Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(18):3689-3696. doi:10.7150/jca.19501 This issue Cite
Review
Strategies for Bispecific Single Chain Antibody in Cancer Immunotherapy
Shu-juan Zhou1, Jia Wei1, Shu Su1, Fang-jun Chen1, Yu-dong Qiu2 ![]() , Bao-rui Liu1
, Bao-rui Liu1 ![]()
1. The Comprehensive Cancer Centre of Drum Tower Hospital, Medical School of Nanjing University & Clinical Cancer Institute of Nanjing University, Nanjing, China;
2. Department of Hepatopancreatobiliary Surgery, The Affiliated Drum Tower Hospital of Medical School of Nanjing University, Nanjing, China.
Received 2017-2-5; Accepted 2017-8-7; Published 2017-10-17
Abstract
Genetic engineering has resulted in more than 50 recombinant bispecific antibody formats over the past two decades. Bispecific scFv antibodies represent a successful and promising immunotherapy platform that retargets cytotoxic T cells to tumor cells, with one scFv directed to tumor-associated antigens and the other to T cells. Based on this antibody construct, strategies for both specific tumor targeting and T cell activation are reviewed here. Three distinct types of tumor antigens are considered to optimize specificity and safety in bispecific scFv based treatment: cancer-testis antigens, neo-antigens and virus-associated antigens. In terms of T cell activation, although CD3 has been widely applied in bispecific scFvs being developed, CD28 and CD137 among co-stimulatory signals are also ideal candidates to be evaluated. Besides, LIGHT and HIV-Tat101 have drawn much attention as their potential roles in modulating antitumor responses.
Keywords: bispecific antibody, cancer-testis antigen, neoantigen, co-stimulatory signals.
Introduction
T cells have been considered to be the most specific and potent killer cells in antitumor activity. However, the antitumor efficiency of T-cells is often insufficient partly due to the immune evasion mechanisms of cancer cells to escape from T cells killing under selective pressure. Several immunotherapies utilizing the cytotoxic potential of T cells to fight against tumors have been rapidly developed and successfully translated into clinical benefits. These include adoptive cell therapies, such as chimeric antigen receptor (CAR) T cells, genetically engineered TCR-T cells, and tumor-infiltrating lymphocytes (TILs). Adoptive therapy of TILs, for instance, consists of collection, ex vivo expansion, and reinfusion of patient-derived in particular mutation specific TILs. Relevant studies include objective regression in a patient with metastatic colorectal cancer after the infusion of TILs that specifically target KRAS G12D mutation [1]. Another highlighted immunotherapy to promote T cell function is the checkpoint blockades of PD-1/PD-L1 and CTLA-4, which have also demonstrated potent efficiency with approval for the treatment of advanced melanoma and NSCLC. Recombinant bispecific antibodies have also been intensively studied to retarget T cells against highly expressed tumor-associated antigens. Although bispecific antibodies are designed to take multiple forms, our review here focuses on a particular bispecific format: single chain variable fragment bispecific antibody.
Development of bispecific scFvs in solid tumors
The concept of recombinant bispecific antibodies was devised more than two decades ago. Over 50 different formats of bispecific antibodies are now available for therapeutic and diagnostic applications [2]. The approval of blinatumomab by the US. FDA in December 2014 highlighted bispecific single chain variable fragment antibody as powerful and promising platform for immunotherapy both in hematologic and solid tumor settings [3, 4]. This bispecific antibody format is engineered by combining scFv domains of two different antibodies on one polypeptide linker chain. One scFv domain is designed to recognize CD3Ɛ on T cells, whereas the other scFv is selected to bind tumor associated antigens. By simultaneously binding to tumor associated antigens (TAAs) and CD3Ɛ, The scFv antibody can retarget surrounding T cells to antigen-expressing tumor cells. This interaction is exclusively relies on TAAs and CD3Ɛ while completely independent of peptide/MHC complex and the antigen specificity of bound T cells [5-8].
In solid tumor indications, three bispecific scFvs have entered phase I clinical trial and at least ten other antibodies are being evaluated in preclinical development against an impressive variety of solid tumors (Table 1). MT-110 (Solitomab, AMG110): the EpCAM/CD3 bispecific scFv antibody aimed for solid malignancies of epithelial origin by targeting the epithelial marker EpCAM. A phase I clinical trial (NCT00635596) to assess safety and tolerability of MT110 in various solid tumors has been completed. The observations of transient decrease in tumor markers and CTC counts as well as necrotic tissue in tumor biopsies suggest anti-tumor activity. Best response of SD is reported in 38% of patients evaluable for anti-tumor response [9]. MT-111 (MEDI-565, AMG 211): the CEA/CD3 bispecific scFv antibody. Two phase I clinical trials of MT-111(NCT01284231 and NCT02291614) has been conducted in advanced gastrointestinal adenocarcinoma. The study NCT01284231 has already been completed. MT-111 was given intravenously over 3 hours on days 1 through 5 in 28-day cycles and no objective responses occurred, with 11 (28%) patients achieving stable disease as best response.[10] MT-112 (BAY2010112): the prostate specific membrane antigen (PSMA)/CD3 antibody intended to treat prostate cancer. Preclinical studies in mice models have demonstrated rapid tumor shrinkage and complete remissions of established human prostate cancer xenografts [11]. A phase I trial (NCT01723475) of MT-112 in subjects with castration-resistant prostate cancer is ongoing.
