Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(18):3675-3681. doi:10.7150/jca.21148 This issue Cite
Research Paper
Cpt1c regulated by AMPK promotes papillary thyroid carcinomas cells survival under metabolic stress conditions
Rui Wang1*, Yajun Cheng2*, Dongwei Su3*, Boshen Gong1, Xiaobo He 1, Xinyu Zhou 1, Zhijun Pang 1, Lingtao Cheng 1, Yuelei Chen 4, Zhenzhen Yao 1 ![]()
1. Department of Biochemistry and Molecular Biology, Second Military Medical University, Shanghai 200433, China;
2. Department of urology, shanghai ninth people hospital, Shanghai Jiaotong University School of Medicine, Shanghai 200011, China.
3. Department of General Surgery (Surgical Breast and Thyroid Section), Changhai Hospital, Second Military Medical University, Shanghai 200433, China
4. Shanghai institute of biochemistry and Cell Biology, shanghai 200031, China
*These authors contributed equally to this article
Received 2017-5-23; Accepted 2017-8-31; Published 2017-10-12
Citation:
Wang R, Cheng Y, Su D, Gong B, He X, Zhou X, Pang Z, Cheng L, Chen Y, Yao Z. Cpt1c regulated by AMPK promotes papillary thyroid carcinomas cells survival under metabolic stress conditions. J Cancer 2017; 8(18):3675-3681. doi:10.7150/jca.21148. https://www.jcancer.org/v08p3675.htm
Other stylesAbstract
Background: Cancer cells have to take metabolic transformation in tumor progression when facing need of increased energy and adequate vascularization. However, molecular mechanism is not fully known. In this study, we showed that expression of carnitine palmitoyltransferase 1C (Cpt1c), as a member of the gate-keeper enzymes , which transferring long-chain fatty acids into mitochondria to further oxidation, which is regulated by AMPK promotes papillary thyroid carcinomas cells survival under metabolic stress conditions.
Methods: Firstly, we used qRT-PCR to detect expression of Cpt1c in papillary thyroid carcinomas tissues compared with paired normal tissues. Secondly, to evaluate whether Cpt1c is induced under metabolic stress, models of hypoxia (0.2% oxygen) and glucose deprivation for cultured papillary thyroid carcinomas cells were established. Lastly, KTC-1 cells were treated with AICAR (as an agonist of AMPK) and Compound C (as an inhibitor of AMPK) to investigate the correlation of AMPK activity with Cpt1c expression under metabolic stress.
Results: Cpt1c is higher in papillary thyroid carcinomas tissues compared with paired normal tissues. Furthermore, Cpt1c up-regulation promotes cancer cell growth and metastasis. In addition, the results showed that Cpt1c expression is induced by metabolic stress, including hypoxia and low glucose treatment. Consistently, Cpt1c can protect cells from cancer cells death caused by hypoxia and low glucose. Lastly, Cpt1c expression is regulated by AMPK activity.
Conclusion: Here we describe that induction of Cpt1c expression facing metabolic stress in papillary thyroid carcinomas is at least partly regulated by AMPK activity and ultimately contribute to development and progression of papillary thyroid carcinomas.
Keywords: Cpt1c, AMPK, metabolic stress, thyroid cancer
Introduction
Thyroid cancers derived from thyroid follicular cells, including differentiated thyroid carcinoma (DTC), medullary carcinoma and anaplastic thyroid carcinomas(ATC)[1]. Papillary thyroid carcinoma (PTC) and follicular thyroid carcinoma (FTC) constitutes well- differentiated TC (DTC), which along with poorly differentiated (PDTC) and undifferentiated thyroid carcinomas (ATC), represents epithelial thyroid cancers also named non-medullary thyroid cancers (NMTC)[2].Though PTC is the most common malignancy among all thyroid cancer types and has a relatively optimistic prognosis diagnosed at early stage with traditional therapies including surgery, thyroidectomy, thyroid-stimulating hormone (TSH) suppression, and radioactive iodine (RAI), it is ineffective against recurrent disease and distant metastatic disease at initiation[3]. Furthermore, ATC cells have other characteristics of high proliferation rate and marked aneuploidy, which results in almost uniformly fatal of ATC patients with a short-lived survival from diagnosis, about 6 months [4]. Patients with advanced PTC or ATC face poor prognosis and need explore novel therapeutics to improve the poor outcome of patients.
