Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(18):3707-3717. doi:10.7150/jca.20239 This issue Cite
Research Paper
Santamarine Inhibits NF-кB and STAT3 Activation and Induces Apoptosis in HepG2 Liver Cancer Cells via Oxidative Stress.
Tahir Mehmood, Amara Maryam, Xiangge Tian, Muhammad Khan ![]() , Tonghui Ma
, Tonghui Ma ![]()
College of Basic Medical Sciences, Dalian Medical University, Dalian, Liaoning 116044, P.R. China.
Received 2017-3-24; Accepted 2017-7-15; Published 2017-10-17
Citation:
Mehmood T, Maryam A, Tian X, Khan M, Ma T. Santamarine Inhibits NF-кB and STAT3 Activation and Induces Apoptosis in HepG2 Liver Cancer Cells via Oxidative Stress.. J Cancer 2017; 8(18):3707-3717. doi:10.7150/jca.20239. https://www.jcancer.org/v08p3707.htm
Other stylesAbstract
Sesquiterpene lactones have long been used in traditional Chinese medicines to treat inflammatory diseases. Recently, sesquiterpene lactone family compounds have been recognized as potential anticancer agents. Thus, it is necessary to explore new sesquiterpene lactones and their antitumor mechanism for cancer treatments. In the present study, we have explored the potential anti-cancer activity of a novel sesquiterpene lactone compound “santamarine” (STM) in HepG2 cells. It inhibited proliferation and induced apoptosis dose-dependently with IC50 ~ 70 μM. Induction of apoptosis was found to be linked with increased reactive oxygen species (ROS) generation, decreased activity of thioredoxin reductase (TrxR), glutathione (GSH) depletion, mitochondrial membrane potential (ΔΨm) dissipation, Bcl-2 family proteins modulation, cytochrome c release, caspases-9, -8 and -3 activation and PARP cleavage. Further mechanistic study demonstrated that STM inhibited the constitutive and TNF-α-induced translocation of NF-кB into nucleus by decreasing phosphorylation of IkB-α. Moreover, STM inhibited STAT3 activation by decreasing phosphorylation at tyrosine705. NAC pretreatment reversed the effect of STM-mediated cell death, NF-кB inhibition and blockage of STAT3 activity, indicating the involvement of oxidative stress in STM-mediated anticancer activity. Further studies are needed to explore the exact molecular mechanism of STM-induced apoptosis to develop it into a lead for treatment of liver cancer in future.
Keywords: Santamarine, HepG2, oxidative stress, apoptosis, NF-κB, STAT3
Introduction
Hepatocellular carcinoma (HCC) is the 5th most common malignant tumor with ~600,000 new diagnosis leading to >250,000 mortalities annually [1]. Surgical resection and liver transplantation are currently most common medical procedures for non-metastatic liver cancers. Sorafenib is the only available first line chemotherapeutic drug for advanced HCC patients [2, 3]. However, no chemotherapeutics have been evidenced better than surgery due to drug resistance and toxicity to normal cells. Therefore, identification of novel bioactive compounds with promising anticancer activity and investigation of their cellular targets are needed for the effective treatment of liver cancer.
A large body of literature evidence has shown that transcription factor NF-кB and STAT3 express constitutively in various cancer cells and play critical roles in cell survival, growth and invasion [4, 5]. Nuclear factor kappa B (NF-κB), being present in almost all types of cells is involved in diverse biological activities such as cell growth, cell adhesion, apoptosis, differentiation, inflammation, oxidative stress responses and immune responses [6-8]. It is well established that constitutive NF-кB activity is a key regulator of cancer cell survival and proliferation as it transcriptionally modulates many anti-apoptotic and pro-survival genes such as Bcl-2 family proteins and survivin [9]. Signal transducers and activators of transcription 3 (STAT3), a member of STAT family, is involved in cell proliferation, drug resistance, tumor invasion and metastasis [10]. Several studies have shown that due to heterogeneous nature of HCC, several pathways including NF-кB and STAT3 are constitutively activated in hepatocellular carcinoma [10-12]. Therefore, inhibition of NF-кB activation and STAT3 blockage might be an effective approach to treat hepatocellular carcinomas and reduce drug resistance.
Sesquiterpene lactones are plant-derived bioactive compounds, which have long been used in traditional medicines to treat inflammatory diseases and many types of cancers such as lung cancer, glioma, pancreatic cancer and hepatocellular carcinoma. Several studies have shown that sesquiterpene lactone compounds induce apoptosis in cancer cells through multiple pathways [13-15]. Thus, it is necessary to explore new sesquiterpene lactones for cancer treatments. STM is a sesquiterpene lactone compound that has been shown to induce anticancer activity by G2/M phase arrest and Caspase-3 activation in L1210 murine leukemia. Here, in this study, we have mechanistically explored the anticancer activity of bioactive sesquiterpene lactone compound “STM” for the first time using HepG2 cells. The anticancer activity of STM has been found to be associated with induction of apoptosis and inhibition of STAT3 and NF-κB activation via oxidative stress.
Materials and Methods
Antibodies and reagents
Santamarine (purity > 95%) was purchased from BioBioPha. Co., Ltd. (Kunming, China). Hoechst 33258, Propidium Iodide (PI), Calcein acetoxymethylester (Calcein-AM), Dimethyl sulfoxide (DMSO), [3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT), Crystal violet staining solution and N-acetyl L-cysteine (NAC) were purchased from Beyotime Institute of Biotechnology (Nanjing, China). IKK-16 (I-кB Kinase Inhibitor) and S31.201 (STAT3 inhibitor) was purchased from Selleckchem. (Shanghai, China). Human TNF-α was purchased from Sino Biological technology (Beijing, China). Dulbecco's Modified Eagle's Medium (DMEM) was purchased from GIBCO (Shanghai, China). Penicillin and Streptomycin was purchased from Solarbio co., Ltd. (Beijing, China). Fetal Bovine Serum (FBS) was purchased from Tissue culture biologicals (TCB, America). Antibodies specific to Bax, bad, Bcl2, cytochrome c, caspase 3, -8, Bid, PARP, TrxR and p50 were purchased from Protein tech. (Wuhan, China). Survivin, p65, histone H3 and IкB-α was purchased from Beyotime (Nanjing, China) while p-IкB-α was purchased by RuiYing, (Nanjing, China). SHP2, Src, p-Src, STAT3, p-STAT3 were purchased from cell signaling technology.