Overview of bispecific scFv antibody in solid tumor indications
| Targets | T cell marker | M Molecule | Indication | Status | Citation |
|---|---|---|---|---|---|
| EpCAM | CD3 | MT110 (AMG110) | Lung, gastric, colorectal, breast, hormone-refractory prostate cancer, and ovarian cancer | Phase I (completed) SD:38% | [9]/NCT00635596 |
| CEA | CD3 | MT-111 (AMG 211) | Gastrointestinal cancer | Phase I (completed) SD:28% | [10]/NCT01284231 |
| Phase I (recruiting) | NCT02291614 | ||||
| PSMA | CD3 | MT-112 (BAY2010112) | Prostate cancer | Phase I (recruiting) | NCT01723475 |
| EGFRVIII | CD3 | / | Glioblastoma | Preclinical | [69] |
| EGFR | CD3 | / | Colorectal cancer | Preclinical | [70] |
| GD2 | CD3 | / | Neuroblastoma, melanoma ovarian carcinoma | Preclinical | [71, 72] |
| MCSP | CD3 | / | Melanoma | Preclinical | [73] |
| ADAM17/PSCA | CD3 | A300E/ | Prostate Cancer | Preclinical | [74, 75] |
| CLDN6 | CD3 | 6PHU3 | ovarian carcinoma | Preclinical | [76] |
| EphA2 | CD3 | / | Colorectal cancer, melanoma ovarian carcinoma, breast cancer | Preclinical | [77] |
| HER2 | CD3 | / | / | Preclinical | / |
Strategies for specific tumor targeting
All tumor antigens targeted in bispecific scFvs constructs above show relative lower expression in normal tissues and consequently show relative lack of specificity. The challenge of specificity, which currently presents a major obstacle for the wider implementation of powerful immunotherapeutic agents such as chimeric antigen receptors and bispecific antibodies, has emerged studies on reagents that recognize or kill cancer cells but as less normal cells as possible. Three distinct types of tumor antigens to optimize bispecific scFv antibody design will be discussed in our review with effort to improve specificity and safety (Figure 1).
Optional strategies for tumor antigens and activation of T cells. To target tumor cells more specifically, three types of tumor antigens are reviewed: cancer-testis antigens, neo-antigens derived from mutation and virus associated antigens. For activation of T cells, co-stimulatory molecules CD28 and CD137, LIGHT and HIV-Tat101 are discussed.
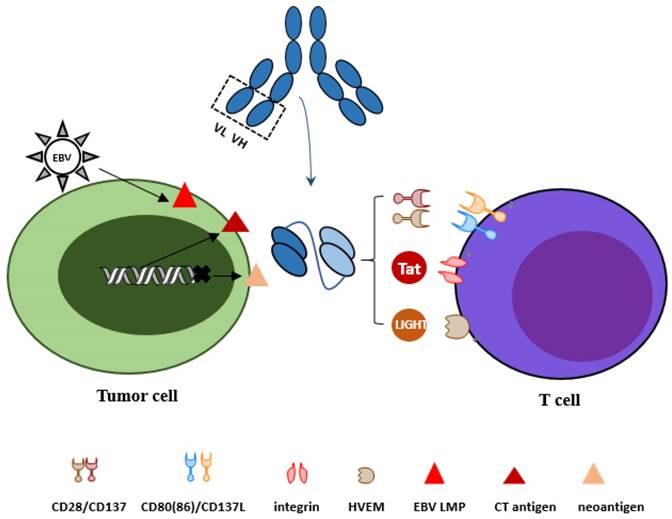
Cancer-testis antigens
Cancer-testis (CT) antigens are promising candidates in cancer immunotherapy. Most of CT antigens are limited to male germ cells among normal tissues, but highly expressed in various types of solid tumors. Testis germ cells are immune-privileged because of the absence of HLA-class I molecules and therefore cannot be recognized by T cells [12]. However, there are some CT antigens found in normal tissues except germ cells. Based on their expression, the CT antigens can be divided into three groups: testis-restricted antigens, such as New York esophageal squamous cell carcinoma 1 (NY-ESO-1), melanoma-associated antigens (MAGE-A1), synovial sarcoma X (SSX2); testis/brain-restricted antigens and testis-selective antigens [13]. Among CT antigens, MAGE-A1 and NY-ESO-1 have been extensively exploited in peptide vaccination and adoptive T cell transfer therapies [12, 14].
When applied in bispecific antibody, cancer-testis antigens are not accessible to monoclonal antibodies because they are typically located in nuclear or cytoplasm. However, endogenous antigens can be processed in the proteasome, and ultimately presented on the cell surface with major histocompatibility complex (MHC) molecules. Therapeutic monoclonal antibodies, called “TCR-like” mAbs, have been developed to recognize peptide/MHC complexes derived from intracellular tumor antigens on the cell surface [15-17]. Such tumor antigens for which specific TCR-like Fab or scFv antibodies have been successfully produced include telomerase catalytic subunit [18], Wilms tumor 1(WT1) [19], phosphatase of regenerating liver 3(PRL-3) [20], and MAGE-A1 and NY-ESO-1in CT antigens.
MAGE -A1 is frequently found in various solid tumors: esophageal squamous cell carcinomas, non-small cell lung carcinomas, metastatic melanomas, head and neck squamous cell carcinomas and bladder carcinomas. Several peptides derived from MAGE-A1 in the context of different HLA-A1 alleles have been identified. The first human antibody developed against class I peptide-MHC complex is the TCR-like Fab fragment directed for the MAGE-A1/HLA-A1 complex [21]. Another derivative Fab fragment against the same complex with an 18-fold improved affinity yet identical peptide specificity has also been identified. Retroviral transfer of chimeric receptors based on these two selected Fab fragments has used to genetically program primary human T lymphocytes. These T cells are examined for tumor cell killing capacities, with the high-affinity Fab fragment showing faster and enhanced anti-tumor responses [22, 23]. However, high affinity scFv Hyb3-γ/CD28 chimeric receptors T cells show only partial specific as no MAGE-A1 peptide loaded target cells were also lysed [24]. Recently, conversion of these two Fab fragments to scFv have been coupled to liposomes so as to specifically target melanoma cells and promote internalization of nanoparticles by these tumor cells [25].