Solid tumors, including thyroid cancer, frequently contain regions without adequate vascularization depleting of oxygen, glucose, and other crucial nutrients and experience metabolic stress[5]. Cancer cell survival under metabolic stress is a pivotal step not only for solid tumor growth but also for metastatic progression by metabolic transformation. It is known that the Warburg effect, as an important way of metabolic transformations, produces more energy to maintain cancer cell survival in tumorigenesis by enhancing glucose uptake[6, 7]. In addition, other metabolism patterns also exist in tumor to promote cancer cell survival by supplying more energy, including glutamine metabolism and aberrant fatty acid metabolism[8]. Transformation of cellular metabolism can be mediated by abnormal expression of oncogene and tumor suppressor gene, which plays an important role in tumorigenesis and development[8]. However, molecular mechanisms of metabolic adaptations of solid tumors are still not fully elucidated.
Recent reports showed that fatty acid oxidation (FAO) emerging in metabolic transformation facing metabolic stress contributes to the tumorigenesis and development by producing more energy[9]. Higher expressions of several enzymes associated with FAO have been identified in tumors[10]. Carnitine palmitoyltransferase (CPT1) isozymes promote lipid metabolism by importing FA into the mitochondria. Cpt1c, as an isoform of CPT1, is high expressed in various tumors and induced to promote cancer cell survival under metabolic stress. However, the expression and role of Cpt1c in papillary thyroid carcinoma have not been reported. In this paper, specimens of papillary thyroid carcinoma patient were collected to analyze the Cpt1c expression. What is more, the roles of CPT1C in papillary thyroid carcinoma cell growth and metastasis or facing metabolic stress were explored.
Materials and Methods
Ethics statement
This study was approved through the Ethics Committee of Second Military Medical University (SMMU) (Shanghai, China). And, informed consent form was received from all participants.
Patients' tissue samples
The human tissue specimens, including 48 papillary thyroid carcinomas tissues and paired normal paracancer tissues, were obtained from patients who accepted surgery treatment at the First Affiliated Hospital (Changhai Hospital) of Second Military Medical University, Shanghai, China. In addition, other relative treatments, especially chemotherapy or radiotherapy, were not applied to treat the patients before surgery.
Cell culture
Human papillary thyroid carcinomas cell lines B-CPAP and KTC-1 were kindly purchased from Stem Cell Bank, Chinese Academy of Sciences, and maintained in RMPI1640 media (Invitrogen, 11875093) with 10% fetal bovine serum (FBS) (Gibco, 10099141), 1% NEAA (Invitrogen 11140050), 1% Glutamax (Invitrogen 35050061), 1%Sodium Pyruvate 100 mM Solution (Invitrogen 11360070) and 100 U/ml penicillin/streptomycin at 37 °C and 5% CO2. Cells with 2 × 105 cells/well were plated into six-well plates and incubated for 12 h in growth medium. After this, cells were treated with hypoxia (0.2% O2) or maintained in glucose-free medium (Gibco), and with or without AICAR (Selleck, S1802) or Compound C (Selleck, S7306) treatment.
Cell transfection
For cell interference, Cpt1c siRNA was synthesized at Shanghai GenePharma Company Limited. The sequences of Cpt1c and negative control siRNA are shown: Cpt1c (Sense: GGCGUCCAAUUAUGUCAGUTT; Antisense: ACUGACAUAAUUGGACGCCTT); In Brief, cells were maintained in 6-well. After 12 h, siRNA were transfected into cells using Roche reagent (06366546001) according to the manufacturer's protocol. For plasmid transfection, pcDNA3.1-Cpt1c was a gift from Michael J. Wolfgang professor, Johns Hopkins University School of Medicine. After12 h culture, cells were transfected with plasmids containing empty vector or pcDNA3.1-CPT1C constructs using Roche reagent according to the manufacturer's protocol.
