Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(2):239-249. doi:10.7150/jca.21336 This issue Cite
Research Paper
Early and Partial Reduction in CD4+Foxp3+ Regulatory T Cells during Colitis-Associated Colon Cancer Induces CD4+ and CD8+ T Cell Activation Inhibiting Tumorigenesis
Jonadab E. Olguín1,2, Itzel Medina-Andrade1, Emmanuel Molina1, Armando Vázquez1, Thalia Pacheco-Fernández1, Rafael Saavedra3, Carlos Pérez-Plasencia1, Yolanda I. Chirino1, Felipe Vaca-Paniagua1,2, Luis E. Arias-Romero1, Emma B. Gutierrez-Cirlos1, Sonia A. León-Cabrera1, Miriam Rodriguez-Sosa1, Luis I. Terrazas1,2 ![]()
1. Unidad de Biomedicina, Facultad de Estudios Superiores Iztacala, Universidad Nacional Autónoma de México (UNAM),
2. Laboratorio Nacional en Salud: Diagnóstico Molecular y Efecto Ambiental en Enfermedades Crónico-degenerativas, UNAM,
3. Departamento de Inmunología, Instituto de Investigaciones Biomédicas, UNAM
Received 2017-6-5; Accepted 2017-10-8; Published 2018-1-1
Abstract
Colorectal cancer (CRC) is the second most commonly diagnosed cancer in women and the third in men in North America and Europe. CRC is associated with inflammatory responses in which intestinal pathology is caused by different cell populations including a T cell dysregulation that concludes in an imbalance between activated T (Tact) and regulatory T (Treg) cells. Treg cells are CD4+Foxp3+ cells that actively suppress pathological and physiological immune responses, contributing to the maintenance of immune homeostasis. A tumor-promoting function for Treg cells has been suggested in CRC, but the kinetics of Treg cells during CRC development are poorly known. Therefore, using a mouse model of colitis-associated colon cancer (CAC) induced by azoxymethane and dextran sodium sulfate, we observed the dynamic and differential kinetics of Treg cells in blood, spleen and mesenteric lymph nodes (MLNs) as CAC progresses, highlighting a significant reduction in Treg cells in blood and spleen during early CAC development, whereas increasing percentages of Treg cells were detected in late stages in MLNs. Interestingly, when Treg cells were decreased, Tact cells were increased and vice versa. Treg cells from late stages of CAC displayed an activated phenotype by expressing PD1, CD127 and Tim-3, suggesting an increased suppressive capacity. Suppression assays showed that T-CD4+ and T-CD8+ cells were suppressed more efficiently by MLN Treg cells from CAC animals. Finally, an antibody-mediated reduction in Treg cells during early CAC development resulted in a better prognostic value, because animals showed a reduction in tumor progression associated with an increased percentage of activated CD4+CD25+Foxp3- and CD8+CD25+ T cells in MLNs, suggesting that Treg cells suppress T cell activation at early steps during CAC development.
Keywords: Regulatory T cells, colorectal cancer, depletion, Activated T cells, CD8+ cells
Introduction
Colorectal cancer (CRC) is one of the most common and deadly cancers in the world [1] and is also globally the second most common cancer in women and the third in men. The incidence rates are higher in developed countries than in developing countries, but the mortality rate is higher in the latter [2-6]. Approximately, one million new cases of CRC emerge per year with more than half a million deaths annually [7]. There is clearly a role for both genetic and epigenetic alterations underlying the development of CRC [8-10], contributing to the formation of immunogenic tumor-specific and tumor-associated antigens [11]. These tumor antigens allow for the identification and elimination of CRC cells by cells of the immune response [7, 12]. However, some self-regulation immunological mechanisms play a role in the avoidance of efficient tumor elimination, inducing immunosuppression. Examples for immunosuppressive cells are tumor-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs) and regulatory T (Treg) cells, which foster tumor progression and the formation of metastases [2, 13]
Treg cells are CD4+ T lymphocytes having a major role in the immunological self-tolerance and immune homeostasis during pathological and physiological immune responses [14]. Treg cells constitutively express the transcription factor Foxp3, which is required during their development and for their suppressive function [15, 16]. They also express constitutively some molecules associated with T cell activation: CD25, CTLA-4, GITR and LAG-3 [17]. Some mechanisms of suppression used by Treg cells have been described, which include: modulation of the expression of costimulatory molecules, secretion of immunosuppressive cytokines, IL-2 deprivation, induction of tryptophan catabolism and cytotoxicity [18].
In recent years, understanding the role that the immune response plays during CRC development has been a growing research of interest [19, 20], given that it is a clear example about how inflammation influences the onset of this type of cancer [21, 22]. Different studies have noted a deleterious role for inflammatory responses where IL-17 as well as STAT6 appear to be implicated [23, 24], but anti-inflammatory cells such as TAMs and MDSCs also appear to play a negative role in CRC development [13, 25]. Indeed, a tumor-promoting role for Treg cells during CRC has recently been described, and the ablation of Treg cells can evoke antitumor responses [26]. However, such ablation early in disease development results in an exacerbated inflammatory response with fatal consequences for the host. The timing of Treg cell ablation is therefore critical. In other studies on different types of cancer, the role of Tregs remains controversial [27-29]. In contrast, in a model of CRC using APCMin/+ mice, the depletion of Treg cells was shown to increase the accumulation of conventional T cells in intestinal tumors [30] However, in these background and pioneer papers, the experimental design was only focused on the final phase of CRC and excluded the possible role for Treg cells during CRC development. In the other hand, the use of monoclonal antibodies against some immune-regulatory molecules has been tested [31]; for example, blocking of the PD-1 pathway has been demonstrated to induce a limited improvement in patients with CRC [32].Therefore, targeting Treg cells could be a possible anti-tumor immunotherapy.
In the present work, we analyzed the kinetics of the recruitment of Treg cells as well as the kinetics of activated T cells (Tact) in spleen, mesenteric lymph nodes (MLN) and blood during the development of CRC by using a mouse model of colitis-associated colon cancer (CAC) induced by azoxymethane (AOM) and dextran sodium sulfate (DSS). Our data indicate that as CAC progressed, the percentage of Treg cells increased in MLN and spleen and became more suppressive, whereas the inflammatory response was ablated. When Treg depletion was performed early during CAC development, an improvement in CD4+ and CD8+ T cell activation was observed, which was associated with a reduction in tumor numbers without side effects on the host.
