dehydrogenase discriminates the CD133 liver cancer stem cell populations[J]. Molecular Cancer Research. 2008;6(7):1146-1153
19. Hermann P C, Huber S L, Herrler T. et al. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer[J]. Cell stem cell. 2007;1(3):313-323
20. Malecka-Panas E, Wlodarski W, Danilewicz M. et al. Expression and Clinical Significance of Cancer Stem Cell Markers CD24, CD44, and CD133 in Pancreatic Ductal Adenocarcinoma and Chronic Pancreatitis:[J]. Disease Markers. 2017;2017(4):3276806
21. Yu H, Lee H, Herrmann A. et al. Revisiting STAT3 signalling in cancer: new and unexpected biological functions[J]. Nature Reviews Cancer. 2014;14(11):736-746
22. Wang P, Xue Y, Han Y. et al. The STAT3-binding long noncoding RNA lnc-DC controls human dendritic cell differentiation[J]. Science. 2014;344(6181):310-313
23. Stuart H T, van Oosten A L, Radzisheuskaya A. et al. NANOG amplifies STAT3 activation and they synergistically induce the naive pluripotent program[J]. Current Biology. 2014;24(3):340-346
24. Yue P, Lopez-Tapia F, Tius M. et al. Small molecule inhibitor of Stat3 induces antitumor cell effects in vitro and antitumor effects in vivo against human glioma or breast cancer model[J]. Cancer Research. 2014;74(19 Supplement):3801-3801
25. Nguyen A V, Wu Y Y, Liu Q. et al. STAT3 in epithelial cells regulates inflammation and tumor progression to malignant state in colon[J]. Neoplasia. 2013;15(9):998-1008
26. Zhang L, Alizadeh D, Van Handel M. et al. Stat3 inhibition activates tumor macrophages and abrogates glioma growth in mice[J]. Glia. 2009;57(13):1458-1467
27. Rokavec M, Öner M G, Li H. et al. IL-6R/STAT3/miR-34a feedback loop promotes EMT-mediated colorectal cancer invasion and metastasis[J]. The Journal of clinical investigation. 2014;124(4):1853-1867
28. Schroeder A, Herrmann A, Cherryholmes G. et al. Loss of androgen receptor expression promotes a stem-like cell phenotype in prostate cancer through STAT3 signaling[J]. Cancer research. 2014;74(4):1227-1237
29. Wei W, Tweardy D J, Zhang M. et al. STAT3 Signaling Is Activated Preferentially in Tumor-Initiating Cells in Claudin-Low Models of Human Breast Cancer[J]. Stem cells. 2014;32(10):2571-2582
30. Don-Doncow N, Escobar Z, Johansson M. et al. Galiellalactone is a direct inhibitor of the transcription factor STAT3 in prostate cancer cells[J]. Journal of Biological Chemistry. 2014;289(23):15969-15978
31. Zhang X, Yue P, Page B D G. et al. Orally bioavailable small-molecule inhibitor of transcription factor Stat3 regresses human breast and lung cancer xenografts[J]. Proceedings of the National Academy of Sciences. 2012;109(24):9623-9628
32. Horiguchi A, Asano T, Kuroda K. et al. STAT3 inhibitor WP1066 as a novel therapeutic agent for renal cell carcinoma[J]. British journal of cancer. 2010;102(11):1592-1599
33. Adachi M, Cui C, Dodge C T. et al. Targeting STAT3 inhibits growth and enhances radiosensitivity in head and neck squamous cell carcinoma[J]. Oral oncology. 2012;48(12):1220-1226
34. Shukrun R, Pode S N, Dekel B. Targeted therapy aimed at cancer stem cells: Wilms' tumor as an example.[J]. Pediatric Nephrology. 2014;29(5):815-823
35. Pode-Shakked N, Dekel B. Wilms tumor-a renal stem cell malignancy?[J]. Pediatric Nephrology. 2011;26(9):1535
36. Kushitani K, Takeshima Y, Amatya VJ, Furonaka O, Sakatani A, Inai K. Immunohistochemical marker panels for distinguishing between epithelioid mesothelioma and lung adenocarcinoma. Pathol Int. 2007;57:190-199