Cell culture
The HepG2 liver cancer cells and Chang normal liver cells were purchased from American Type Culture Collection (ATCC, USA) and cultured in DMEM supplemented with 10% FBS, 100 µg/mL streptomycin and 100 units/mL penicillin and maintained at 37 °C with 5% CO2 in humidified atmosphere.
Determination of cell viability
MTT assay was performed to determine the toxicity of STM on HepG2 and Chang cells as described by us previously [16]. Briefly, HepG2 and Chang cells were seeded and cultured in 96 well cell culture plates and treated with STM in the presence or absence of S31.201 and IKK-16 for 24 h. Cells were then incubated with 10 µL MTT (5 mg/mL) reagent at 37 °C for 4 h. Subsequently, 150 µL DMSO was added to dissolve farmazan crystals and absorbance was measured at 570 nm by Synergy neo HTS multi-mode microplate reader, BioTec. The percentage of cell viability was calculated as follows: Cell viability (%) = (A570 sample - A570 blank) / (A570 control - A570 blank) × 100.
Observation of Morphological Changes
To observe cell morphological changes, HepG2 and Chang cells were treated with 50 & 100 µM of STM in the presence or absence of NAC for 24 h and morphological changes were observed by microscope (Leica, DMIL LED).
Live/Dead Assay
Further toxicity of STM on HepG2 cells was determined by live and dead cell assay as described by us previously [15]. Briefly, HepG2 cells were treated with 50 & 100 µM of STM in the presence or absence of NAC for 24 h. Subsequently, cells were collected, washed with phosphate buffered saline (PBS) and incubated for 20 minutes with PBS solution containing 2 µM calcein-AM and 4 µM PI in the dark at room temperature. Finally, 100 cells from five different areas were counted randomly using microscope (Leica, DMIL LED) for the percentage of live and dead cells.
Colony forming assay
HepG2 cells exposed to different concentrations of STM for 4 h were seeded and cultured in DMEM with 1% FBS in 6-well culture plate (300/well). After two weeks on appearance of colonies, cells were washed twice with PBS, fixed with 4% paraformaldehyde (PFA) for 20 minutes. Cells were then stained with crystal violet for 15 minutes at room temperature. Extra stain was removed by washing with PBS until background was clear. Stained colonies were rinsed by methanol to dissolve crystal violet and O.D. was calculated at 595 nm by fluorescent spectrophotometer (Synergy neo HTS multi-mode microplate reader, BioTek).
Nuclear morphological changes by Hoechst 33258 staining
HepG2 cells were treated with 50 & 100 µM of STM in the presence or absence of NAC for 24 h. Following treatment, the cells were collected, washed with PBS and fixed with 4% PFA for 30 min at room temperature. Subsequently, cells were washed again with PBS and then incubated with Hoechst 33258 (20 μg/mL) in dark at room temperature for 20 minutes. Finally, the cells were washed and re-suspended in PBS to observe nuclear morphological changes under fluorescence microscope (Leica, DMI 4000B).
Measurement of Reactive Oxygen Species (ROS)
Reactive oxygen species assay kit (Beyotime, Nanjing, China) was used to determine the intracellular changes in ROS generation. Briefly, HepG2 Cells were treated with different concentrations of STM in the presence or absence of NAC for different time intervals. The cells were collected, washed with PBS and incubated with 2′, 7′-dichlorofluorescein-diacetate (DCFC-DA) for 20 minutes. Subsequently, dichlorofluorescein (DCF) fluorescence distribution was detected at 488 nm (ex. λ) and at 525 nm (em. λ) according to manufacturer's instructions by fluorescent spectrophotometer (Synergy neo HTS multi-mode microplate reader, BioTek).
Measurement of mitochondrial membrane potential (ΔΨm)
Mitochondrial membrane potential was measured by mitochondrial membrane potential assay kit with JC-1 (Beyotime, Nanjing, China). Briefly, HepG2 cells treated with 50 and 100 µM of STM in the presence or absence of NAC and washed with serum free DMEM and incubated with JC-1 working solution at 37 °C. After 20 minutes, cells were collected, washed and re-suspended in JC-1 dyeing buffer. Subsequently, fluorescence distributions of JC-1 monomer (ex. λ 490 nm & em. λ 530 nm) and j-aggregates (ex. λ 525 nm & em. λ 590 nm) were measured by fluorescent spectrophotometer (Synergy neo HTS multi-mode microplate reader, BioTek) according to manufacturer's instructions.
Measurement of Intracellular reduced glutathione (GSH)
Intracellular level of GSH was determined by reduced glutathione assay kit (Nanjing Jiancheng, China). Briefly, HepG2 cells were treated with 50 and 100 µM of STM. Following 24 h STM treatment, the intracellular GSH was measured by fluorescent spectrophotometer (Synergy neo HTS multi-mode microplate reader, BioTek) at 405 nm according to manufacturer's instructions.
Thioredoxin reductase (TrxR) assay
Intracellular TrxR activity was determined by thioredoxin reductase assay kit (Nanjing Jiancheng, China). Briefly, HepG2 cells were treated with 50 & 100 µM of STM. Following 24 h STM treatment, the intracellular TrxR activity was measured by fluorescent spectrophotometer (Synergy neo HTS multi-mode microplate reader, BioTek) at 412 nm according to manufacturer's instructions.
Western blot analysis
Proteins were extracted from control and STM treated HepG2 cells as described previously [15]. Nuclear and cytosolic proteins were extracted by nuclear and cytosolic protein extraction kit (Beyotime, Nanjing, China) according to manufacturer's instructions. Protein concentrations were measured using enhanced BCA protein Assay kit (Beyotime, Nanjing, China) by spectrophotometer (Synergy neo HTS multi-mode microplate reader, BioTek) according to manufacturer's instructions. 50 μg of proteins were resolved on 10 % sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidine difluoride (PVDF) membranes. After blocking with 5% (w/v) nonfat milk for 1 h and washing three times with Tris-buffered saline-tween (TBST) solution, membranes were incubated overnight at 4 ºC with Bax (1:1000), Bad (1:1000), Bcl-2 (1:1000), Survivin (1:1000), TrxR (1:1000) Cytochrome c (1:500), cleaved Caspase-9 (1:1000), cleaved Caspase-3 (1:1000), Caspase-8 (1:1000), BID (1:1000), PARP (1:1000), IkB-α (1:1000), p-IkB-α (1:1000), p50 (1:500), p65 (1:1000), SHP2 (1:1000), p-SRC (1:1000), SRC (1:1000), STAT3 (1:1000), p-STAT3 (1:1000) and GAPDH (1:2000) antibodies. After washing with TBST, the blots were incubated with peroxidase-conjugated affiniPure goat anti-rabbit or goat anti-mouse secondary antibodies for 1 h at room temperature. After washing with TBST, signals were detected using ECL plus chemiluminescence kit by DNR bioimaging system MicroChemi 4.2. ImageJ software was used to quantify changes in protein expressions and data are presented in graphical format.