NY-ESO-1 is highly expressed in solid tumors, including neuroblastoma, synovial sarcoma, melanoma, cervical cancer and epithelial ovarian cancer. Epitopes derived from NY-ESO-1 have been identified, including three peptides presented by the HLA-A2 molecule and at least 21 distinctive epitopes presented by at least five HLA class II alleles [26]. A Fab fragment that specifically recognizes 157-165 peptide/HLA-A0201 complex with high affinity (KD=60 nM) has been reported [27]. Similarly, another fragment specific to 157-165 peptide/HLA-A0201 with 20-fold affinity improvement by mutations has also been produced. T cells with recombinant single-chain TCRs specifically recognized and killed NY-ESO-1/HLA-A1+ T2 cells, with the scFv fragment of higher affinity showing higher level of IFN-γ release and cytotoxicity to peptide 157-165/HLA-A0201+ cells.[15] NY-ESO-1-specific CAR-T cells also showed anti-tumor effects in xenograft mode with endogenously NY-ESO-1 positive myeloma cells [28].
Neo-antigens
Neo-antigens are the consequences of genetic mutations accumulated by cancer cells during the tumorigenesis process. It is now feasible to identify mutations present within the protein encoding part of the genome with deep-sequencing technologies [29, 30]. Somatic mutations are the most optimized targets for cancer immunotherapy because they can only be found in tumor tissues, not in any normal cells. Similar to CT antigens, most of epitopes derived from somatic mutations lies in the interior of tumor cells [31]. However, 10-20% of these mutations are predicted to present on cell surface bound to common HLA types [32]. We review our emerging interest on the development of antibodies that recognize mutant peptide/HLA complexes.
There are few studies about antibodies specific for neo-antigens. It has been reported that scFvs specific for the epitopes derived from two commonly mutated oncogenes KRAS and EGFR, illustrated in the context of two common HLA types A2 and A3 respectively, have been obtained [33].These two targets have been selected out because of the mutation frequency and HLA alleles types. KRAS G12V mutation can be commonly found in tumor cells, particularly in 45% pancreatic adenocarcinomas, 14% lung adenocarcinomas, and 13% colorectal adenocarcinomas [34]. The mutant peptide containing the G12V mutation is predicted to be presented by the most common HLA allele in ethnic groups HLA-A2. The EGFR L858R mutation, located in the cytoplasmic domain of tumor cells, is found in ∼10% of lung adenocarcinomas [35]. This mutation resulted in the epitope presented by the HLA-A3 allele. ScFvs can specifically bind to and efficiently lyse T2 or T2A3 cells displaying mutant peptides in the context of HLA. The work reviewed above provided us the inspiration to generate antibody reagents that recognize mutation epitopes so as to kill mutant cancer cells specifically but not any normal cells. However, it still remains urgently to see whether and to what extent these antibody reagents can bind to tumor cells with low amounts of mutant peptides on the cell surface.
Virus associated antigens
Relevant antigens derived from oncogenic viruses provide attractive targets for tumors in which specific aetiology has been suggested. The present review focuses on Epstein-Barr virus (EBV) targeting in gastric carcinomas. About 10% (1.3% to 20.1%) of gastric carcinomas are EBV positive, which called EBV-associated gastric cancer (EBVaGC) [36, 37]. In EBVaGC, EBV is demonstrated to be exclusively restricted in gastric cancer cells and no detectable EBV is observed in normal gastric mucosa, intestinal metaplasia or stromal cells.[38] According to the viral RNAs and proteins detected during the latent phase, three major latency programs are commonly described [39]. EBV positive gastric carcinomas belong to latency program I, which includes the expression of EBV-encoded small RNAs (EBERs), transcripts from the BamHI A region(BARF0) and EBV nuclear antigens EBNA1 [40]. Somewhat different from conventional latency 1, latent membrane protein LMP2A is also expressed in about half of the EBVaGC cases.
In EBV-associated lymphoproliferative diseases (LPDs) and nasopharyngeal carcinoma (NPC), adoptive immunotherapy including allogenic or autologous EBV-specific CTLs transfer has been intensively investigated [41]. When EBV associated antigens are utilized for bispecific antibodies, trans-membrane LMPs expressed in cells infected but not in normal cells are prior candidate targets to be considered. This idea has been encouraged by the identification of specific antibody against LMP1 and the second generation CAR-T containing anti-LMP1 scFv to target LMP1 positive nasopharyngeal carcinoma cells [42]. The anti-LMP1 CAR-T displays specific cytolytic action in response to NPC cells overexpressing LMP1 in vitro and significantly inhibits growth of LMP1 expressing NPC cells in a subcutaneous xenograft model by intratumoral injection. Similarly, LMP2A is considered to be the optimal target for EBVaGC, which plays a crucial role in EBV-induced gastric carcinogenesis on cell proliferation and migration [43].
Strategies for activation of T cells
To date, anti-CD3 scFv has been widely utilized in bispecific scFvs being developed to target T cells. Co-stimulatory signals are also crucial for further regulating T cell activation [44]. It remains to be explored whether targeting these immunomodulatory molecules is sufficient for scFv antibody to mediate T cell activation, proliferation and cytotoxicity. We reviewed several leading candidates to be included in designs: CD28, CD137 among co-stimulatory molecules and immune-manipulating molecules like LIGHT and HIV-Tat101 (Figure 1).