Cell proliferation and viability assay
For cell proliferation assay, the cells (2 × 104cells/well) were plated in 96-well plates. At 0, 24, 48 and 72 h post-transfection, cells were collected and cell proliferation was evaluated by CCK-8 assay kit (Beyotime, China). For Cell viability assay, the cells (5 × 104 cells/well) were plated in 96-well plates and treated with various concentrations of Glucose or different time of hypoxia (0.2% O2). Next, CCK-8 assay kit (Beyotime, China) was applied into determining the cell viability.
Cell cycle and apoptosis analysis
Cells were harvested at 48 h post-transfection. For cell cycle assay, the collected cells were fixed in 70% ethanol at 4 ℃ for overnight. The cells were subsequently incubated with propidium iodide (PI) in the presence of RNase A. Finally, the cell cycle was analyzed using Flow cytometry. For Cell apoptosis analysis, cells were washed twice with PBS, and then resuspended in buffer with propidium iodide (PI) and annexin V. After lucifugal incubation at room temperature for 15 min, the stained cells were transferred to a BD LSR Fortessa Cell Analyzer (BD Biosciences, San Jose, CA, USA), determining the fluorescence emission at 488 nm excitation.
Cell migration assay
After transfection, cancer cells(1 × 104 cells/0.1 mL in serum-free medium) were transferred into the upper chamber, and the lower chamber was full of 0.6 mL of medium with 20%FBS. After 24 hours, cotton swab was used to remove the cells adhering to the upper surface of the well, and migrated cells on the lower surface were fixed and stained with 0.4% crystal violet. Next, image was measured and statistically analyzed.
Western blot
The harvest cells were lysed in cell lysis buffer (Beyotime, China) to obtain the whole cell lysates. The protein concentration was assessed using a protein assay reagent (BCA, Beyotime, China). Equal protein from samples was electrophoresed on SDS-PAGE and then transferred to the nitrocellulose (NC) membrane. Furthermore, the NC membrane was incubated with antibodies against Cpt1c (Abcam, ab87773), β-actin (Proteintech, 60008), total and phosphorylated FAK (pY172, Cell Signaling Technology, USA) at 4℃ overnight, and secondary horseradish peroxidaseconjugated antibody. The quantification of the Western blot was evaluated using the Quantity One software.
qRT-PCR
The total RNAs isolated from patient tissue specimens were treated with a standard TRIzol protocol (Ambion) RNA quality A260/A280 ratio and spectrophotometry was used to assess RNA quantity. And then, cDNA was synthesized with the PrimeScriptTMRT Master Mix Kit (Takara, RR036A) and qPCR was carried out using GoTaq® qPCR Master Mix (Promega, A6002) according to the manufacturer's protocol. The primer sequences were used as followed: Cpt1c: forward primer (GGATGGCACTGAAGAGGAAA), reverse primer (TCCTGGAAAAGGCATCTCTC); Actin: forward primer (GGGACCTGACTGACTACCTC), reverse primer (TCATACTCCTGCTTGCTGAT).
Statistical evaluation
Data are expressed as mean ± SD. A Student's T test or one-way ANOVA for experiments were used to perform statistical analyses. Differences were considered to be significant at a value of P < 0.05. All data shown are from at least three independent experiments.
Results
Cpt1c expression is higher in papillary thyroid carcinomas and promotes cancer cell growth and metastasis
To evaluate the Cpt1c expression in papillary thyroid carcinomas tissue and adjacent normal tissues, qRT-PCR was performed to detect mRNA level in 48 paired samples. Cpt1c expression was higher in papillary thyroid carcinomas samples than in matched normal tissue (34/48, 71%) (P<0.05, Figure 1A). In order to address the potential biological role of Cpt1c in papillary thyroid carcinoma cells, Cpt1c siRNA was synthetized and transfected into KTC-1 and B-CPAP cell lines. The down-regulated expression of Cpt1c was confirmed through Western blot (Figure 1B). The effects of Cpt1c knockdown on the proliferation of papillary thyroid carcinomas cells were evaluated using CCK-8 kit. Though there is no significant differences at 24 h after transfection (P>0.05), Cpt1c significantly inhibited cell growth in Cpt1c-siRNA transfected cells relative to control at 48 and 72 h (P < 0.05) (Figure 1C). Moreover, cell apoptosis and the cell cycle influenced by Cpt1c down-regulation were assessed using flow cytometry. As shown in figure 1D and 1E, respectively, Cpt1c silencing induced papillary thyroid carcinomas cells apoptosis (P<0.05) and cell cycle arrest in G1/S phase (P<0.05) compared with control. In addition, it also inhibited papillary thyroid carcinomas cells migration as shown in transwell migration assays (P<0.05) (Figure 1F). These results suggested that Cpt1c expression is higher in papillary thyroid carcinomas and promote cancer cells growth and metastasis.