Materials and methods
Mice
Eight-ten-week-old female Foxp3EGFP knock-in female BALB/c mice (C.Cg-Foxp3tm2Tch/J, The Jackson Laboratory) were bred in our animal house and maintained in microisolator cages according to institutional guidelines.
CRC induction and sacrifice days
Foxp3EGFP mice were treated to induce CAC as described [33]. Briefly, mice were injected with azoxymethane at 12.5 mg per kg of weight. Then, 7, 29, and 51 days after AOM injection, 2% dextran sodium sulfate was added to the drinking water for a duration of 7 days. Animals were sacrificed at 15, 37 and 73 days after AOM injection.
Immunofluorescence and flow cytometry
Splenocytes, blood and mesenteric nodules (MLNs) (107 cells/ml) were incubated (30 min, 4°C, darkness) with the indicated mAbs in FACS Sheath (Becton Dikinson®). Cells were washed once, resuspended in FACS Sheath (BD®) and analyzed by flow cytometry using a FACSAria Fusion (BD®) or Attune NxT (ThermoFisher®) cytometer. A detailed analysis of each experiment is indicated in the figure legends. Five thousand gated events were captured and analyzed. Data were analyzed using the FlowJo software V X (Tree Star).
Monoclonal antibodies (mAbs)
The following fluorochrome-conjugated mAbs were used: anti-CD4-Pacific Blue, or -APC (GK1.5), anti-CD8-Brilliant Violet 605 (53-6.7), anti-CD25-APC/Cy7 (PC61), anti-CD127-PE/Cy7 (A7R34), anti-CD279 (PD-1)-PE and anti-CD366 (TIM-3)-APC (B8.2C12) from Biolegend.
Histology
Colon tissue samples were collected, set in absolute ethanol, and later processed and embedded in paraffin for histopathological analysis. Then, 5-μm-thick sections were stained with hematoxylin and eosin (H&E), and inflammatory changes were evaluated in 5 sections from each sample. Three samples of each experimental group from 3 different experiments were analyzed.
Quantification of cytokines in the supernatant
For quantification of cytokines in the supernatant, splenocytes or MLN cells (1x105 cells/ml) were incubated with the anti-CD3 antibody (5 μg/ml) in complete RPMI medium in each well of a 96-well plate (Costar) in a humidified atmosphere containing 5% CO2 in air at 37°C. At 48 hours, the supernatants were harvested and stored at -20°C until required for analyses. Cytokines were quantified using LEGENDplex™ Mouse Th17 Panel (Biolegend®) following the instructions provided by the manufacturer.
RT-PCR assay for the determination of cytokine gene expression in the colon
Colons from different groups of mice were obtained and processed for RT-PCR as previously described [30]. The RNA was purified using a PureLink™ RNA Mini Kit (ThermoFisher) following the manufacturer's instructions. The cDNA was amplified using SuperScript™ First-Strand Synthesis System for RT-PCR (Invitrogen) for IL-7, TGF-β and IL-2-specific primers as described [34]. The gene expression was normalized to the expression of a reference gene (GAPDH) as described [34]. The primers used to amplify the genes were as follows: for IL-2, IL-2-F (AGCAGCACCTGGAGCAGCTG) and IL-2-R (GTCCACCACAGTTGCTGACT); for IL-7, IL-7-F (GCCTGTCACATCATCTGAGTGCC) and IL-7-R (CAGGAGGCATCCAGGAACTTCTG); for TGF-β, TGF-β-F (GCCCTTCCTGCTCCTCAT) and TGF-β-R (TTGGCATGGTAGCCCTTG); and for GAPDH, GAPDH-F (CTCATGACCACAGTCCATGC) and GAPDH-R (CACATTGGGGGTAGGAACAC) (Sigma).
Purification of Treg cells and suppression assays
CD4+ cells from splenocytes of healthy or CAC Foxp3EGFP mice were first enriched by positive selection using anti-CD4 microbeads by MACS, following the instructions provided by the manufacturer (Milteny Biotec). CD4+ T cells were sorted in a FACSaria Fusion cytometer (Beckton Dickinson), and the CD4+ EGFP+ cells (CD4+Foxp3+) and CD4+EGFP- cells (CD4+Foxp3-) were obtained. The purity of CD4+Foxp3+ (Treg) cells was ≥ 95%. Treg CD4+EGFP+ cells were incubated in complete RPMI medium in each well of a 96-well plate (Costar) with anti-CD3 antibody (5 μg/ml) in a 1:1, 1:3, 1:5 and 1:10 ratio of splenocytes from healthy mice in a humidified atmosphere containing 5% CO2 in air at 37°C. Splenocytes used as suppressed cells were stained with Cell Trace Violet ™ (ThermoFisher®) following the manufacturer's instructions. After 72 hours of incubation, cells were harvested, stained for CD4 and CD8 molecules, and immediately analyzed by flow cytometry.
Anti-CD25 antibody injection
PC61 Hybridoma (ATCC TIB-222) secreting rat IgG1 against murine IL-2α chain receptor was grown in CD Hybridoma medium (GIBCO) supplemented with 4% L-glutamine. Monoclonal antibodies (mAbs) were purified from hybridoma culture supernatants by ammonium sulfate (45%) precipitation. After dialysis against PBS for 2 days, the antibody concentration was quantified by spectrophotometry at 280 nm. Antibodies were stored at -20°C until they were used [35]. Then, 200 μg PC61 antibody was intraperitoneally injected into mice 37 days after AOM injection.
Statistical analysis
Statistical differences between groups were determined by One-Way ANOVA with Tukey's Multiple Comparison test. All statistical analyses were performed using PRISM 5 software (GraphPad).