Co-stimulatory molecules: CD28, CD137
CD28
The classical signal 2 is delivered by CD28 on binding to the ligands CD80 (B7-1) or CD86 (B7-2) on APCs. CD28 is constitutively expressed on the membrane of most resting CD4 T cells and about half of the CD8 T cells [45]. As for agonistic mAbs, anti-CD28 mAbs have been applied in sustained ex vivo expansion of cultured T cells. However, the further use of antibodies against CD28 has been discouraged as a result of a series of acute and serious adverse events in a phase I clinical trial where super agonist anti-CD28 mAb was tested systemically [46]. So localized or targeted use of anti-CD28 mAb is emerged to minimize the risks and promote the antitumor immunity, which can be achieved by bispecific antibodies targeting CD28 co-stimulation to tumor tissues. The research work from Spain has engineered a multidrug resistant-associated protein 1 (MRP1)/CD28 bivalent aptamer which is able to bind MRP1-expressing chemotherapy-resistant tumors and deliver the CD28 costimulatory signal to tumor infiltrating lymphocytes. Melanoma bearing mice systemically treated with MRP1-CD28 bivalent aptamer show reduced tumor growth and improved survival. Notably, both IFN-ү, TNF-α and immunosuppressive cytokines IL-10 significantly increased during the treatment, so a combination of Gvax, P60 and MRP1-CD28 bispecific aptamer were used to inhibit tumor growth [47]. The promising results from MRP1-CD28 bispecific aptamer make CD28 still a promising candidate in bispecific antibody constructs.
CD137
Also known as 4-1BB, CD137 is expressed on activated T cells including CD8 T cells, CD4 T cells, Tregs, and NKT cells [45]. Ligation of CD137 on T cells induces cell activation and enhanced effector functions. Upregulation of CD137 on recently activated T cells has been used to identify and isolate virus, tumor and neo-antigens reactive T cells from both peripheral blood and TIL in adoptive immunotherapy [48].
Agonistic mAbs are the leading therapeutic modality for harnessing the antitumor effects of CD137. Two anti-CD137 mAbs are currently in the clinical trials, Urelumab and PFZ-05082566 [49]. In bispecific settings, antitumor effects have been observed with bispecific oligonucleotide aptamers containing an agonistic CD137 aptamer and PSMA or VEGF aptamer. The PSMA-CD137 aptamer conjugate is a first-generation prototype bispecific aptamers. Systemic delivery of PSMA/CD137 aptamer inhibits tumor growth in both subcutaneously implanted tumor-bearing models and lung metastasis models, exhibiting a superior therapeutic index compared to CD137 antibodies [50]. Given that most receptors, including PSMA, are internalized once engaged by their ligands, research work from the same team turns to target the costimulatory ligands to products secreted into the tumor stroma such as VEGF, osteopontin (OPN) and metalloproteases instead of to tumor associated antigens expressed on the tumor cells. VEGF/CD137 aptamer conjugate inhibits multiple unrelated tumors in subcutaneous, postsurgical lung metastasis and autochthonous tumor models [51]. What's more, an anticalin-based HER2/CD137 bispecific antibody is being developed by Pieris Pharmaceuticals [52]. We are looking forward to the antitumor outcomes of this bsAb.
Immune-manipulating molecules: LIGHT and HIV-Tat101
LIGHT
LIGHT (the abbreviation of homologous to lymphotoxins, shows inducible expression, and competes with herpes simplex virus glycoprotein D for herpes virus entry mediator, a receptor expressed by T lymphocytes) has been verified to manipulate the anti-tumor immune responses. There are three already recognized receptors of LIGHT: lymphotoxin β receptor (LTβR), herpes viral entry mediator (HVEM) and DcR3. LIGHT affect immune responses in different ways when bound to different receptors. Interactions with LTβR on the infiltrating stroma cells can facilitate the homing of naive lymphocytes into the tumor stroma by inducing the expression of chemokine like CCL21 and the adhesion molecules like MAdCAM-1 [53]. The second receptor, HVEM, is expressed on both naive T cells and T cells at the end of activation. LIGHT acts as a strong CD28-independent costimulatory molecule on T cells by binding to HVEM [54]. HVEM transduces signal pathways via TRAFs to activate anti-tumor immune response. The HVEM cytoplasmic portion interacts with TNFR-associated factor family, especially TRAF5, which leads to synergistic activation of NF-κB. NF-κB activation can be further boost when there is cooperation with TCR signaling. Overexpression of HVEM also induced marked activation of Jun N-terminal kinase (JNK) and the Jun-containing transcription factor activator protein-1 (AP-1) [55]. These activities indicate that forced LIGHT expression in the tumor microenvironment may act as potent therapeutic strategies by recruiting and priming T cells.
Studies from Yangxin Fu and colleagues show that LIGHT-conditioned microenvironment not only selectively expands antigen specific tumor infiltrating lymphocytes (TILs), but also induces an increased infiltration of naive T lymphocytes as a consequence of the upregulation of chemokine and adhesion molecules. Changes happened in LIGHT-conditioned microenvironment finally lead to effective eradication of established tumors both in situ and at distal sites in vivo. [53] Except for injection of LIGHT-expressing tumors, forced focal LIGHT expression in the tumor microenvironment can be achieved through other various delivery methods. The same group has made efforts by intratumoral inoculation of Ad-LIGHT, an adenovirus that expresses LIGHT [56], and injection of LIGHT-expressing mesenchymal stem cells (MSC-L), which show potent tropism to tumor tissues [57]. With respect to bispecific antibody constructs, specifically targeting delivery of LIGHT to antigen expressing tumor tissues can be achieved by generating scFv-LIGHT fusion protein, with the supporting outcome that anti-EGFR-LIGHT induced complete regression in EGFR-expressing tumors, established protective T cell immunity to tumor challenges and overcame resistance to checkpoint blockade [58].
HIV Tat101
HIV-1 Tat 101 is a 101-amino acid protein produced after the HIV-1 infection [59]. In addition to affect viral replication and infectivity, Tat101 can be released out of the infected cells and then affect the functionality of neighboring immune cells, including DCs, CD4 T cells and CD8 T cells.
Immature dendritic cells are firstly infected by HIV after mucosal exposure. Monocyte-derived dendritic cells (MDDC) efficiently and selectively take up exogenous Tat protein in a dose, time, and temperature-dependent fashion. Upon uptake, Tat induces MDDC maturation by increasing expression of MHC and costimulatory molecules and MDDC activation by producing Th1-type cytokines, including IL-12, TNF-α, and β-chemokines [60].