Statistical Analysis
Figure 1
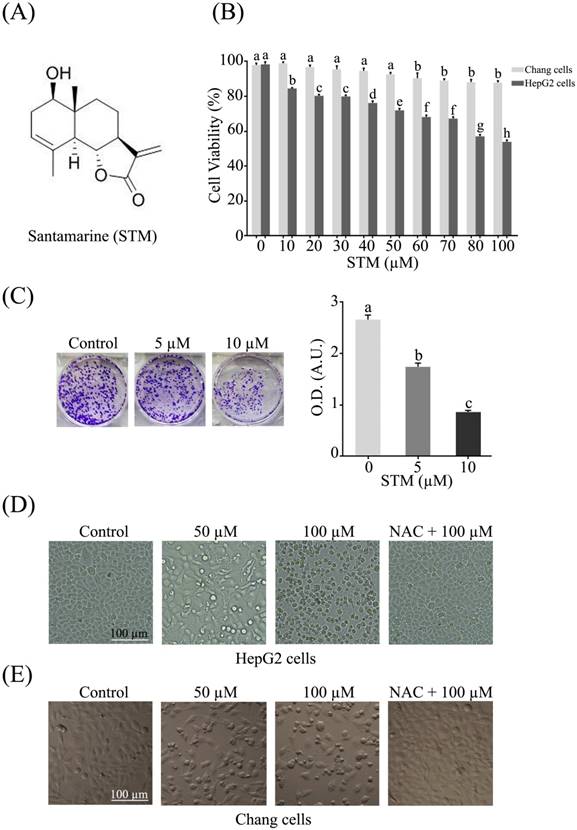
Structure of STM and its effect on growth and morphology of HepG2 and Chang normal liver cells. (A) Chemical structure of STM. (B) Effect of STM on growth inhibition of HepG2 and Chang cells. HepG2 and Chang cells were treated with indicated concentration of STM for 24 h and cell viability was measured by MTT assay. Data are expressed as mean ± SD of three independent experiments. Columns not sharing the same superscript letters differ significantly (p<0.05). (C) HepG2 cells exposed to different concentrations of STM were seeded in 6-well culture plate and cultured in DMEM with 1% FBS for fourteen days. Colonies were rinsed with methanol and optical density (O.D.) in arbitrary units (A.U.) was determined at 595 nm by fluorescent spectrophotometer. (D) HepG2 cells were treated with indicated concentrations of STM in the presence or absence of 3 mM NAC for 24 h to study morphological changes by phase contrast microscopy. Scale bar = 100 µM. (E) Chang cells were treated with indicated concentrations of STM in the presence or absence of 3 mM NAC for 24 h to study morphological changes by phase contrast microscopy. Scale bar = 100 µM.
The results are expressed as Mean ± SD form three different independent experiments and are statistically compared with untreated control group or compared within treated groups using one-way “ANOVA” followed by “Tukey's Multiple Comparison Test”. Columns not sharing the same superscript letters are statistically significant at P < 0.05.
Results
Anti-proliferative effect of STM against HepG2 cells
The anti-proliferative activity of STM against HepG2 and Chang liver cells was evaluated by MTT assay. The data showed that STM inhibited the proliferation of HepG2 cells in dose-dependent manner with IC50 value ~70 µM. Interestingly, STM showed relatively less cytotoxic activity against Chang normal liver cells at concentrations which are toxic to HepG2 liver cancer cells as shown in figure 1B. Furthermore, anti-proliferative nature of STM on HepG2 cells was determined by colony forming assay. The data showed that STM reduces the formation of colonies in dose-dependent manner (Figure 1C).
STM induces morphological changes and cell death in HepG2 cells
HepG2 and Chang liver cells were treated with 50 and 100 µM of STM in the presence or absence of NAC and cellular morphology was observed under phase contrast microscope. The data showed that STM induced conspicuous changes in cellular morphology such as cell shrinkage and loss of cellular architecture in HepG2 cells (Figure 1D). Cells were found detached and floating in medium. Pretreatment of cells with NAC, a ROS scavenger, protected the cell from toxic effect of STM. However, Chang normal liver cells were found relatively resistant to STM at the given concentrations as shown in figure 1E. Live/dead assay was performed to know the toxicity of STM. The data showed that STM induced cell death in dose-dependent manner in HepG2 cells while the effect of STM was reversed by NAC pretreatment (Figure 2A). Moreover, Hoechst-33258 stain was applied to evaluate the effect of STM on nuclear fragmentation (apoptosis marker) in HepG2 cells. The data showed that STM induced DNA fragmentation in HepG2 cells in a dose-dependent manner. However, NAC pretreatment attenuated the effect of STM and protected the DNA from STM-induced DNA damage (Figure 2B). Collective data from MTT, live/dead assay and Hoechst-33258 staining clearly show the ROS-based anticancer activity of STM in HepG2 cells.