Figure 1
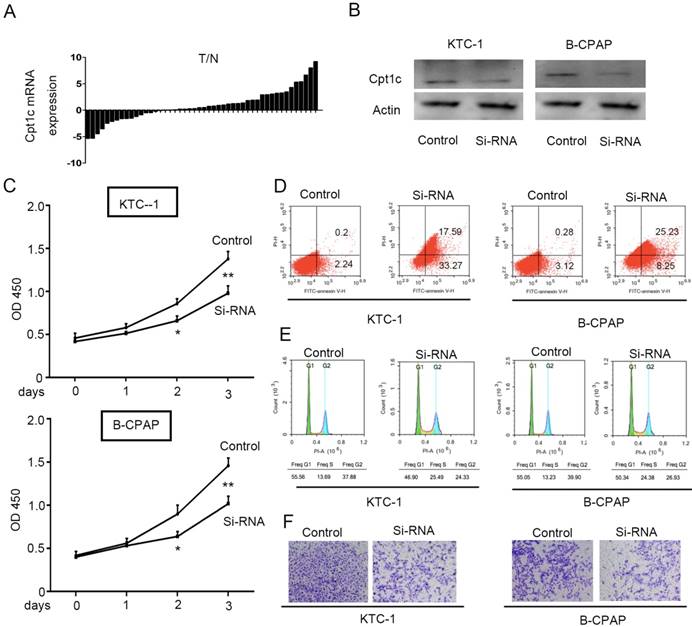
Cpt1c expressions is higher in papillary thyroid carcinomas and Cpt1c promotes cancer cell growth and metastasis. (A): Cpt1c expression is analyzed in 48 paired papillary thyroid carcinomas samples by qRT-PCR. (B) Transfect efficiency of Cpt1c siRNA is evaluated by Western blot. (C) Cpt1c siRNA effect on cell viability is measured by CCK-8 at day 1, 2 and 3. (D) and (E) cell apoptosis and cell cycle are analyzed by FCS after transfecting 48h. (F) The cell migration was assessed using transwell assays. *P < 0.05, **P < 0.01 VS control.
Cpt1c is induced under metabolic stress and down-regulation of Cpt1c promotes cancer cells death facing metabolic stress
To evaluate whether Cpt1c is induced under metabolic stress, models of hypoxia (0.2% oxygen) and glucose deprivation for cultured cancer cells were established. We found that Cpt1c was induced time-dependently under depleting of O2 by qRT-PCR evaluation (Figure 2A). Meanwhile, glucose deprivation also significantly increased Cpt1c expression after 48h concentration-dependently (Figure 2B). Next, we measured whether the viability of cancer cells facing metabolic stress was influenced by Cpt1c expression. The results showed that depletion of Cpt1c promoted the cancer cells death under hypoxia compared with NC (Figure 2C). Consistently, glucose deprivation also induced relatively more death in KTC-1 and B-CPCP cell lines with down-regulation of Cpt1c compared with control (Figure 2D). These results suggested that Cpt1c is induced under metabolic stress to increase cell survival facing metabolic stress.
Increasing the Cpt1c expression promotes cancer cell survival under metabolic stress
To further verify the effect of Cpt1c on promoting cancer cell survival facing metabolic stress, Cpt1c plasmid vector was constructed and transfected into KTC-1 cells. Figure 3A showed that Cpt1c was significantly over-expressed in KTC-1 cells. Next, we found that Cpt1c over-expression promoted the cancer cells survival under hypoxia compared with vector (Figure 3B). In addition, Cpt1c over-expression promoted the cancer cells survival under glucose deprivation (Figure 3C). Above results further verified that Cpt1c is induced under metabolic stress to increase cell survival under metabolic stress.