For CD4 T cells, exogenous Tat protein interacts with one or more of surface receptors on CD4 T cells, including integrin receptors [61]. Tat activates ERK/MAPK and PI3K/ Akt pathways through its RGD domain. These pathways further activate the induction of T-bet, Eomes and Blimp-1, which finally enhance the secretion of IFN-γ, perforins and granzymes [62]. Studies from several groups support that interactions of Tat with CD4 T cells synergistically stimulates proliferation and IL-2 superproduction following stimulation of the CD3 and CD28 receptors [63, 64]. In CD8 T cells, Tat favors multi-functionalities of CD8 T cells when activated with antiCD3/CD28, including IL-2 and INF-γ expression, proliferation and cytotoxic activity of both antigen specific naive and memory CTL cells [62]. It has also shown that Tat prolongs the duration of CD8 T cells responses with earlier start and longer last time. However, antigen specific CD8+ T cells induced by Tat show affected cytokine release during the response. It is indicated that Tat causes part functionality loss of CD8 T cells except for hyperactivation [65]. It has been reported that Tat hyperactivates T cells by blocking the nicotinamide adenine dinucleotide (NAD+)-dependent deacetylase SIRT1, which potentiates NF-κB transcriptional activity through deacetylating lysine 310 in the p65 subunit of NF-kB specifically [66]. It remains to see whether Tat101 will show negative impacts in terms of impaired functionality, exhaustion, and susceptibility to apoptosis as observed in chronic HIV infection.
Perspective
Bispecific scFv antibodies in solid tumor indications have not yet been able to demonstrate credible therapeutic effects as indicated in phase I clinical trials. In addition to these alternative strategies for tumor associated antigens with improved specificity and activation of T cells, combinations of bispecific antibodies with checkpoint inhibitors or other immune modulating agents may offer beneficial synergistic effects to overcome immunosuppressive tumor microenvironment. Recent studies have shown the induction of PD-L1 expression after bispecific scFv antibody administration. Dual blockades of PD-1 and PD-L1 have been reported to maximize T cell cytotoxicity mediated by CEA/CD3 bispecific antibody [67]. Another major obstacle for immunotherapy in solid tumors is the absence of the T cell infiltration in tumor microenvironment. Recent study demonstrates that lack of spontaneous immune infiltration in solid tumors is unlikely due to lack of antigens. So it's of great importance to improve T cell infiltration into tumors therefore be able to facilitate clinical response to immunotherapy [68]. In summary, bispecific scFv antibody represents a promising platform for cancer immunotherapy and more evaluation and development are needed for their further application in solid tumors.
Abbreviations
scFv: single chain variable fragment; CT antigen: cancer-testis antigen; EpCAM: epithelial cell adhesion molecule; CEA: carcinoembryonic antigen; PSMA: prostate specific membrane antigen; EGFRVIII: epidermal growth factor receptor variant III; HER2: human epidermal growth factor receptor-2; GD2: disialoganglioside; MCSP: melanoma-associated chondroitin sulfate proteoglycan; ADAM17: a disintegrin and metalloproteinase 17; PSCA: prostate stem cell antigen; CLDN6: claudin 6; EphA2: ephrin type A receptor 2.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Tran E, Robbins PF, Lu YC, Prickett TD, Gartner JJ, Jia L. et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N Engl J Med. 2016;375:2255-62
2. Kontermann RE, Brinkmann U. Bispecific antibodies. Drug Discov Today. 2015;20:838-47
3. Topp MS, Gökbuget N, Stein AS, Zugmaier G, O'Brien S, Bargou RC. et al. Safety and activity of blinatumomab for adult patients with relapsed or refractory B-precursor acute lymphoblastic leukaemia: a multicentre, single-arm, phase 2 study. The Lancet Oncology. 2015;16:57-66
4. Goebeler ME, Bargou R. Blinatumomab: a CD19/CD3 bispecific T cell engager (BiTE) with unique anti-tumor efficacy. Leuk Lymphoma. 2016;57:1021-32
5. Klinger M, Benjamin J, Kischel R, Stienen S, Zugmaier G. Harnessing T cells to fight cancer with BiTE® antibody constructs-past developments and future directions. Immunological reviews. 2016;270:193-208
6. Stieglmaier J, Benjamin J, Nagorsen D. Utilizing the BiTE (bispecific T-cell engager) platform for immunotherapy of cancer. Expert Opin Biol Ther. 2015;15:1093-9
7. Suryadevara CM, Gedeon PC, Sanchez-Perez L, Verla T. Are BiTEs the "missing link" in cancer therapy? Oncoimmunology. 2015;4:e1008339
8. Huehls AM, Coupet TA, Sentman CL. Bispecific T-cell engagers for cancer immunotherapy. Immunol Cell Biol. 2015;93:290-6
9. Fiedler WM, Wolf M, Kebenko M, Goebeler M-E, Ritter B, Quaas A. et al. A phase I study of EpCAM/CD3-bispecific antibody (MT110) in patients with advanced solid tumors. ASCO Annual Meeting Proceedings. 2012;30:2504