Figure 2
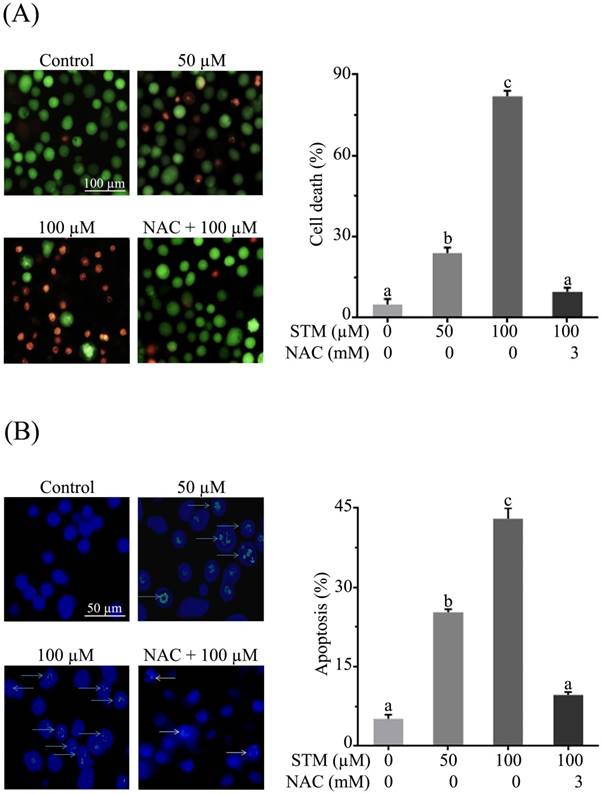
Effect of STM on cell death and DNA fragmentation in HepG2 cells. (A) HepG2 cells were treated with indicated concentrations of STM in the presence or absence of 3 mM NAC for 24 h to determine cell death by live/dead cell assay using fluorescent probe calcein-AM and PI. Scale bar = 100 µM; (B) HepG2 cells were treated with indicated concentrations of STM in the presence or absence of 3 mM NAC for 24 h to examine nuclear morphological changes by Hoechst 33258 staining. Scale bar = 50 µM. 100 nuclei were counted from five different areas randomly for percentage of fragmented nuclei (apoptosis) in each group. Data are expressed as mean ± SD of three independent experiments. Columns not sharing the same superscript letters differ significantly (p<0.05).
STM induces ROS generation
As supplementation of NAC attenuated the effect of STM in HepG2 cells, so we measured the level of ROS. The data showed that STM induced ROS generation in dose-dependent manner. STM started inducing ROS generation from 5 µM which reached to a significant value at 10 µM and so on (Figure 3A). STM also induced ROS generation in time-dependent manner. It started inducing ROS generation as early as 15 minutes of drug treatment which reached to its maximum value in 2 h and then decreased gradually to the level of control group in 12 h of treatment. However, NAC pretreatment reversed the effect of STM as shown in figure 3B.
STM reduces mitochondrial membrane potential (ΔΨm)
As mitochondrial membrane potential (ΔΨm) is disrupted with induction of ROS, therefore, we determined the effect of STM on ΔΨm. The data showed that STM significantly reduced ΔΨm in a dose-dependent manner whereas the NAC pretreatment attenuated its effect (Figure 4A).
STM diminishes intracellular GSH
Induction of ROS changes redox homeostasis which ultimately disturbs antioxidant systems of the cells. GSH is an important antioxidant which prevents cells from oxidative stress. Its level is depleted due to ROS generation which leads to apoptotic cell death. Therefore, we determined the intracellular level of GSH. The data showed that STM significantly depleted the level of intracellular GSH in dose-dependent manner (Figure 4B).
Figure 3
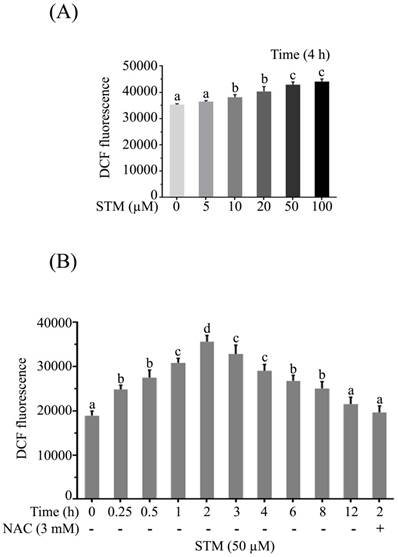
Effect of STM on intracellular ROS generation in HepG2 cells. HepG2 cells were treated with indicated concentrations of STM in the presence or absence of NAC for indicated time periods and reactive oxygen species assay kit was used to determine (A) dose-dependent and (B) time-dependent intracellular changes in ROS generation by spectrophotometer according to kit instructions. Data are expressed as mean ± SD of three independent experiments. Columns not sharing the same superscript letters differ significantly (p<0.05).
STM inhibits intracellular Thioredoxin reductase (TrxR) activity
Thioredoxin reductase maintains redox homeostasis and protects the cells from oxidative damage. Therefore, we measured the activity of TrxR. The data showed that STM reduced the TrxR activity (Figure 4C). Subsequently, the relative expression level of TrxR was determined by western blot. The data showed that STM reduced TrxR level in dose-dependent manner as shown in figure 4D.
Figure 4
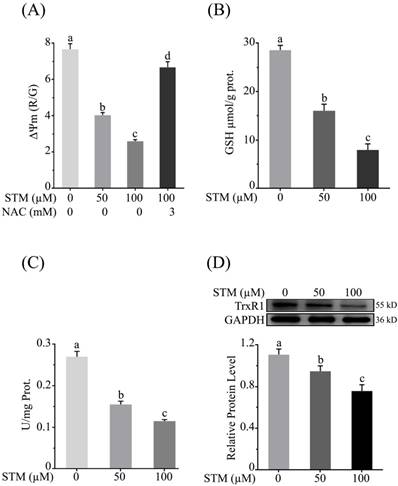
Effect of STM on mitochondrial membrane potential and antioxidant systems in HepG2 cells. Cells were treated with indicated concentrations of STM in the presence or absence of NAC for 24 h and were analyzed by spectrophotometer for the analysis of (A) mitochondrial membrane potential (B) intracellular level of GSH and (C) TrxR activity according to instructions of kits. (D) Intracellular level of TrxR was determined by western blot. Data are expressed as mean ± SD of three independent experiments. Columns not sharing the same superscript letters differ significantly (p<0.05).
STM induces mitochondrial mediated (intrinsic) apoptosis
As loss of ΔΨm is an important marker of apoptosis which is regulated by Bcl-2 family proteins, therefore, we measured the expression of Bcl-2 family proteins by western blot. We found that STM up-regulated the pro-apoptotic Bax and Bad proteins while down-regulated the anti-apoptotic Bcl-2 protein with proclamation of cytochrome c form inner membrane of mitochondria into cytosol in dose-dependent manner (Figure 5A). Survivin being a member of inhibitors of apoptosis protein (IAP) family is expressed in tumors and it modulates cell division and apoptosis. Therefore, we determined the expression of survivin in HepG2 cells. STM treatment down regulated the expression of survivin as shown in figure 5A.