Cpt1c expression is regulated by AMPK activity
Figure 2
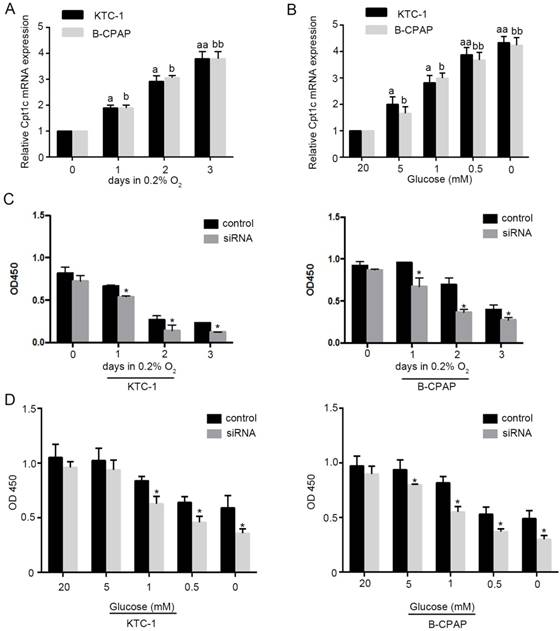
Cpt1c is induced under metabolic stress and down-regulation of Cpt1c promotes cancer cells death facing metabolic stress. (A): KTC-1 cells were cultured in hypoxia for 0, 1, 2 and 3 day, and Cpt1c expression was evaluated by qRT-PCR. (B): B-CPAP cells were cultured in low glucose (20, 5, 1, 0.5 and 0 mM) for 48h, and Cpt1c expression was evaluated by qRT-PCR. (C): KTC-1 cells and B-CPAP cells with Cpt1c siRNA and control were cultured in hypoxia for for 0, 1, 2 and 3 day , and cell viability was measured by CCK-8. (D) KTC-1 cells and B-CPAP cells with Cpt1c siRNA and control were cultured in low glucose (20, 5, 1, 0.5 and 0 mM) for 48h, and cell viability was measured by CCK-8. *P < 0.05, **P < 0.01 VS control.
Though Cpt1c plays a vital role in papillary thyroid carcinomas cells facing metabolic stress, molecular mechanism of Cpt1c expression induced by metabolic stress is not known and it need to be explored. AMPK was known to be activated to limit energy consumption and produce more energy in process of metabolic transformation[11]. Glucose deprivation not only promoted the Cpt1C expression, but also significantly increased AMPK activity after 48h in a concentration-dependent manner (Figure 4A). To investigate the correlation between AMPK activity and Cpt1c expression under metabolic stress, we evaluated whether AICAR, as an agonist of AMPK, treatment could induce the Cpt1c expression in KTC-1 cells. Accompanying the increase of AMPK activity induced by AICAR, Cpt1c expression was also increased in KTC-1 cells (Figure 4B). On this basis, Compound C, as an inhibitor of AMPK, was used to culture KTC-1 cells with depriving of O2 and glucose. We found that Cpt1c expression induced by metabolic stress could be impaired by Compound C and with concentration dependence (Figure 4C and 4D). These data indicated that the induction of CPT1C expression facing metabolic stress is at least partly regulated by AMPK activity.
Discussion
In this study, we demonstrate that Cpt1c expression is higher in papillary thyroid carcinomas tissues compared with paired normal tissues. Furthermore, Cpt1c silencing suppresses cancer cells growth and metastasis. In addition, the results showed that Cpt1c expression is induced by metabolic stress, including hypoxia and glucose deprivation. And Cpt1c can protect cells from cell death induced by hypoxia and low glucose. Lastly, we found that AICAR (an agonist of AMPK) treatment could induce the Cpt1c expression, and inhibition of AMPK activity can impair Cpt1c expression induced by metabolic stress. In a word, induction of CPT1C expression facing metabolic stress in papillary thyroid carcinomas is at least partly regulated by AMPK activity and ultimately contributes to development and progression of papillary thyroid carcinomas.