37. von Gise A, Zhou B, Honor LB, Ma Q, Petryk A, Pu WT. WT1 regulates epicardial epithelial to mesenchymal transition through beta-catenin and retinoic acid signaling pathways. Dev Biol. 2011;356:421-431
38. Plönes T, Fischer M, Höhne K. et al. Turning back the Wheel: Inducing Mesenchymal to Epithelial Transition via Wilms Tumor 1 Knockdown in Human Mesothelioma Cell Lines to Influence Proliferation, Invasiveness, and Chemotaxis[J]. Pathology & Oncology Research. 2017:1-8
39. Royer-Pokora B, Busch M, Beier M. et al. Wilms tumor cells with WT1 mutations have characteristic features of mesenchymal stem cells and express molecular markers of paraxial mesoderm[J]. Hum Mol Genet. 2010May1;19(9):1651-68 doi: 10.1093/hmg/ddq042. Epub 2010 Jan 27
40. Inoue K, Tamaki H, Ogawa H. et al. Wilms' Tumor Gene (WT1) Competes With Differentiation-Inducing Signal in Hematopoietic Progenitor Cells[J]. Blood. 1998;91(8):2969-76
41. Visvader J E, Lindeman G J. Cancer stem cells: current status and evolving complexities[J]. Cell stem cell. 2012;10(6):717-728
42. Pallini R, Ricci-Vitiani L, Banna G L. et al. Cancer stem cell analysis and clinical outcome in patients with glioblastoma multiforme[J]. Clinical Cancer Research. 2008;14(24):8205-8212
43. Boumahdi S, Driessens G, Lapouge G. et al. SOX2 controls tumour initiation and cancer stem-cell functions in squamous-cell carcinoma[J]. Nature. 2014;511(7508):246-250
44. Hassan K A, Wang L, Korkaya H. et al. Notch pathway activity identifies cells with cancer stem cell-like properties and correlates with worse survival in lung adenocarcinoma[J]. Clinical cancer research. 2013;19(8):1972-1980
45. Li Y, Kong D, Ahmad A. et al. Pancreatic cancer stem cells: emerging target for designing novel therapy[J]. Cancer letters. 2013;338(1):94-100
46. De Beça F F, Caetano P, Gerhard R. et al. Cancer stem cells markers CD44, CD24 and ALDH1 in breast cancer special histological types[J]. Journal of clinical pathology. 2013;66(3):187-191
47. Yamashita T, Honda M, Nio K. et al. The evolution of diverse cancer stem cells in human liver cancer[J]. Cancer Research. 2013;73(8 Supplement):262-262
48. Abdullah L N, Chow E K H. Mechanisms of chemoresistance in cancer stem cells[J]. Clinical and Translational Medicine. 2013;2(1):1-9
49. Richardson G D, Robson C N, Lang S H. et al. CD133, a novel marker for human prostatic epithelial stem cells[J]. Journal of cell science. 2004;117(16):3539-3545
50. Singh S K, Hawkins C, Clarke I D. et al. Identification of human brain tumour initiating cells[J]. Nature. 2004;432(7015):396-401
51. Silva I A, Bai S, McLean K. et al. Aldehyde dehydrogenase in combination with CD133 defines angiogenic ovarian cancer stem cells that portend poor patient survival[J]. Cancer research. 2011;71(11):3991-4001
52. Shmelkov S V, Butler J M, Hooper A T. et al. CD133 expression is not restricted to stem cells, and both CD133+ and CD133- metastatic colon cancer cells initiate tumors[J]. Journal of Clinical Investigation. 2008;118(6):2111-2120
53. Dah-C D, Hwan-Wun L, Yu-H C. et al. Expression of CD133 in endometrial cancer cells and its implications[J]. Journal of Cancer. 2017;8(11):2142-2153
54. Meng X, Li M, Wang X. et al. Both CD133+ and CD133- subpopulations of A549 and H446 cells contain cancer-initiating cells. Cancer Sci. 2009;100(6):1040-1046
55. Blazek ER, Foutch JL, Maki G. Daoy medulloblastoma cells that express CD133 are radioresistant relative to CD133- cells, and the CD133+ sector is enlarged by hypoxia. Int J Radiat Oncol Biol Phys, 2007, 67(1): 1-5.8 Platet N, Liu SY, Atifi ME, et al. Influence of oxygen tension on CD133 phenotype in human glioma cell cultures. Cancer Lett. 2007;258(2):286-290