In the execution of both “intrinsic” and “extrinsic” apoptotic pathways, activation of caspases, a family of cysteine aspartyl-specific proteases and cleavage of poly (ADP-ribose) polymerase (PARP), a proteolytic substrate of caspases-3 are important markers of apoptosis. Therefore, we determined the expression of cleaved caspase-9, -3 and PARP-1. The data showed that treatment of HepG2 cells with STM resulted in dose-dependent increase in the cleaved forms of caspase-9, -3 and PARP-1 (Figure 5A). Bcl-2 family proteins modulation, mitochondrial membrane potential disruption and ultimately release of cytochrome c into cytosol indicate that STM-induced apoptosis is associated with mitochondrial apoptosis pathway.
Figure 5
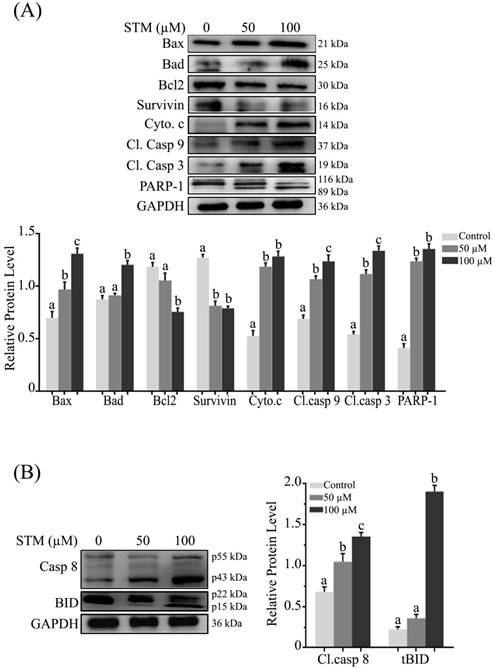
Effect of STM on apoptosis regulators; (A) Intrinsic and (B) extrinsic apoptosis regulators. HepG2 cells were treated with indicated concentrations of STM for 24 h and proteins extracts (total & cytosolic/nuclear proteins) were prepared and analyzed by immunoblotting. Data are expressed as Mean ± SD of three independent experiments. Columns not sharing the same superscript letter within the group differ significantly (p<0.05).
STM induces extrinsic apoptosis
Activation of caspase-8 and truncation of Bid are characteristics features of extrinsic apoptosis. Therefore, we determined the expression of caspase-8 and bid. The data showed that treatment of HepG2 cells with STM significantly cleaved caspase-8 in dose-dependent manner with concomitant truncation of Bid at high concentration (Figure 5B) which showed the involvement of extrinsic pathway with STM treatment.
STM inhibits constitutive and inducible NF-кB activation in HepG2 cells
As induction of oxidative stress inhibits activation of NF-кB [15, 17], therefore, we determined NF-кB expression in nuclear fraction. The data showed that treatment of HepG2 cells with STM inhibited the translocation of NF-кB-p65 and -p50 from cytoplasm to nucleus which was associated with inhibition of phosphorylation of IкB-α (Figure 6A). In order to confirm if STM-induced NF-кB inhibition were ROS mediated, we evaluated NF-кB-p65 expression in nuclear fractions in the presence of NAC. We found that NAC pretreatment reversed STM-induced NF-кB inhibition in HepG2 cells (Figure 6B). To further confirm STM-induced NF-кB inhibition, we decided to compare its affect with commercially available IкB Kinase (IKK) inhibitor, IKK-16. The data showed that STM inhibited NF-кB translocation as that of IKK-16 (Figure 6C). Now we were interested to know if STM could reduce TNF-α-induced NF-кB translocation from cytoplasm to nucleus, we determined the expression of TNF-α-induced NF-кB-p65 with STM and IKK-16 treatment. The data showed that STM and IKK-16 abridged TNF-α-induced NF-кB translocation from cytoplasm to nucleus (Figure 6D). Collective data clearly demonstrated that STM inhibited both constitutive and TNF-α-induced activation of NF-кB through ROS generation.
STM inhibits STAT3 activation in HepG2 cells
Figure 6
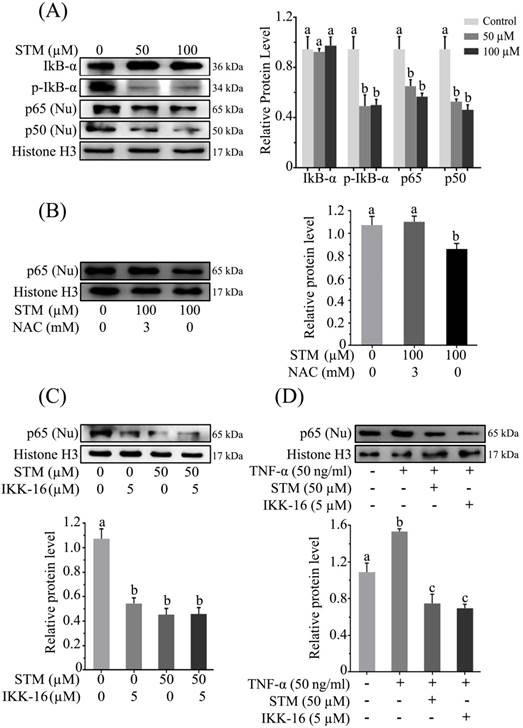
Effect of STM on NF-кB activity and NF-кB regulators in HepG2 cells. HepG2 cells were treated with indicated concentrations of STM in the presence or absence of NAC for 24 h and proteins extracts (total/nuclear proteins) were prepared and analyzed by immunoblotting (A) to evaluate the effect of STM on NF-кB regulators and NF-кB activity (B) in the presence or absence of NAC (3mM). Data are expressed as Mean ± SD of three independent experiments. Columns not sharing the same superscript letter within the group differ significantly (p<0.05). (C) Effect of STM (50 µM) and IKK-16 (5 µM), an IкB kinase (IKK) inhibitor, on NF-кB activity after 24 h treatment. Nuclear proteins extracts were prepared and analyzed by immunoblotting. Data are expressed as Mean ± SD of three independent experiments. Columns not sharing the same superscript letter differ significantly (p<0.05). (D) Effect of STM on TNF-α-induced NF-кB activity. Cells were pretreated with TNF-α (50ng/ml) for 30 minutes followed by 24 h STM (100 µM) and IKK-16 (5 µM) treatment. Nuclear proteins extracts were prepared and analyzed by immunoblotting. Data are expressed as mean ± SD of three independent experiments. Columns not sharing the same superscript letters differ significantly (p<0.05).