Thyroid cancer, as the most common endocrine malignancy, has a rapidly increasing incidence in world-wide recently [12]. PTC, as the most common type of thyroid cancer, accounts for 70-90 % of all thyroid malignancies[13]. In order to meet the requirement of increased energy, macromolecular substrate for high rate proliferation in cancer cells, high rates of glycolysis commonly exist in cell interior[14]. Meanwhile, due to poor tumor vasculature, tumor microenvironment with limited nutrients or hypoxia lead to an adaptive metabolic program in cancer cells to survival and growth[15]. For instance, in order to maintain cell survival under metabolic stress, it may transform their metabolic phenotypes in cancer cells was spontaneously shifted from glycolysis to fatty acid oxidation (FAO; also known as β-oxidation)[16]. Therefore, it is potential to find novel prevention or therapeutic targets by targeting the energy metabolism pathways of tumors. Camarda R et al [17] reported that pharmacologic inhibition of FAO catastrophically decreased energy production in MYC-overexpressing breast cancer cells and inhibited tumor growth. And etomoxir, as an inhibitor of FAO, promoted cancer cell death and impaired ATP depletion by decreasing NADPH production and increasing reactive oxygen species in glioblastoma[18]. In addition, Hossain F et al [19] reported that Inhibition of FAO metabolic pathways also can Modulate Immunosuppressive functions of myeloid-derived suppressor cells to enhance cancer therapies efficiency. These findings support a valuable role of targeting FAO metabolic pathway for cancer therapy.
Figure 3
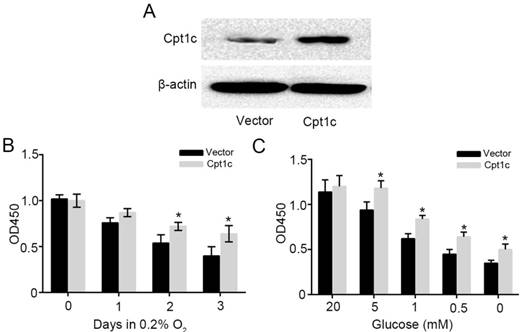
increasing the Cpt1c expression promotes cancer cell survival facing metabolic stress. (A): Cpt1c overexpressed in KTC-1 cells was confirmed by western blot. (B): KTC-1 cells with Cpt1c and control were cultured in hypoxia for 0, 1, 2 and 3 day, and cell viability was measured by CCK-8. (C): KTC-1 cells with Cpt1c and control were cultured in low glucose (20, 5, 1, 0.5 and 0 mM) for 48h, and cell viability was measured by CCK-8.*P < 0.05, **P < 0.01 VS control.
Figure 4
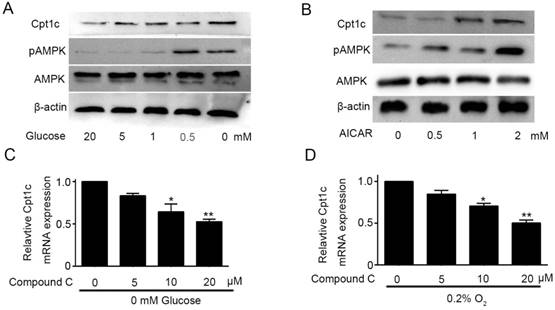
Cpt1c expression is regulated by AMPK activity. (A): AMPK activity was evaluated in KTC-1 cells under deprivation of glucose (20, 5, 1, 0.5 and 0 mM) for 48h. (B): KTC-1 cells were cultured in AICAR (0, 0.5, 1 and 2mM) after 48h and Cpt1c expression was analyzed by western blot. (C): KTC-1 cells were treated for 48 h with different concentration of compound C (0, 5, 10 and 20μM) in the absence of glucose (0 mM), and Cpt1C expression was detected by qRT-PCR. (D): KTC-1 cells were treated for 48 h with different concentration of compound C (0, 5, 10 and 20μM) in the absence of oxygen (O2%), and Cpt1C expression was assessed by qRT-PCR.*P < 0.05, **P < 0.01 VS control.