Results
Induction of CAC in BALB/c Foxp3EGFP mice
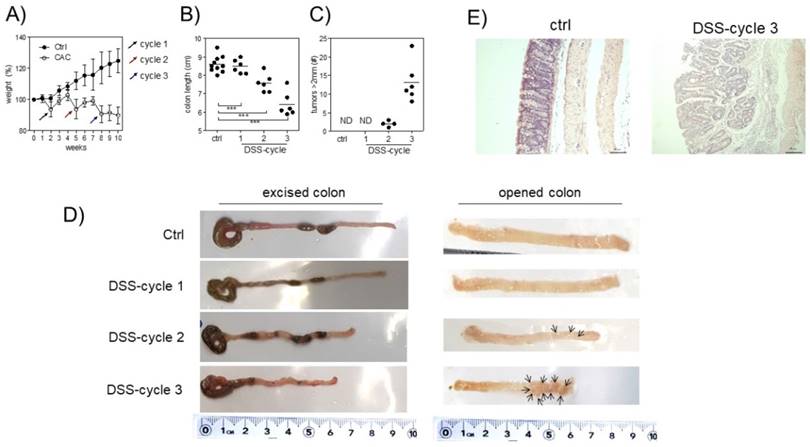
A tumor-promoting role for Treg cells during CAC was previously described [26]. Since we are interested in understanding the role of Treg cells during CAC development, we treated Foxp3EGFP mice with AOM and 3 DSS cycles, as described in the material and methods. First, we observed differences in weight. Animals under CAC treatment exhibited a reduction in weight at the end of every DSS cycle (Fig. 1A), which was remarkable until 10 weeks. Foxp3EGFP mice showed a reduction in the colon length at each DSS cycle (Fig. 1B and D). Additionally, they showed increased tumor numbers mainly after the third round of DSS (Fig. 1C and D). Histological analyses by H&E staining showed that CAC-induced mice showed increased inflammatory infiltrate with a severe destruction of intestinal muscle and mucosa (Fig. 1E). These results demonstrate a progressive development of CAC in every DSS cycle, which concludes in the formation of tumors and a reduction in the length of the colon, being a hallmark for colorectal cancer. Thus, Foxp3EGFP mice are as susceptible to develop CAC as regular BALB/c mice.
Different kinetics of Treg cells in blood, spleen and MLNs during CAC
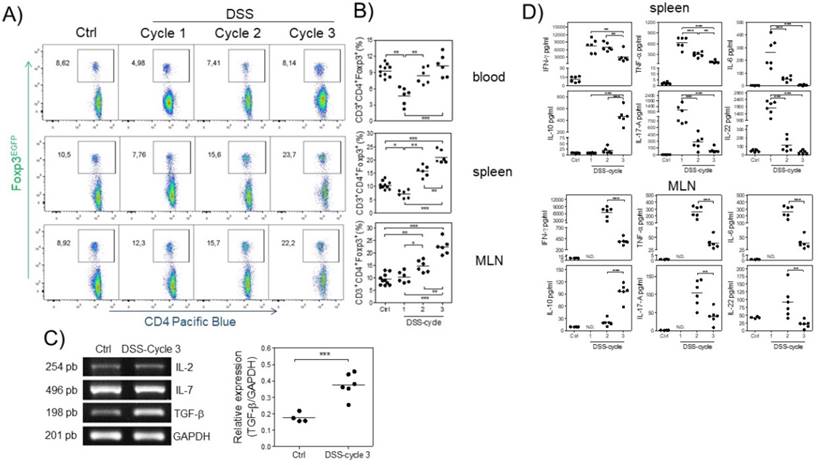
Next, we analyzed the kinetics of Treg cells at 15, 37 and 73 days (at the end of each DSS cycle) after AOM injection by flow cytometry during the development of CAC. We first observed a significant decrease in the percentage of Treg cells at the first DSS cycle in the blood and spleen but not in MLNs (Fig. 2A and B). During the second DSS cycle, there was no difference in the percentage of Treg cells in the blood or spleen of control animals, but a slightly increased percentage in MLNs was observed (Fig. 2A and B). Treg cells in the CAC-induced mice were maintained at a similar percentage as those in control animals in the blood during the third DSS cycle, but, remarkably, a significant increase in the percentage of Treg cells in the spleen and MLNs (Fig. 2A and B) was observed. These results showed different kinetics of Treg cells dependent on the site analyzed.
The increase in Treg cells is probably the result of cytokines promoting their development in a specific site. To determine whether some critical cytokines such as IL-7, TGF-β and IL-2, which are involved in Treg cell development and survival, were altered during CAC, we analyzed their gene expression in total colon samples by RT-PCR in both control and CAC-induced mice. As shown in figure 2C, the expression of IL-2 and IL-7 was not significantly different between control and CAC mice. In contrast, a significant increase in the transcript levels of TGF-β was observed in the CAC-induced mice (Fig. 2C).
To determine whether these differential kinetics of the recruitment of Treg cells had an impact on the local immune response, we isolated spleen cells as well as MLN cells from naïve and CAC-induced mice and stimulated them with a polyclonal stimulus such as plate-bound anti-CD3 antibody, and we detected an early production of several inflammatory cytokines in the splenocytes of CAC-induced mice, with higher levels of IFN-γ, TNF-α, IL-17, IL-6 and IL-22 during the first DSS cycle (Fig. 2D). As CAC progressed, a weaker production of these cytokines was displayed with a significant reduction in all of these cytokines at the end of the third DSS cycle, including a total abrogated production of IL-17, IL-6 and IL-22. In contrast, the IL-10 production in spleen cell cultures displayed an inverse behavior, with low levels of IL-10 early in CAC induction, which increased as CAC progressed (Fig. 2D). Similar kinetics were observed with the MLN cell culture, where the higher levels of the same inflammatory cytokines dropped towards the end of the third DSS cycle (Fig. 2D). Similarly, the IL-10 levels were significantly increased at the last DSS cycle. These results suggest that during CAC progression, a modulation of the inflammatory response is caused by an increased suppressor immune response distinguished by elevated TGF-β, IL-10 and Treg cells.