10. Pishvaian M, Morse MA, Mcdevitt J, Norton JD, Ren S, Robbie G. et al. Phase 1 Dose Escalation Study of MEDI-565, a Bispecific T-Cell Engager that Targets Human Carcinoembryonic Antigen, in Patients With Advanced Gastrointestinal Adenocarcinomas. Clinical Colorectal Cancer. 2016;15:345-51
11. Friedrich M, Raum T, Lutterbuese R, Voelkel M, Deegen P, Rau D. et al. Regression of Human Prostate Cancer Xenografts in Mice by AMG 212/BAY2010112, a Novel PSMA/CD3-Bispecific BiTE Antibody Cross-Reactive with Non-Human Primate Antigens. Molecular Cancer Therapeutics. 2012;11:2664-73
12. Gjerstorff MF, Andersen MH, Ditzel HJ. Oncogenic cancer/testis antigens: prime candidates for immunotherapy. Oncotarget. 2015;6:15772-87
13. Hofmann O, Caballero OL, Stevenson BJ, Chen YT, Cohen T, Chua R. et al. Genome-wide analysis of cancer/testis gene expression. Proc Natl Acad Sci U S A. 2008;105:20422-7
14. Meek DW, Marcar L. MAGE-A antigens as targets in tumour therapy. Cancer Lett. 2012;324:126-32
15. Stewart-Jones G, Wadle A, Hombach A, Shenderov E, Held G, Fischer E. et al. Rational development of high-affinity T-cell receptor-like antibodies. Proc Natl Acad Sci U S A. 2009;106:5784-8
16. Salter AI, Riddell SR. A BiTE from cancer's intracellular menu. Nat Biotechnol. 2015;33:1040-1
17. Chang AY, Gejman RS, Brea EJ, Oh CY, Mathias MD, Pankov D. et al. Opportunities and challenges for TCR mimic antibodies in cancer therapy. Expert Opinion on Biological Therapy. 2016;16:979-87
18. Lev A, Denkberg G, Cohen CJ, Tzukerman M, Skorecki KL, Chames P. et al. Isolation and characterization of human recombinant antibodies endowed with the antigen-specific, major histocompatibility complex-restricted specificity of T cells directed toward the widely expressed tumor T-cell epitopes of the telomerase catalytic subu. Cancer Research. 2002;62:3184-94
19. Dao T, Yan S, Veomett N, Pankov D, Zhou L, Korontsvit T. et al. Targeting the intracellular WT1 oncogene product with a therapeutic human antibody. Sci Transl Med. 2013;5:176
20. Guo K, Li J, Tang JP, Tan CP, Hong CW, Alaidaroos AQ. et al. Targeting intracellular oncoproteins with antibody therapy or vaccination. Science Translational Medicine. 2011;3:1338-46
21. Chames P, Hufton SE, Coulie PG, Uchanska-Ziegler B, Hoogenboom HR. Direct selection of a human antibody fragment directed against the tumor T-cell epitope HLA-A1-MAGE-A1 from a nonimmunized phage-Fab library. Proceedings of the National Academy of Sciences. 2000;97:7969-74
22. Willemsen R, Debets R, Hart E, Hoogenboom H, Bolhuis R, Chames P. A phage display selected fab fragment with MHC class I-restricted specificity for MAGE-A1 allows for retargeting of primary human T lymphocytes. Gene therapy. 2001;8:1601-8
23. Chames P, Willemsen RA, Rojas G, Dieckmann D, Rem L, Schuler G. et al. TCR-like human antibodies expressed on human CTLs mediate antibody affinity-dependent cytolytic activity. The Journal of Immunology. 2002;169:1110-8
24. Willemsen RA, Ronteltap C, Chames P, Debets R, Bolhuis RLH. T Cell Retargeting with MHC Class I-Restricted Antibodies: The CD28 Costimulatory Domain Enhances Antigen-Specific Cytotoxicity and Cytokine Production. The Journal of Immunology. 2005;174:7853-8
25. Saeed M, van Brakel M, Zalba S, Schooten E, Rens JA, Koning GA. et al. Targeting melanoma with immunoliposomes coupled to anti-MAGE A1 TCR-like single-chain antibody. Int J Nanomedicine. 2016;11:955-75
26. Esfandiary A, Ghafouri-Fard S. New York esophageal squamous cell carcinoma-1 and cancer immunotherapy. Immunotherapy. 2015;7:411-39
27. Held G, Matsuo M, Epel M, Gnjatic S, Ritter G, Lee SY. et al. Dissecting cytotoxic T cell responses towards the NY-ESO-1 protein by peptide/MHC-specific antibody fragments. Eur J Immunol. 2004;34:2919-29
28. Schuberth PC, Jakka G, Jensen SM, Wadle A, Gautschi F, Haley D. et al. Effector memory and central memory NY-ESO-1-specific re-directed T cells for treatment of multiple myeloma. Gene Therapy. 2012;20:386-95
29. Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348:69-74
30. Tran E, Turcotte S, Gros A, Robbins PF, Lu Y, Dudley ME. et al. Cancer Immunotherapy Based on Mutation-Specific CD4+ T Cells in a Patient with Epithelial Cancer. Science. 2014;344:641-5
31. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546-58
32. Segal NH, Parsons DW, Peggs KS, Velculescu V, Kinzler KW, Vogelstein B. et al. Epitope landscape in breast and colorectal cancer. Cancer Research. 2008;68:889-92
33. Skora AD, Douglass J, Hwang MS, Tam AJ, Blosser RL, Gabelli SB. et al. Generation of MANAbodies specific to HLA-restricted epitopes encoded by somatically mutated genes. Proc Natl Acad Sci U S A. 2015;112:9967-72
34. Tran E, Ahmadzadeh M, Lu Y-C, Gros A, Turcotte S, Robbins PF. et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science. 2015;350:1387-90