Adenosine monophosphate-activated protein kinase (AMPK), as a sensor of various cellular energy homeostasis under metabolic stress conditions, has received much attention in cancer therapy[20]. Although AMPK activated by pharmacologic activator reduced risk and/or mortality in certain types of cancers[21], especially those of the breast, pancreas, and prostate, among patients with type II diabetes and inhibits cancer cell proliferation in vitro, we showed that AMPK activity increased under metabolic stress conditions in papillary thyroid carcinomas cells. Consistent with our results, AMPK is also activated in various tumor microenvironments characterized by low oxygen and glucose deprivation, such as colorectal cancer[22], hepatocellular carcinoma[23] and lung cancer [10]. AMPK activation is intimately correlated with enhanced FAO[23]. Therefore, AMPK agonists combated with FAO inhibitors may be an effective way to cancer therapy.
FAO is controlled by CPT1 via importing FA into the cell mitochondria[24]. Gene-targeting studies have demonstrated that Cpt1c, as an import uniform of Cpt1, is not essential in mice but that the animals with Cpt1c gene knock out exhibit reduced FAO[25]. Others showed that CPT1C is mainly located in the endoplasmic reticulum instead of mitochondria and CPT1C does not regulate FAO[26,27]. Furthermore, Carrasco et al [28] found that CPT1C increases ceramide levels control dendritic spine maturation and cognition, instead of FAO. Although the approach of metabolism regulated by Cpt1c needs to be further explored, Cpt1c expression could be mediated by metabolism approach transformation. Zaugg Ket al[10] showed that activity of AMPK strengened by metabolic stress including hypoxia and glucose deprivation induced the Cpt1c expression to promote the cell survial under this condition. Furthermore,a repressing open reading frame located in Cpt1c 5′UTR is relieved under metabolic stress, and increased its expression [29]. In addition, Cpt1c is a novel p53-target gene, and p53 plays a vital role in Cpt1c expression induced by metabolic stress [30].
In this present study, we showed that Cpt1c has a higher expression in papillary thyroid carcinomas compared with paired normal tissues and can be induced under metabolic stress condition in papillary thyroid carcinomas cells. In addition, Cpt1c can protect papillary thyroid carcinomas cells from cell death induced by hypoxia and low glucose. Hence, Cpt1c may be a targeting gene for cancer therapy. Furthermore, induction of Cpt1c expression facing metabolic stress in papillary thyroid carcinomas is at least partly regulated by AMPK activity. Therefore, AMPK agonists combined with Cpt1c inhibitor to treat papillary thyroid carcinomas may be a novel way.
Acknowledgements
We thank Michael J. Wolfgang professor, Johns Hopkins University School of Medicine, for providing PcDNA3.1-Cpt1c as a gift to our laboratory. We also thank Stem Cell Bank, Chinese Academy of Sciences for kindly providing KTC-1 and B-CPAP cells lines.
Funding
This work was supported by the National Natural Science Foundation of China (No.81372862)
Competing Interests
The authors have declared that no competing interest exists.
References
1. Nucera C, Nehs MA, Mekel M. et al. A novel orthotopic mouse model of human anaplastic thyroid carcinoma. Thyroid: official journal of the American Thyroid Association. 2009;19:1077-84
2. Rusinek D, Krajewska J, Jarzab M. Mouse models of papillary thyroid carcinoma - short review. Endokrynologia Polska. 2016;67:212-23
3. Vanden Borre P, Gunda V, McFadden DG. et al. Combined braf(v600e)- and src-inhibition induces apoptosis, evokes an immune response and reduces tumor growth in an immunocompetent orthotopic mouse model of anaplastic thyroid cancer. Oncotarget. 2014;5:3996-4010
4. Baudin E, Schlumberger M. New therapeutic approaches for metastatic thyroid carcinoma. Lancet Oncol. 2007;8:148-56
5. Zhang Z, Rahme GJ, Chatterjee PD. et al. Id2 promotes survival of glioblastoma cells during metabolic stress by regulating mitochondrial function. Cell Death Dis. 2017;8:e2615