Treg cells from MLNs and the spleen display an activated phenotype
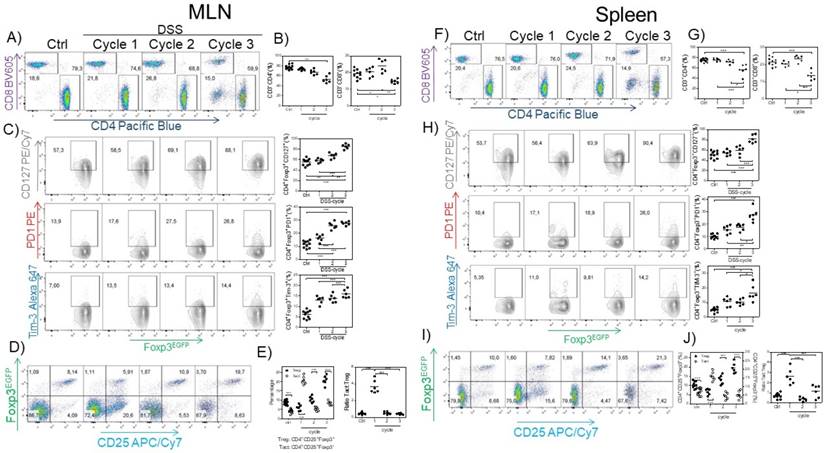
Next, we searched for possible differences in the CD4+ and CD8+ cell population in the spleen and MLNs induced by CAC. We observed a reduction in the percentage of CD3+CD4+ and CD3+CD8+ T cells in the spleen and MLNs (Fig. 3A, B, F and G) in the third DSS cycle. To determine whether the Tregs from the CAC-induced mice displayed some different markers compared to those displayed by Tregs from the control mice, we analyzed the expression of some markers associated with activation in Treg cells. As shown in figure 3C and H, as CAC progressed, we observed a higher expression of PD-1, CD127 and TIM-3 on CD3+CD4+Foxp3+ cells from both the spleen and MLNs, which was more remarkable towards the end of the experiment (Fig. 3C and H). We also analyzed the percentage of CD3+CD4+Foxp3+ Treg cells expressing CD25 so that we could distinguish between Treg and Tact cells. We detected an inverse correlation between Treg and Tact cells as CAC progressed, given that early in CAC induction, Tact cells reached 21% in the MLN (Fig. 3 D and E) and 15% in the spleen at the same time of analysis (Fig. 3 I and J). However, as CAC progressed, the percentages of Tact cells dropped significantly until they were less than 10% in both the MLN and spleen (Fig. 3D, E, I and J). Thus, when Tact cells were increased early in the development of CAC, Treg cells were decreased in the spleen and MLNs (Fig. 3D, E, I and J), whereas the increased percentages of Treg cells detected during the second and third DSS cycles were associated with lower levels of Tact cells (Fig. 3D, E, I and J). When we evaluated the ratio of Tact:Treg cells, we confirmed this fact (Fig. 3E and J). These results suggest that Treg cells from CAC-induced mice display an activated phenotype that may more strongly down-modulate the activation of CD4+ and CD8+ T cells during CAC development.
Induction of CRC in Foxp3EGFP mice. A) Percentage of weight loss. The mean weight of mice on day 0 of treatment was taken as 100% and was compared with the weekly weight until the end of the experiment. B) Length and C) number of tumors in the colon 73 days after AOM injection. D) Macroscopic images of the colon length and number of tumors 73 days after AOM injection (arrows are pointing the tumors). E) Hematoxylin and eosin staining of the colon to investigate the morphology of healthy and CAC-induced mice 73 days after AOM injection. Total data (A, B, C) and representative (D and E) of 2 different experiments with at least 3 mice per group per day of the analysis. * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001; one-way ANOVA and Tukey's multiple comparison test.
Different kinetics of Treg cells in blood, spleen and MLNs during CAC development. At 15, 37 and 73 days after AOM injection, Treg cells were analyzed, shown in a A) representative dot plot, in B) total blood, spleen and MLN from Foxp3EGFP mice. The lymphocyte region was first defined by FSC and SSC characteristics and further subgated based on CD3 and CD4 expression. Five thousand events from either subgate were captured. C) Representative (IL-2, IL-7 and TGF-β) and total (TGF-β) RT-PCR data from colon samples. D) 1x105 spleen and MLN cells were stimulated with α-CD3 for 48 hours to analyze the expression of IFN-γ, TNF-α, IL-6, IL-10, IL-17A and IL-22 cytokines using Th17 LEGENDPlex (Biolegend). Data from 2 different experiments with at least 3 mice per group per day of the analysis. * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001; one-way ANOVA and Tukey's multiple comparison test.
Treg cells from the MLN and spleen display an activated phenotype. At 15, 37 and 73 days after AOM injection, CD4+, CD8+ (A-B MLN, F-G spleen) and Treg cells were analyzed for PD-1, TIM-3 and CD127 expression in the MLN (C) and spleen (H). CD3+CD4+Foxp3+ was the gate for C and H. Representative (left) and total (right) data. Additionally, the Treg and Tact percentages were analyzed in the MLN (D, E) and spleen (I, J). Ratio of Treg cells (3E right) was calculated from data obtained from figure 3E left. The lymphocyte region was first defined by FSC and SSC characteristics and further subgated based on CD3 and CD4 or CD8 expression. Five thousand events from either subgate were captured. Data from 2 different experiments with at least 3 mice per group per day of the analysis. * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001; one-way ANOVA and Tukey's multiple comparison test.
Treg cells from CAC-induced mice display an increased suppressive capacity over T-CD4+ and T-CD8+ cells
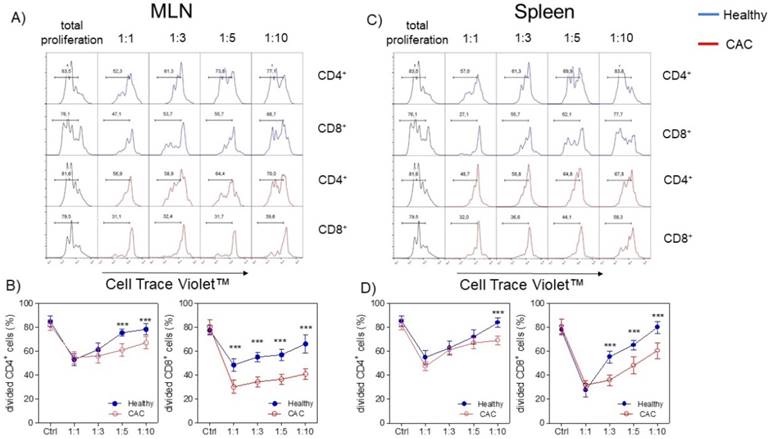
Given that the expression of PD1, CD127 and TIM-3 on Treg cells has been associated with a higher suppressive activity [36-40], we decided to evaluate whether Treg cells isolated from the late stage of CAC development may show a different suppressive capacity. We evaluated the suppressive capacity of Tregs sorted from splenocytes from healthy and CAC-induced mice over naïve splenocytes stimulated with α-CD3, which were exposed to different ratios of Treg cells. When total splenocytes were stimulated with α-CD3, they proliferated strongly as expected (Fig. 4A, B, C and D). When Treg cells came from healthy mice and the Treg:splenocytes ratio was diluted, we observed a higher proliferative capacity of CD4+ and CD8+ cells (Fig. 4A, B, C and D). However, CAC-derived Treg cells strongly inhibited the proliferative capacity of CD4+ cells but mainly CD8+ cells from both the MLN and spleen (Fig. 4A, B and C). These results confirm that Treg cells from CAC-induced mice have an increased suppressive capacity over CD4+ and CD8+ T cells with a more remarkable effect on CD8+ T cells.