As ROS generation has been shown to inhibit STAT3 activation [18], therefore, we determined the expression of p-STAT3, STAT3 and its regulators such as SHP2 and p-Src/Src in HepG2 cells treated with STM. The data showed that STM inhibited the activation of STAT3 at Tyr-705 in a dose-dependent manner (Figure 7A). Activation of STAT3 is regulated by various up-stream regulators including protein tyrosine phosphatases and tyrosine Kinases. Therefore, we measured the expressions of SHP-2 and p-Src/Src in STM-treated and control cells. The data demonstrated that STM did not change the expression of SHP2 (Figure 7B) while it inhibited the phosphorylation and expression of Src (Figure 7C). Finally, to confirm if STM-induced STAT3 inhibition was indeed ROS mediated, we evaluated STAT3 and p-STAT3 expression in the presence of NAC. The data showed clearly that STM-induced STAT3 inhibition was reversed by NAC pretreatment in HepG2 cells (Figure 7D).
To know the additive or synergistic effect of STM with inhibitors (IKK-16 & S31.201) we determined the cytotoxicity of STM (50 µM) with IKK-16 (5 µM) and S31.201 (200 µM) against HepG2 cells. The data showed that the cytotoxicity of STM in the presence of IKK-16 and S31.201 was significantly increased (Figure 8).
Discussion
Sesquiterpene lactones are plant-derived bioactive molecules and have long been used to treat inflammatory diseases in traditional Chinese medicines. In recent years sesquiterpene lactone compounds have been shown to exert anti-tumor activity in various human cancers including lung cancer, glioma, pancreatic cancer and hepatocellular carcinoma through various distinct mechanisms [19]. One of the most important key characteristics of sesquiterpene lactone family compounds is to induce oxidative stress due to the presence of α-methylene-γ-lactone moiety [19, 20]. It has been shown by many studies that oxidative stress induced by sesquiterpene lactones increases apoptosis and inhibits the activation of many transcription factors including NF-кB and STAT3 [19-21]. STM being a member of sesquiterpene lactone family contains α-methylene-γ-lactone moiety. Therefore, we hypothesized that it could induce oxidative stress resulting in apoptosis. As expected, STM induced oxidative stress and inhibited growth and enhanced apoptotic cell death in HepG2 cells.
Figure 7
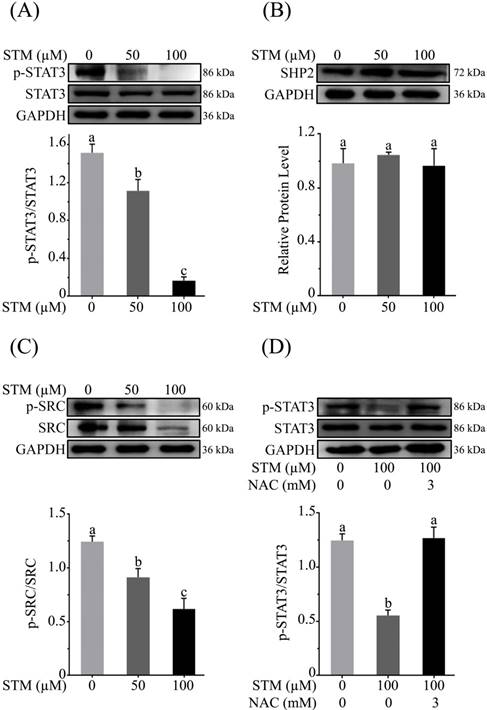
Effect of STM on STAT3 activity and STAT3 regulators in HepG2 cells. HepG2 cells were treated with indicated concentrations of STM in the presence or absence of NAC for 24 h and proteins extracts were prepared and analyzed by immunoblotting to determine the effect of STM on expression of (A) STAT3, p-STAT3, (B) SHP2, (C) Src & p-Src. (D) HepG2 cells were treated with 100 µM of STM in the presence or absence of NAC (3 mM) for 24 h and proteins extracts were prepared and analyzed by immunoblotting to determine the effect of STM on expression of STAT3, p-STAT3 in the presence or absence of NAC. Data are expressed as Mean ± SD of three independent experiments. Columns not sharing the same superscript letter within the group differ significantly (p<0.05).
Apoptosis is regulated by two alternative mechanisms including mitochondrial mediated or “intrinsic” apoptosis and death receptor mediated or “extrinsic” apoptosis. In the execution of both intrinsic and extrinsic apoptosis, caspases are key regulators of apoptosis [22, 23]. It is well established that oxidative stress triggers mitochondrial mediated or intrinsic apoptosis by modulating Bcl-2-family proteins. Bcl-2 family proteins are further divided into pro-apoptotic proteins such as Bax, Bad and anti-apoptotic proteins such as Bcl-2 [24]. Once activated, pro-apoptotic protein Bax is translocated to outer mitochondrial membrane where it forms mitochondrial permeability transition pores and release cytochrome c. Oxidative stress also inhibits anti-apoptotic protein Bcl-2 [25, 26]. Henceforth, in this study we found that STM dissipated ΔΨm accompanied by up-regulation of Bax and Bad and down-regulation of Bcl-2 with concomitant release of cytochrome c form inner membrane of mitochondria to cytosol. Release of cytochrome c activates caspase-9 followed by activation of caspase-3, which ensures the execution of apoptosis by PARP cleavage [25]. STM increased the expression of cleaved caspase-9, -3 and PARP-1 dose-dependently. These findings showed clearly that STM-induced cell death is associated with intrinsic apoptosis in HepG2 cells. Several studies have shown that sesquiterpene lactone compounds induce extrinsic or death receptor-mediated apoptosis by activating caspase-8. Once activated, caspase-8 induces either activation of caspase-3 directly or truncation of Bid to tBid indirectly. Finally, tBid translocates to mitochondrial membranes and enhances permeabilization which results in further release of cytochrome c [26, 27]. All these cellular processes cause extrinsic apoptosis. Here, we found that STM activated capase-8 in dose-dependent manner with bid cleavage at high concentration. These findings showed that STM induced extrinsic apoptosis as well in HepG2 cells.