35. D'Angelo SP, Pietanza MC, Johnson ML, Riely GJ, Miller VA, Sima CS. et al. Incidence of Exon 19 Deletions and L858R in Tumor Specimens From Men and Cigarette Smokers With Lung Adenocarcinomas. Journal of Clinical Oncology. 2011;29:2066-70
36. Chen J-N, He D, Tang F, Shao C-k. Epstein-Barr virus-associated gastric carcinoma: a newly defined entity. Journal of clinical gastroenterology. 2012;46:262-71
37. Cho J, Kang MS, Kim KM. Epstein-Barr Virus-Associated Gastric Carcinoma and Specific Features of the Accompanying Immune Response. J Gastric Cancer. 2016;16:1-7
38. Truong CD, Feng W, Li W, Khoury T, Li Q, Alrawi S. et al. Characteristics of Epstein-Barr virus-associated gastric cancer: a study of 235 cases at a comprehensive cancer center in U.S.A. J Exp Clin Cancer Res. 2009;28:14
39. Taylor GS, Long HM, Brooks JM, Rickinson AB, Hislop AD. The immunology of Epstein-Barr virus-induced disease. Annu Rev Immunol. 2015;33:787-821
40. De Oliveira DE, Mullercoan BG, Pagano JS. Viral Carcinogenesis Beyond Malignant Transformation: EBV in the Progression of Human Cancers. Trends in Microbiology. 2016;24:649-64
41. Masmoudi A, Toumi N, Khanfir A, Kallel-Slimi L, Daoud J, Karray H. et al. Epstein-Barr virus-targeted immunotherapy for nasopharyngeal carcinoma. Cancer Treat Rev. 2007;33:499-505
42. Tang X, Zhou Y, Li W, Tang Q, Chen R, Zhu J. et al. T cells expressing a LMP1-specific chimeric antigen receptor mediate antitumor effects against LMP1-positive nasopharyngeal carcinoma cells in vitro and in vivo. J Biomed Res. 2014;28:468-75
43. Shinozaki-Ushiku A, Kunita A, Fukayama M. Update on Epstein-Barr virus and gastric cancer (review). Int J Oncol. 2015;46:1421-34
44. Mahoney KM, Rennert PD, Freeman GJ. Combination cancer immunotherapy and new immunomodulatory targets. Nat Rev Drug Discov. 2015;14:561-84
45. Melero I, Hervas-Stubbs S, Glennie M, Pardoll DM, Chen L. Immunostimulatory monoclonal antibodies for cancer therapy. Nat Rev Cancer. 2007;7:95-106
46. Hünig T. The storm has cleared: lessons from the CD28 superagonist TGN1412 trial. Nature Reviews Immunology. 2012;12:317-8
47. Soldevilla MM, Villanueva H, Casares N, Lasarte JJ, Bendandi M, Inoges S. et al. MRP1-CD28 bi-specific oligonucleotide aptamers: target costimulation to drug-resistant melanoma cancer stem cells. Oncotarget. 2016;7:23182-96
48. Ye Q, Song DG, Poussin M, Yamamoto T, Best A, Li C. et al. CD137 accurately identifies and enriches for naturally-occurring tumor-reactive T cells in tumor. Clinical Cancer Research. 2014;20:44-55
49. Sanmamed MF, Pastor F, Rodriguez A, Perez-Gracia JL, Rodriguez-Ruiz ME, Jure-Kunkel M. et al. Agonists of Co-stimulation in Cancer Immunotherapy Directed Against CD137, OX40, GITR, CD27, CD28, and ICOS. Semin Oncol. 2015;42:640-55
50. Pastor F, Kolonias D, McNamara JO 2nd, Gilboa E. Targeting 4-1BB costimulation to disseminated tumor lesions with bi-specific oligonucleotide aptamers. Mol Ther. 2011;19:1878-86
51. Schrand B, Berezhnoy A, Brenneman R, Williams A, Levay A, Kong LY. et al. Targeting 4-1BB costimulation to the tumor stroma with bispecific aptamer conjugates enhances the therapeutic index of tumor immunotherapy. Cancer Immunol Res. 2014;2:867-77
52. Makkouk A, Chester C, Kohrt HE. Rationale for anti-CD137 cancer immunotherapy. Eur J Cancer. 2016;54:112-9
53. Yu P, Lee Y, Liu W, Chin RK, Wang J, Wang Y. et al. Priming of naive T cells inside tumors leads to eradication of established tumors. Nat Immunol. 2004;5:141-9
54. Tamada K, Shimozaki K, Chapoval AI, Zhu G, Sica G, Flies D. et al. Modulation of T-cell-mediated immunity in tumor and graft-versus-host disease models through the LIGHT co-stimulatory pathway. Nature medicine. 2000;6:283-9
55. Wang Y, Zhu M, Miller M, Fu YX. Immunoregulation by tumor necrosis factor superfamily member LIGHT. Immunol Rev. 2009;229:232-43
56. Yu P, Lee Y, Wang Y, Liu X, Auh S, Gajewski TF. et al. Targeting the primary tumor to generate CTL for the effective eradication of spontaneous metastases. The Journal of Immunology. 2007;179:1960-8
57. Zou W, Zheng H, He TC, Chang J, Fu YX, Fan W. LIGHT delivery to tumors by mesenchymal stem cells mobilizes an effective antitumor immune response. Cancer Res. 2012;72:2980-9
58. Tang H, Wang Y, Chlewicki Lukasz K, Zhang Y, Guo J, Liang W. et al. Facilitating T Cell Infiltration in Tumor Microenvironment Overcomes Resistance to PD-L1 Blockade. Cancer Cell. 2016;29:285-96
59. Roy CN, Khandaker I, Furuse Y, Oshitani H. Molecular characterization of full-length Tat in HIV-1 subtypes B and C. Bioinformation. 2015;11:151
60. Fanales-Belasio E, Moretti S, Nappi F, Barillari G, Micheletti F, Cafaro A. et al. Native HIV-1 Tat protein targets monocyte-derived dendritic cells and enhances their maturation, function, and antigen-specific T cell responses. The Journal of Immunology. 2002;168:197-206
61. Secchiero P, Zella D, Curreli S, Mirandola P, Capitani S, Gallo RC. et al. Pivotal role of cyclic nucleoside phosphodiesterase 4 in Tat-mediated CD4+ T cell hyperactivation and HIV type 1 replication. Proc Natl Acad Sci U S A. 2000;97:14620-5