56. Hashimoto O, Shimizu K, Semba S. et al. Hypoxia induces yumor aggressiveness and the expansion of CD133-positive cells in a hypoxia-induciblefactor-1α-dependent manner in pancreatic cancer cells. Pathobiology. 2011;78(4):181-192
57. Bidlingmaier S, Zhu X, Liu B. The utility and limitations of glycosylated human CD133 epitopes in defining cancel stem cells. J Mol Med. 2008;86(9):1025-1032
58. Bidlingmaier S, Zhu X, Liu B. The utility and limitations of glycosylated human CD133 epitopes in defining cancer stem cells[J]. J Mol Med. 2008;86:1025-1032
59. Xie T, Wei D, Liu M. et al. Stat3 activation regulates the expression of matrix metalloproteinase-2 and tumor invasion and metastasis[J]. Oncogene. 2004;23(20):3550-3560
60. Siveen K S, Sikka S, Surana R. et al. Targeting the STAT3 signaling pathway in cancer: role of synthetic and natural inhibitors[J]. Biochimica et Biophysica Acta (BBA)-Reviews on Cancer. 2014;1845(2):136-154
61. Liu W H, Chen M T, Wang M L. et al. Cisplatin-selected resistance is associated with increased motility and stem-like properties via activation of STAT3/Snail axis in atypical teratoid/rhabdoid tumor cells[J]. Oncotarget. 2015;6(3):1750
62. Czabotar P E, Lessene G, Strasser A. et al. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy[J]. Nature reviews Molecular cell biology. 2014;15(1):49-63
63. Marzo I, Brenner C, Zamzami N. et al. The permeability transition pore complex: a target for apoptosis regulation by caspases and Bcl-2-related proteins[J]. The Journal of experimental medicine. 1998;187(8):1261-1271
64. Volkmann N, Marassi F M, Newmeyer D D. et al. The rheostat in the membrane: BCL-2 family proteins and apoptosis[J]. Cell Death & Differentiation. 2014;21(2):206-215
65. Renault T T, Teijido O, Antonsson B. et al. Regulation of Bax mitochondrial localization by Bcl-2 and Bcl-x L: keep your friends close but your enemies closer[J]. The international journal of biochemistry & cell biology. 2013;45(1):64-67
66. Rahmani M, Aust M M, Attkisson E. et al. Dual inhibition of Bcl-2 and Bcl-xL strikingly enhances PI3K inhibition-induced apoptosis in human myeloid leukemia cells through a GSK3-and Bim-dependent mechanism[J]. Cancer research. 2013;73(4):1340-1351
67. Levine B, Sinha S C, Kroemer G. Bcl-2 family members: dual regulators of apoptosis and autophagy[J]. Autophagy. 2008;4(5):600-606
68. Pei X Y, Sanderson M W, Youssefian L E. et al. Bcl-2 Antagonism Potentiates MEK1/2/Chk1 Inhibitor Lethality in Multiple Myeloma Cells Overexpressing Bcl-2 through a Stat3-Dependent Mechanism[J]. Blood. 2014;124(21):4763-4763
Author contact
![]() Corresponding authors: Jing Wang, Department of Periodontology, School of Stomatology, Lanzhou University, 199 Donggang Western Road, Lanzhou Gansu 730000, China. Phone: 0931-8915051, Fax: 0931-8915051, E-mail: lzukqwjcom and Jian Chen, Department of Pediatric Surgery, The First Hospital of Lanzhou University, 1 Donggang Western Road, Lanzhou Gansu 730000, China. E-mail address:chenjianlanyicom.
Corresponding authors: Jing Wang, Department of Periodontology, School of Stomatology, Lanzhou University, 199 Donggang Western Road, Lanzhou Gansu 730000, China. Phone: 0931-8915051, Fax: 0931-8915051, E-mail: lzukqwjcom and Jian Chen, Department of Pediatric Surgery, The First Hospital of Lanzhou University, 1 Donggang Western Road, Lanzhou Gansu 730000, China. E-mail address:chenjianlanyicom.