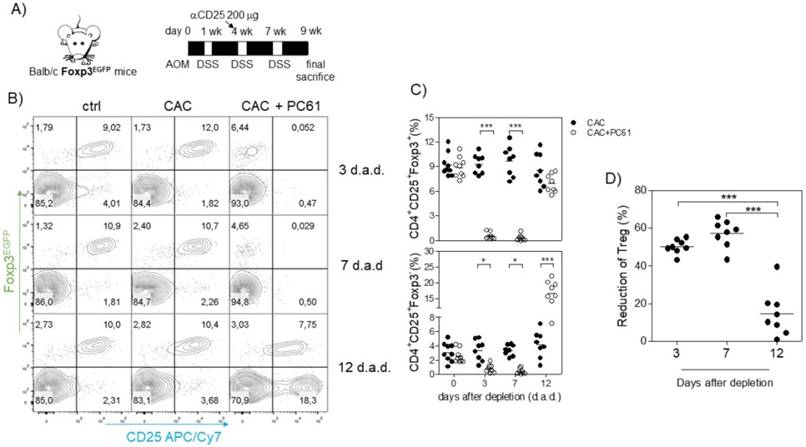
Injection of the α-CD25 antibody during the second DSS cycle impacts Treg and not Tact cells
Because we observed an inverse correlation between Tact and Treg cells such that when Treg cells were increased, Tact cells were decreased and vice versa (Fig. 3D, E, I and J), and we also observed an increased suppressive capacity of Treg cells from CAC-induced mice in vitro, we inferred that the increased percentage of Treg cells may modulate both CD4+ and CD8+ cells in vivo. For this reason, we focused on reducing the percentage of Treg cells during CAC development. The injection of the anti-CD25 antibody PC61 has been largely used as a mechanism to deplete Treg cells in vivo; however, it has some disadvantages, as it can eliminate Tact cells as well as Treg cells [35]. However, on the basis that Tact cells only increase transiently after the first DSS cycle (Fig. 3D, E, I and J), we decided to perform an intraperitoneal injection of 200 μg anti-CD25 PC61 before the second DSS cycle (Fig. 5A) and analyzed the percentage of Treg cells and the reduction in Tact in the blood. We found that Treg cells in CAC-induced mice receiving PC61 antibody were reduced by 50.19±3.78% at 3 days after injection and 57.7±7.20% at 7 days after injection (Fig. 5A and D) compared to that in the untreated mice. However, we observed only a 14.58±12.08% reduction in Treg cells 12 days after PC61 injection (Fig. 5A and D). We previously reported that the depletion of Treg cells with 200 μg PC61 is transient, and the reduction of Treg cells lasts approximately 14 days [41]. We only observed a slight decrease in the percentage of Tact cells 3 and 7 days after PC61 injection, but surprisingly, we found a significant increase in Tact cells 12 days after PC61 injection (Fig. 5B and C). These results suggest that Treg cells from CAC-induced mice prevent an increased percentage of Tact CD4+CD25+Foxp3- cells. We did not need to inject more PC61 antibody at the third DSS cycle because the levels of Tact cells remained high (data not shown), and if we injected PC61 at this time, a major depletion of Tact and no Treg cells would probably be observed.
Early reduction in Treg cells with the anti-CD25 antibody in CAC-induced mice increases the percentage of CD8+CD25+ cells
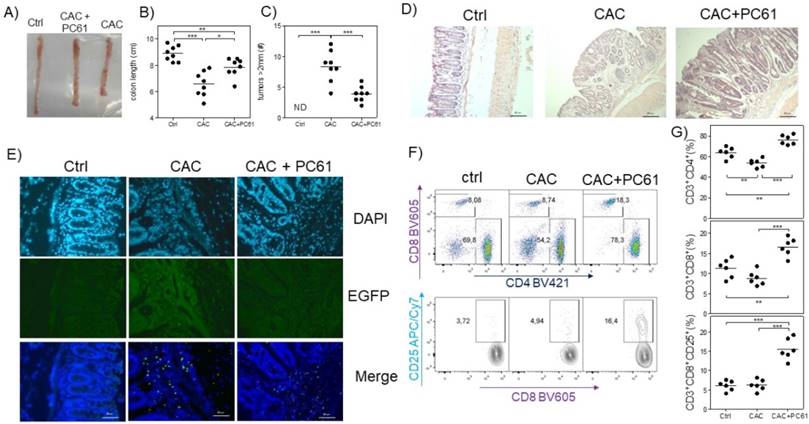
Finally, we analyzed whether PC61 injection had an effect on the health of CAC-induced mice at the end of the third DSS cycle. We observed that CAC-induced mice that received the PC61 antibody showed a less pronounced reduction in colon length, and had significantly fewer tumors than the untreated mice (Fig. 6A, B and C). Additionally, by H&E staining, we observed an increased inflammatory infiltrate in the colon but only in the mucosa and the villi but not in the intestinal muscle in CAC-induced mice injected with the PC61 antibody (Fig. 6D). Injection of the PC61 antibody decreased the presence of Treg cells in the colon (Fig. 6E). Furthermore, the Treg cell depletion induced by PC61 increased the percentages of T-CD4+ and T-CD8+ cells in the MLNs of CAC-induced mice (Fig. 6D). Moreover, a higher expression of CD25 in CD8+ T cells was observed in CAC-induced mice treated with PC61 (Fig. 6E). These results confirm that Treg cells play a role in the modulation of T-CD4+ and T-CD8+ cells and that the reduction in Treg cells is vital during CAC.
Discussion
Treg cells have largely been known to be able to down-modulate immune responses during different diseases or infections [42, 43]; however, the kinetics by which this population appears is much less known. Moreover, the mechanism by which these cells become more suppressive in different pathologies such as cancer is still mainly unknown. In the present study, we investigated the kinetics and the role of Treg cells during the development of colorectal cancer. Notwithstanding that a tumor-promoting function for Treg cells during CRC had been demonstrated in previous studies [26], here, we found that during CAC development there are several conditions favoring an increased percentage of Treg cells, which is reflected in the formation of tumors.