Transcription factors such as NF-кB and STAT3 are constitutively activated in HCC and play an important role in propagation and cell survival. NF-кB family is composed of 5 members as RelA/p65, RelB, c-Rel, p50 & p52 [28, 29]. Activation of NF-кB is usually associated with phosphorylation of inhibitors of κBs (IκBs). Once IκB-α is phosphorylated, NF-кB becomes free, forms dimer and is transported to nucleus where it initiates transcription of target genes [29, 30]. In this study we found that STM inhibited the activation of both constitutive and TNF-α-induced NF-кB by reducing the phosphorylation of IκB-α.
Figure 8
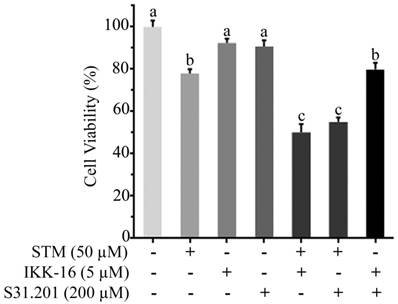
Effect of STM, IKK-16 and S31.201 on growth inhibition of HepG2 cells. HepG2 cells treated with indicated concentrations of STM with or without IKK-16 and S31.201 for 24 h and cell viability was determined by MTT assay. Data are expressed as mean ± SD of three independent experiments. Columns not sharing the same superscript letters differ significantly (p<0.05).
Signal transducers and activators of transcription 3 (STAT3), a member of STAT protein family, is aberrantly activated in several cancers such as lymphoma, breast, lung, ovarian, prostate, and liver cancers [26]. Phosphorylation of STAT3 at Tyr-705 triggers the activation of STAT3 which is mediated by cytoplasmic kinases such as Src family kinases [31, 32]. Upon activation, STAT3 is translocated into nucleus where it regulates the expressions of several essential genes responsible for cell proliferation, differentiation, tumor invasion, metastasis, angiogenesis, apoptosis and immune response [33, 34]. Here, we found that STM inhibited the STAT3 activity by decreasing phosphorylation at Tyr-705 which was mediated by decreased phosphorylation of Src kinase.
In conclusion, our data provide evidence for the first time that STM induces oxidative stress in HepG2 cells resulting in ΔΨm dissipation, increased bax/bcl-2 ratio, cytochrome c release and activation of caspase-9, -3 and PARP-1 cleavage. It also induces extrinsic apoptosis via activation of caspase-8 and Bid cleavage. Apoptotic effect of STM was further enhanced by IKK-16 and S31.201. STM inhibits the activation of transcription factors, NF-кB and STAT3, in HepG2 cells via oxidative stress. Further studies are needed to explore the exact molecular mechanism of STM-induced apoptosis and validate its efficacy in experimental animal models to develop it into a lead for treatment of liver cancer in future.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Zhao JA, Sang MX, Geng CZ. et al. A novel curcumin analogue is a potent chemotherapy candidate for human hepatocellular carcinoma. Oncol Lett. 2016;12:4252-4262
2. Xue F, Liu Y, Chu H. et al. eIF5A2 is an alternative pathway for cell proliferation in cetuximab-treated epithelial hepatocellular carcinoma. Am J Transl Res. 2016;8:4670-4681
3. Xue F, Liu Y, Zhang H. et al. Let-7a enhances the sensitivity of hepatocellular carcinoma cells to cetuximab by regulating STAT3 expression. Onco Targets ther. 2016;9:7253-7261
4. Lin WL, Lai DY, Lee YJ. et al. Antitumor progression potential of morusin suppressing STAT3 and NFκB in human hepatoma SK-Hep1 cells. Toxicol Lett. 2015;232:490-498
5. Ghosh S, Febin Prabhu Dass J. Non-canonical pathway network modelling and ubiquitination site prediction through homology modelling of NF-κB. Gene. 2016;581:48-56
6. Grivennikov SI, Karin M. Dangerous liaisons: STAT3 and NF-κB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev. 2010;21:11-19
7. Gilmore TD, Herscovitch M. Inhibitors of NF-[kappa]B signaling: 785 and counting. Oncogene. 2006;25:6887-6899
8. Ghosh S, Dass JFP. Study of pathway cross-talk interactions with NF-κB leading to its activation via ubiquitination or phosphorylation: A brief review. Gene. 2016;584:97-109
9. Hussain AR, Ahmed SO, Ahmed M. et al. Cross-Talk between NFkB and the PI3-Kinase/AKT Pathway Can Be Targeted in Primary Effusion Lymphoma (PEL) Cell Lines for Efficient Apoptosis. PLoS ONE. 2012;7:e39945
10. Bao C, Li Y, Huan L. et al. NF-κB signaling relieves negative regulation by miR-194 in hepatocellular carcinoma by suppressing the transcription factor HNF-1α. Sci Signal. 2015;8:ra75-ra75
11. Chung JS, Lee S, Yoo YD. Constitutive NF-κB activation and tumor-growth promotion by Romo1-mediated reactive oxygen species production. Biochem Biophys Res Commun. 2014;450:1656-1661
12. Zuo M, Li C, Lin J. et al. LLL12, a novel small inhibitor targeting STAT3 for hepatocellular carcinoma therapy. Oncotarget. 2015;6:10940-10949
13. Khan M, Li T, Ahmad Khan MK. et al. Alantolactone Induces Apoptosis in HepG2 Cells through GSH Depletion, Inhibition of STAT3 Activation, and Mitochondrial Dysfunction. BioMed Res Int. 2013;2013:719858
14. Lagoutte R, Serba C, Abegg D. et al. Divergent synthesis and identification of the cellular targets of deoxyelephantopins. Nat Commun. 2016;7:12470
15. Mehmood T, Maryam A, Zhang H. et al. Deoxyelephantopin induces apoptosis in HepG2 cells via oxidative stress, NF-κB inhibition and mitochondrial dysfunction. BioFactors. 2017;43:63-72
16. Khan M, Ding C, Rasul A. et al. Isoalantolactone Induces Reactive Oxygen Species Mediated Apoptosis in Pancreatic Carcinoma PANC-1 Cells. Int J Biol Sci. 2012;8:533-547