62. Sforza F, Nicoli F, Gallerani E, Finessi V, Reali E, Cafaro A. et al. HIV-1 Tat affects the programming and functionality of human CD8(+) T cells by modulating the expression of T-box transcription factors. AIDS. 2014;28:1729-38
63. Kwon H-S, Brent MM, Getachew R, Jayakumar P, Chen L-F, Schnolzer M. et al. Human immunodeficiency virus type 1 Tat protein inhibits the SIRT1 deacetylase and induces T cell hyperactivation. Cell host & microbe. 2008;3:158-67
64. Sforza F, Nicoli F, Gallerani E, Finessi V, Reali E, Cafaro A. et al. HIV-1 Tat affects the programming and functionality of human CD8+ T cells by modulating the expression of T-box transcription factors. Aids. 2014;28:1729-38
65. Nicoli F, Finessi V, Sicurella M, Rizzotto L, Gallerani E, Destro F. et al. The HIV-1 Tat protein induces the activation of CD8+ T cells and affects in vivo the magnitude and kinetics of antiviral responses. PLoS One. 2013;8:e77746
66. Kwon HS, Brent MM, Getachew R, Jayakumar P, Chen LF, Schnolzer M. et al. Human immunodeficiency virus type 1 Tat protein inhibits the SIRT1 deacetylase and induces T cell hyperactivation. Cell Host Microbe. 2008;3:158-67
67. Osada T, Patel SP, Hammond SA, Osada K, Morse MA, Lyerly HK. CEA/CD3-bispecific T cell-engaging (BiTE) antibody-mediated T lymphocyte cytotoxicity maximized by inhibition of both PD1 and PD-L1. Cancer Immunol Immunother. 2015;64:677-88
68. Spranger S, Luke JJ, Bao R, Zha Y, Hernandez KM, Li Y. et al. Density of immunogenic antigens does not explain the presence or absence of the T-cell-inflamed tumor microenvironment in melanoma. Proc Natl Acad Sci U S A. 2016;113:E7759-E68
69. Choi BD, Kuan C, Cai M, Archer GE, Mitchell DA, Gedeon PC. et al. Systemic administration of a bispecific antibody targeting EGFRvIII successfully treats intracerebral glioma. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:270-5
70. Lutterbuese R, Raum T, Kischel R, Hoffmann P, Mangold S, Rattel B. et al. T cell-engaging BiTE antibodies specific for EGFR potently eliminate KRAS- and BRAF-mutated colorectal cancer cells. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:12605-10
71. Yu AL, Gilman AL, Ozkaynak MF, London WB, Kreissman SG, Chen HX. et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J Med. 2010;363:1324-34
72. Cheng M, Santich BH, Xu H, Ahmed M, Huse M, Cheung NK. Successful engineering of a highly potent single-chain variable-fragment (scFv) bispecific antibody to target disialoganglioside (GD2) positive tumors. Oncoimmunology. 2016;5:e1168557
73. Torisuitakura H, Schoellhammer HF, Sim M, Irie RF, Hausmann S, Raum T. et al. Redirected lysis of human melanoma cells by a MCSP/CD3-bispecific BiTE antibody that engages patient-derived T cells. Journal of Immunotherapy. 2011;34:597-605
74. Feldmann A, Arndt C, Topfer K, Stamova S, Krone F, Cartellieri M. et al. Novel humanized and highly efficient bispecific antibodies mediate killing of prostate stem cell antigen-expressing tumor cells by CD8+ and CD4+ T cells. J Immunol. 2012;189:3249-59
75. Yamamoto K, Trad A, Baumgart A, Huske L, Lorenzen I, Chalaris A. et al. A novel bispecific single-chain antibody for ADAM17 and CD3 induces T-cell-mediated lysis of prostate cancer cells. Biochem J. 2012;445:135-44
76. Stadler CR, Bahr-Mahmud H, Plum LM, Schmoldt K, Kolsch AC, Tureci O. et al. Characterization of the first-in-class T-cell-engaging bispecific single-chain antibody for targeted immunotherapy of solid tumors expressing the oncofetal protein claudin 6. Oncoimmunology. 2016;5:e1091555
77. Hammond SA, Lutterbuese R, Roff S, Lutterbuese P, Schlereth B, Bruckheimer E. et al. Selective targeting and potent control of tumor growth using an EphA2/CD3-Bispecific single-chain antibody construct. Cancer Res. 2007;67:3927-35
Author contact
![]() Corresponding authors: Baorui Liu, MD, Ph.D, Comprehensive Cancer Centre of Drum Tower Hospital, Medical School of Nanjing University, Clinical Cancer Institute of Nanjing University, 321 Zhongshan Road, Nanjing 210008, China Tel: +86-25-83107081 Fax: +86-25-83317016 E-mail: baoruiliuedu.cn Yudong Qiu, Ph.D, Department of Hepatopancreatobiliary Surgery, The Affiliated Drum Tower Hospital of Medical School of Nanjing University, 321 Zhongshan Road, Nanjing 210008, People's Republic of China Tel: + 86-25-83106666 Fax: + 86-25-83105929 Email: yudongqiu510com
Corresponding authors: Baorui Liu, MD, Ph.D, Comprehensive Cancer Centre of Drum Tower Hospital, Medical School of Nanjing University, Clinical Cancer Institute of Nanjing University, 321 Zhongshan Road, Nanjing 210008, China Tel: +86-25-83107081 Fax: +86-25-83317016 E-mail: baoruiliuedu.cn Yudong Qiu, Ph.D, Department of Hepatopancreatobiliary Surgery, The Affiliated Drum Tower Hospital of Medical School of Nanjing University, 321 Zhongshan Road, Nanjing 210008, People's Republic of China Tel: + 86-25-83106666 Fax: + 86-25-83105929 Email: yudongqiu510com