Our data show that in each site analyzed, there was a different kinetic of Treg cell recruitment and activation. Interestingly, in the MLNs, which are located near to the colon where the tumors develop, there was no reduction in the Treg cell percentage during the first DSS cycle, and there was an outstanding increase in Treg cells during the second and third cycles. A tumor-promoting function for Treg cells during CAC had been demonstrated, where the complete ablation of Treg cells using the DEREG model was shown to result in a reduction of the number of tumors during CAC, but these animals died around the third DSS cycle due to an exacerbated intestinal inflammation [26]. Our data showed similar results; however, importantly, in our experiments using the PC61 antibody, the animals did not die or show any sign of sickness. This result is probably the consequence of a limited reduction (approximately 50%) of Treg cells during the second DSS cycle followed by a Treg cell percentage similar to that in healthy mice. The use of monoclonal antibodies against different regulatory molecules or cytokines has been widely used in patients over the last few years with some success in different pathologies; for example, recently, the use of monoclonal antibodies as an early treatment during CRC has been reported [44]. Pembrolizumab, a monoclonal antibody directed against PD-1, has been used in mismatch-repair deficiency in CRC patients [45]. Also some combination of treatment included monoclonal antibodies directed against CTLA-4, PD-1 and PDL-1 has been reported [44].Thus, it is possible that we could have a better chance to treat CRC in humans by reducing Treg cells instead a complete ablation of them. We are aware about the difficulties to deplete specifically Treg cells in humans, because activated T cells can also express Foxp3 [46]; as well Treg cells may express some activation markers shared with Tact cells, including CD25. Despite these disadvantages, we hope that future research will clarify the real possibility or not to use monoclonal antibodies or combined therapy to eliminate or reduce Treg cells in humans in order to analyze their impact on colon cancer.
Part of our data confirms previous work published in the literature, where Treg cells from mice with CAC have an activated phenotype [26, 30]. However, our data demonstrate that the activation phenotype is gradual and dependent on each DSS cycle or the chronicity of the inflammatory stimulus. Our data showed a gradual increase in the expression of PD-1, TIM-3 and CD127 molecules during CAC. PD-1 expressed on Treg cells has been demonstrated to favor the promotion of Treg cell development and function, inhibiting pathogenic self-reactive T cells [47]. Most likely, this is one of the causes for an increased percentage of CD8+ cells given the reduction in Treg cells observed. Also, PD-1 expression has been identified as a marker for induced Treg cells [48, 49]; thus, CAC development may promote a greater transformation of Tact cells to induced Treg cells. TIM-3 is an inhibitory molecule that has emerged as a key regulator of dysfunctional or exhausted T-CD8+ cells in cancer [36, 38]. However, intratumoral Treg cells have been demonstrated to express both TIM-3 and PD-1, which together induce a higher suppressive effect [39]. CD127 is the α chain for the IL-7 receptor [50]. A role for IL-7 in Treg cell maintenance has been demonstrated. Importantly, natural Treg cells only express a low level of CD127 [40], but memory and induced Treg cells require IL-7 and no IL-2 for their maintenance in peripheral tissues [51, 52]. These observations are in accordance with our results because we detected an increased suppressor capacity for CAC-induced Treg cells associated with an increased expression of PD-1, TIM-3 and CD127. However, we only observed an increased percentage of CD127 in Treg cells and not for the IL-7 gene in the colon samples. Most likely, a more exhaustive investigation of the IL-7 cytokine is necessary.
Remarkably, the percentage of Tact cells is dynamic and dependent on the percentage of Treg cells present in the different tissues. We have already published some articles confirming this fact but under other inflammatory contexts such as infection with the protozoa T. gondii [34, 35, 41]. However, in such cases, the inflammatory process is so severe that it leads to death during the acute phase of the infection. In the case of CAC, there is a chronic inflammatory process modulated at an early stage by increased percentages of Treg cells, but this modulation is exacerbated and prevents the immune system cells from exerting their effector function on transformed cells. Other populations of cells of the immune system that have a role in modulating the immune response have been reported, such as TAMs with an anti-inflammatory profile, as well as neutrophils, which present surface markers that induce immunosuppression [2, 7, 13]. Analyzing whether there is a synergistic involvement between Treg cells, TAMs and suppressor neutrophils and determining if the Treg cells have a role in promoting the development of the other cell populations to induce a suppressive microenvironment or if the anti-inflammatory microenvironment produced by the presence of TAMs and suppressor neutrophils favors the migration and induction of Treg cells are crucial.
Treg cells from CAC-induced mice suppress CD4+ and CD8+ cells more efficiently. Cells were treated as described in the materials and methods. A, C) Representative data and B, D) total divided CD4+ and CD8+ cells stimulated with the α-CD3 antibody. The lymphocyte region was first defined by FSC and SSC characteristics and further subgated based on CD4 expression. Five thousand events from either subgate were captured. Data at 73 days post-AOM administration of 2 experiments with at least 3 mice per group per day of the analysis. * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001; one-way ANOVA and Tukey's multiple comparison test.
Injection of the PC61 antibody transiently reduces the percentage of Treg cells in CAC-induced mice. A) On the same day as the administration of DSS into the drinking water in the second cycle, mice were injected with 200 μg of anti-CD25 PC61 intraperitoneally. At 3, 7 and 12 days after the antibody injection, mice were bled to analyze the percentage of Treg and Tact cells. B) Representative data and C) total percentage of Treg (CD4+CD25+Foxp3+) and Tact (CD4+CD25+Foxp3-) cells. D) The reduction in Treg cells was calculated by expressing the Treg cells 3, 7, and 12 days after PC61 depletion as a percentage of those on day 0. The lymphocyte region was first defined by FSC and SSC characteristics and further subgated based on CD4 expression. Five thousand events from either subgate were captured. Data from 2 experiments with at least 3 mice per group per day of the analysis. * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001; one-way ANOVA and Tukey's multiple comparison test.