17. Gong K, Li W. Shikonin, a Chinese plant-derived naphthoquinone, induces apoptosis in hepatocellular carcinoma cells through reactive oxygen species: A potential new treatment for hepatocellular carcinoma. Free Radic Biol Med. 2011;51:2259-2271
18. Zhang J, Ahn KS, Kim C. et al. Nimbolide-Induced Oxidative Stress Abrogates STAT3 Signaling Cascade and Inhibits Tumor Growth in Transgenic Adenocarcinoma of Mouse Prostate Model. Antioxid Redox Signal. 2016;24:575-89
19. Gach K, Długosz A, Janecka A. The role of oxidative stress in anticancer activity of sesquiterpene lactones. Naunyn Schmiedebergs Arch Pharmacol. 2015;388:477-486
20. Ding Y, Wang H, Niu J. et al. Induction of ROS Overload by Alantolactone Prompts Oxidative DNA Damage and Apoptosis in Colorectal Cancer Cells. Int J Mol Sci. 2016;17:558
21. Mathema VB, Koh YS, Thakuri BC. et al. Parthenolide, a Sesquiterpene Lactone, Expresses Multiple Anti-cancer and Anti-inflammatory Activities. Inflammation. 2012;35:560-565
22. Su M, Chung HY, Li Y. Deoxyelephantopin from Elephantopus scaber L. induces cell-cycle arrest and apoptosis in the human nasopharyngeal cancer CNE cells. Biochem Biophys Res Commun. 2011;411:342-347
23. Khan M, Maryam A, Qazi JI. et al. Targeting Apoptosis and Multiple Signaling Pathways with Icariside II in Cancer Cells. Int J Biol Sci. 2015;11:1100-1112
24. Bennett A, Sloss O, Topham C. et al. Inhibition of Bcl-xL sensitizes cells to mitotic blockers, but not mitotic drivers. Open Biology. 2016;6:160134
25. Farha AK, Dhanya SR, Mangalam SN. et al. Deoxyelephantopin impairs growth of cervical carcinoma SiHa cells and induces apoptosis by targeting multiple molecular signaling pathways. Cell Biol Toxicol. 2014;30:331-343
26. Ma Y, Zhu B, Yong L. et al. Regulation of Intrinsic and Extrinsic Apoptotic Pathways in Osteosarcoma Cells Following Oleandrin Treatment. Int J Mol Sci. 2016;17:1950
27. Di W, Khan M, Rasul A. et al. Isoalantolactone inhibits constitutive NF-kappaB activation and induces reactive oxygen species-mediated apoptosis in osteosarcoma U2OS cells through mitochondrial dysfunction. Oncol Rep. 2014;32:1585-1593
28. Yang ST, Yen CJ, Lai CH. et al. SUMOylated CPAP is required for IKK-mediated NF-κB activation and enhances HBx-induced NF-κB signaling in HCC. J Hepatol. 2013;58:1157-1164
29. Zhao L, Zhang Y. miR-342-3p affects hepatocellular carcinoma cell proliferation via regulating NF-κB pathway. Biochem Biophys Res Commun. 2015;457:370-377
30. Lanucara F, Lam C, Mann J. et al. Dynamic phosphorylation of RelA on Ser42 and Ser45 in response to TNFα stimulation regulates DNA binding and transcription. Open Biology. 2016;6:160055
31. Zhang W, Ma T, Li S. et al. Antagonizing STAT3 activation with benzo[b]thiophene 1, 1-dioxide based small molecules. Eur J Med Chem. 2017;125:538-550
32. Nam S, Wen W, Schroeder A. et al. Dual inhibition of Janus and Src family kinases by novel indirubin derivative blocks constitutively-activated Stat3 signaling associated with apoptosis of human pancreatic cancer cells. Mol Oncol. 2013;7:369-378
33. Subramaniam A, Shanmugam MK, Perumal E. et al. Potential role of signal transducer and activator of transcription (STAT)3 signaling pathway in inflammation, survival, proliferation and invasion of hepatocellular carcinoma. Biochim Biophys Acta. 2013;1835:46-60
34. Siveen KS, Sikka S, Surana R. et al. Targeting the STAT3 signaling pathway in cancer: Role of synthetic and natural inhibitors. Biochim Biophys Acta. 2014;1845:136-154
Author contact
![]() Corresponding authors: Tonghui Ma, College of Basic Medical Sciences, Dalian Medical University, Dalian, Liaoning 116044, P.R. China. Tel: +86-411-86110278, Fax: +86-411-86110378 Email: tonghuimaedu.cn and Muhammad Khan, College of Basic Medical Sciences, Dalian Medical University, Dalian, Liaoning 116044, P.R. China. Tel: +86-411- 86118842. Email: khan_zoologistcom
Corresponding authors: Tonghui Ma, College of Basic Medical Sciences, Dalian Medical University, Dalian, Liaoning 116044, P.R. China. Tel: +86-411-86110278, Fax: +86-411-86110378 Email: tonghuimaedu.cn and Muhammad Khan, College of Basic Medical Sciences, Dalian Medical University, Dalian, Liaoning 116044, P.R. China. Tel: +86-411- 86118842. Email: khan_zoologistcom
Citation styles
APA
Mehmood, T., Maryam, A., Tian, X., Khan, M., Ma, T. (2017). Santamarine Inhibits NF-кB and STAT3 Activation and Induces Apoptosis in HepG2 Liver Cancer Cells via Oxidative Stress.. Journal of Cancer, 8(18), 3707-3717. https://doi.org/10.7150/jca.20239.
ACS
Mehmood, T.; Maryam, A.; Tian, X.; Khan, M.; Ma, T. Santamarine Inhibits NF-кB and STAT3 Activation and Induces Apoptosis in HepG2 Liver Cancer Cells via Oxidative Stress.. J. Cancer 2017, 8 (18), 3707-3717. DOI: 10.7150/jca.20239.
NLM
Mehmood T, Maryam A, Tian X, Khan M, Ma T. Santamarine Inhibits NF-кB and STAT3 Activation and Induces Apoptosis in HepG2 Liver Cancer Cells via Oxidative Stress.. J Cancer 2017; 8(18):3707-3717. doi:10.7150/jca.20239. https://www.jcancer.org/v08p3707.htm
CSE
Mehmood T, Maryam A, Tian X, Khan M, Ma T. 2017. Santamarine Inhibits NF-кB and STAT3 Activation and Induces Apoptosis in HepG2 Liver Cancer Cells via Oxidative Stress.. J Cancer. 8(18):3707-3717.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.