Injection of the PC61 antibody decreases the tumor number by the increased percentage of CD4+ and CD8+ cells during CAC. Mice that were treated as described in figure 5 were sacrificed 73 days after administration of AOM. A) Representative data of the colon morphology of the different groups. Total data of the B) length of the colon and C) number of tumors in the different groups. D) H&E histology to detect morphological changes in the different groups. E) Confocal immunofluorescence of the different groups showing the expression of green fluorescent protein in the cells (Tregs) from the colon tissue. F) Representative dot plot data and G) total percentage of CD4+ and CD8+ T cells and CD8+CD25+ T cells from the MLN of the different experimental groups. The lymphocyte region was first defined by FSC and SSC characteristics and further subgated based on CD3 and CD4 expression. Five thousand events from either subgate were captured. Data from 2 experiments with at least 3 mice per group per day of the analysis. * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001; one-way ANOVA and Tukey's multiple comparison test.
Finally, our suppression assays demonstrated that Treg cells isolated from MLNs of CAC-induced mice more efficiently suppress CD4+ and CD8+ cells than Treg cells from healthy mice. These data, together with those obtained by reducing the percentage of Treg cells with the injection of the anti-CD25 antibody, suggest that Treg cells are clear suppressors of CD8+ cells. CD8 cells are a population that play a major role in the elimination of tumors [7, 13, 37]; therefore, when Treg cells are reduced, CD8+ T cells are expected to be able to recover their role in reducing the number of tumors. However, in our experiments, we observed that CAC-induced mice under treatment with anti-CD25 continued having tumors. This may be because 200 μg of the anti-CD25 antibody only removed approximately 50% of CD4+ Foxp3+ cells [35, 41]. In the literature, a different strategy has been used, which is the use of a transgenic mouse that expresses the diphtheria toxin receptor after the Foxp3 promoter; in these mice, up of 90% of Treg cells are eliminated with low doses of diphtheria toxin [53], but, as we have discussed above, these experiments concluded in the death of the animals before the conclusion of the third cycle of AOM induction [17]. Perhaps, with the use of the anti-CD25 antibody during induction of the second DSS cycle, we have found a way to modulate the action of the Treg cells that favors a protective response elicited by CD4+ and CD8+ cells.
Abbreviations
CRC: colorectal cancer; Tact: activated T cells; Treg: regulatory T cells; CAC: colitis-associated colon cancer; MLN: mesenteric lymph nodes; TAMs: tumor associated-macrophages; MDSCs: myeloid-derived suppressor cells; EGFP: enhanced green fluorescent protein.
Acknowledgements
This work was supported by grant 280013 from CONACYT (Mexico) and grant IN220316 from Programa de Apoyo a Proyectos de Investigación Científica y Tecnológica, Dirección General de Asuntos de Personal Académico (DGAPA)-UNAM. We thank MVZ Georgina Díaz for her expert advice and help in the care of the animals. J.E.O. is a recipient of a postdoctoral fellowship from Programa de Becas Posdoctorales DGAPA-UNAM.
Author contribution
Conception and experimental design: JEO, IMA, and LIT. Experimental performance: JEO, IMA, EM, AV TP and YICh. Data analysis: JEO, IMA, EM, CPP, EBG and AV. Interpretation of the results: JEO, SALC, MRS, LEAR, FVP and LIT. Paper writing: JEO, MRS, RS and LIT.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Tariq K, Ghias K. Colorectal cancer carcinogenesis: a review of mechanisms. Cancer biology & medicine. 2016;13:120-35
2. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M. et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. International journal of cancer. 2015;136:E359-86
3. Aggarwal BB. Inflammation, a silent killer in cancer is not so silent!. Current opinion in pharmacology. 2009;9:347-50
4. Aggarwal BB, Gehlot P. Inflammation and cancer: how friendly is the relationship for cancer patients? Current opinion in pharmacology. 2009;9:351-69
5. Jemal A, Center MM, DeSantis C, Ward EM. Global patterns of cancer incidence and mortality rates and trends. Cancer epidemiology, biomarkers & prevention: a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2010;19:1893-907
6. Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA: a cancer journal for clinicians. 2010;60:277-300
7. de Vries NL, Swets M, Vahrmeijer AL, Hokland M, Kuppen PJ. The Immunogenicity of Colorectal Cancer in Relation to Tumor Development and Treatment. International journal of molecular sciences. 2016:17
8. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436-44
9. Mantovani A, Marchesi F, Portal C, Allavena P, Sica A. Linking inflammation reactions to cancer: novel targets for therapeutic strategies. Advances in experimental medicine and biology. 2008;610:112-27
10. Mantovani A, Pierotti MA. Cancer and inflammation: a complex relationship. Cancer letters. 2008;267:180-1
11. Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015;160:48-61
12. Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science. 2011;331:1565-70
13. Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nature immunology. 2010;11:889-96
14. Ohkura N, Kitagawa Y, Sakaguchi S. Development and maintenance of regulatory T cells. Immunity. 2013;38:414-23
15. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nature immunology. 2003;4:330-6
16. Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057-61
17. Sakaguchi S. Naturally arising CD4+ regulatory t cells for immunologic self-tolerance and negative control of immune responses. Annual review of immunology. 2004;22:531-62
18. Miyara M, Sakaguchi S. Natural regulatory T cells: mechanisms of suppression. Trends in molecular medicine. 2007;13:108-16
19. Deschoolmeester V, Baay M, Lardon F, Pauwels P, Peeters M. Immune Cells in Colorectal Cancer: Prognostic Relevance and Role of MSI. Cancer microenvironment: official journal of the International Cancer Microenvironment Society. 2011;4:377-92
20. Tougeron D, Fauquembergue E, Latouche JB. [Immune response and colorectal cancer]. Bulletin du cancer. 2013;100:283-94
21. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883-99
22. Lasry A, Zinger A, Ben-Neriah Y. Inflammatory networks underlying colorectal cancer. Nature immunology. 2016;17:230-40
23. Ernst M, Putoczki T. IL-17 cuts to the chase in colon cancer. Immunity. 2014;41:880-2
24. Leon-Cabrera SA, Molina-Guzman E, Delgado-Ramirez YG, Vazquez-Sandoval A, Ledesma-Soto Y, Perez-Plasencia CG. et al. Lack of STAT6 Attenuates Inflammation and Drives Protection against Early Steps of Colitis-Associated Colon Cancer. Cancer immunology research. 2017;5:385-96
25. Zhang B, Wang Z, Wu L, Zhang M, Li W, Ding J. et al. Circulating and tumor-infiltrating myeloid-derived suppressor cells in patients with colorectal carcinoma. PloS one. 2013;8:e57114
26. Pastille E, Bardini K, Fleissner D, Adamczyk A, Frede A, Wadwa M. et al. Transient ablation of regulatory T cells improves antitumor immunity in colitis-associated colon cancer. Cancer research. 2014;74:4258-69
27. Roychoudhuri R, Eil RL, Restifo NP. The interplay of effector and regulatory T cells in cancer. Current opinion in immunology. 2015;33:101-11
28. Wang K, Vella AT. Regulatory T Cells and Cancer: A Two-Sided Story. Immunological investigations. 2016;45:797-812
29. Whiteside TL. What are regulatory T cells (Treg) regulating in cancer and why? Seminars in cancer biology. 2012;22:327-34
30. Akeus P, Langenes V, Kristensen J, von Mentzer A, Sparwasser T, Raghavan S. et al. Treg-cell depletion promotes chemokine production and accumulation of CXCR3(+) conventional T cells in intestinal tumors. European journal of immunology. 2015;45:1654-66
31. Buque A, Bloy N, Aranda F, Castoldi F, Eggermont A, Cremer I. et al. Trial Watch: Immunomodulatory monoclonal antibodies for oncological indications. Oncoimmunology. 2015;4:e1008814
32. Liu J, Zhang S, Hu Y, Yang Z, Li J, Liu X. et al. Targeting PD-1 and Tim-3 Pathways to Reverse CD8 T-Cell Exhaustion and Enhance Ex Vivo T-Cell Responses to Autologous Dendritic/Tumor Vaccines. Journal of immunotherapy. 2016;39:171-80
33. Tanaka T, Kohno H, Suzuki R, Yamada Y, Sugie S, Mori H. A novel inflammation-related mouse colon carcinogenesis model induced by azoxymethane and dextran sodium sulfate. Cancer science. 2003;94:965-73
34. Olguin JE, Fernandez J, Salinas N, Juarez I, Rodriguez-Sosa M, Campuzano J. et al. Adoptive transfer of CD4(+)Foxp3(+) regulatory T cells to C57BL/6J mice during acute infection with Toxoplasma gondii down modulates the exacerbated Th1 immune response. Microbes and infection. 2015;17:586-95
35. Tenorio EP, Fernandez J, Olguin JE, Saavedra R. Depletion with PC61 mAb before Toxoplasma gondii infection eliminates mainly Tregs in BALB/c mice, but activated cells in C57BL/6J mice. FEMS immunology and medical microbiology. 2011;62:362-7
36. Fourcade J, Sun Z, Benallaoua M, Guillaume P, Luescher IF, Sander C. et al. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. The Journal of experimental medicine. 2010;207:2175-86
37. Ngiow SF, von Scheidt B, Akiba H, Yagita H, Teng MW, Smyth MJ. Anti-TIM3 antibody promotes T cell IFN-gamma-mediated antitumor immunity and suppresses established tumors. Cancer research. 2011;71:3540-51
38. Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. The Journal of experimental medicine. 2010;207:2187-94
39. Sakuishi K, Ngiow SF, Sullivan JM, Teng MW, Kuchroo VK, Smyth MJ. et al. TIM3+FOXP3+ regulatory T cells are tissue-specific promoters of T-cell dysfunction in cancer. Oncoimmunology. 2013;2:e23849
40. Simonetta F, Chiali A, Cordier C, Urrutia A, Girault I, Bloquet S. et al. Increased CD127 expression on activated FOXP3+CD4+ regulatory T cells. European journal of immunology. 2010;40:2528-38
41. Tenorio EP, Olguin JE, Fernandez J, Vieyra P, Saavedra R. Reduction of Foxp3+ cells by depletion with the PC61 mAb induces mortality in resistant BALB/c mice infected with Toxoplasma gondii. Journal of biomedicine & biotechnology. 2010;2010:786078
42. Belkaid Y, Tarbell K. Regulatory T cells in the control of host-microorganism interactions (*). Annual review of immunology. 2009;27:551-89
43. Mills KH. Regulatory T cells: friend or foe in immunity to infection? Nature reviews Immunology. 2004;4:841-55
44. Boland PM, Ma WW. Immunotherapy for Colorectal Cancer. Cancers. 2017:9
45. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD. et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. The New England journal of medicine. 2015;372:2509-20
46. Kmieciak M, Gowda M, Graham L, Godder K, Bear HD, Marincola FM. et al. Human T cells express CD25 and Foxp3 upon activation and exhibit effector/memory phenotypes without any regulatory/suppressor function. Journal of translational medicine. 2009;7:89
47. Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunological reviews. 2010;236:219-42
48. Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK. et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. The Journal of experimental medicine. 2009;206:3015-29
49. Haxhinasto S, Mathis D, Benoist C. The AKT-mTOR axis regulates de novo differentiation of CD4+Foxp3+ cells. The Journal of experimental medicine. 2008;205:565-74
50. Zaunders JJ, Levy Y, Seddiki N. Exploiting differential expression of the IL-7 receptor on memory T cells to modulate immune responses. Cytokine & growth factor reviews. 2014;25:391-401
51. Gratz IK, Campbell DJ. Organ-specific and memory treg cells: specificity, development, function, and maintenance. Frontiers in immunology. 2014;5:333
52. Gratz IK, Truong HA, Yang SH, Maurano MM, Lee K, Abbas AK. et al. Cutting Edge: memory regulatory t cells require IL-7 and not IL-2 for their maintenance in peripheral tissues. Journal of immunology. 2013;190:4483-7
53. Lahl K, Loddenkemper C, Drouin C, Freyer J, Arnason J, Eberl G. et al. Selective depletion of Foxp3+ regulatory T cells induces a scurfy-like disease. The Journal of experimental medicine. 2007;204:57-63
Author contact
![]() Corresponding author: Luis I Terrazas, PhD. Mailing address: Unidad de Biomedicina, Facultad de Estudios Superiores Iztacala, Avenida de los Barrios # 1, Tlalnepantla, Estado de México, CP 54090. Phone: (+52 1) 5556239794. E-mail: literrazasmx
Corresponding author: Luis I Terrazas, PhD. Mailing address: Unidad de Biomedicina, Facultad de Estudios Superiores Iztacala, Avenida de los Barrios # 1, Tlalnepantla, Estado de México, CP 54090. Phone: (+52 1) 5556239794. E-mail: literrazasmx