6. El Mjiyad N, Caro-Maldonado A, Ramirez-Peinado S. et al. Sugar-free approaches to cancer cell killing. Oncogene. 2011;30:253-64
7. Kim JW, Tchernyshyov I, Semenza GL. et al. Hif-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3:177-85
8. Ford JH. Saturated fatty acid metabolism is key link between cell division, cancer, and senescence in cellular and whole organism aging. Age (Dordr). 2010;32:231-37
9. Jeon SM, Chandel NS, Hay N. Ampk regulates nadph homeostasis to promote tumour cell survival during energy stress. Nature. 2012;485:661-5
10. Zaugg K, Yao Y, Reilly PT. et al. Carnitine palmitoyltransferase 1c promotes cell survival and tumor growth under conditions of metabolic stress. Genes & development. 2011;25:1041-51
11. Cameron KO, Kurumbail RG. Recent progress in the identification of adenosine monophosphate-activated protein kinase (ampk) activators. Bioorg Med Chem Lett. 2016;26:5139-48
12. Bikas A, Jensen K, Patel A. et al. Glucose-deprivation increases thyroid cancer cells sensitivity to metformin. Endocrine-related cancer. 2015;22:919-32
13. Franceschi S, Lessi F, Panebianco F. et al. Loss of c-kit expression in thyroid cancer cells. PloS one. 2017;12:e0173913
14. Kim NH, Cha YH, Lee J. et al. Snail reprograms glucose metabolism by repressing phosphofructokinase pfkp allowing cancer cell survival under metabolic stress. Nat Commun. 2017;8:14374
15. Xiao L, Liu N, Zhang G. et al. Late-course adaptive adjustment based on metabolic tumor volume changes during radiotherapy may reduce radiation toxicity in patients with non-small cell lung cancer. PloS one. 2017;12:e0170901
16. Samudio I, Harmancey R, Fiegl M. et al. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. The Journal of clinical investigation. 2010;120:142-56
17. Camarda R, Zhou AY, Kohnz RA. et al. Inhibition of fatty acid oxidation as a therapy for myc-overexpressing triple-negative breast cancer. Nature medicine. 2016;22:427-32
18. Pike LS, Smift AL, Croteau NJ. et al. Inhibition of fatty acid oxidation by etomoxir impairs nadph production and increases reactive oxygen species resulting in atp depletion and cell death in human glioblastoma cells. Biochimica et biophysica acta. 2011;1807:726-34
19. Hossain F, Al-Khami AA, Wyczechowska D. et al. Inhibition of fatty acid oxidation modulates immunosuppressive functions of myeloid-derived suppressor cells and enhances cancer therapies. Cancer Immunol Res. 2015;3:1236-47
20. Faubert B, Vincent EE, Poffenberger MC. et al. The amp-activated protein kinase (ampk) and cancer: Many faces of a metabolic regulator. Cancer Lett. 2015;356:165-70
21. Sarnowska E, Balcerak A, Olszyna-Serementa M. et al. [amp-activated protein kinase (ampk) as therapeutic target]. Postepy Hig Med Dosw (Online). 2013;67:750-60
22. Lee H, Oh ET, Choi BH. et al. Nqo1-induced activation of ampk contributes to cancer cell death by oxygen-glucose deprivation. Sci Rep. 2015;5:7769
23. Inokuchi-Shimizu S, Park EJ, Roh YS. et al. Tak1-mediated autophagy and fatty acid oxidation prevent hepatosteatosis and tumorigenesis. The Journal of clinical investigation. 2014;124:3566-78
24. Shi W, Hu S, Wang W. et al. Skeletal muscle-specific cpt1 deficiency elevates lipotoxic intermediates but preserves insulin sensitivity. J Diabetes Res. 2013;2013:163062
25. Casals N, Zammit V, Herrero L. et al. Carnitine palmitoyltransferase 1c: From cognition to cancer. Prog Lipid Res. 2016;61:134-48
26. Wolfgang MJ, Kurama T, Dai Y. et al. The brain-specific carnitine palmitoyltransferase-1c regulates energyhomeostasis. Proc Natl Acad Sci U S A. 2006;103:7282-7
27. Sierra AY, Gratacós E, Carrasco P. et al. CPT1c is localized in endoplasmic reticulum of neurons and has carnitine palmitoyltransferase activity. J Biol Chem. 2008;283:6878-85
28. Carrasco P, Sahún I, McDonald J. et al. Ceramidelevels regulated by carnitine palmitoyltransferase 1C control dendritic spinematuration and cognition. J Biol Chem. 2012;287:21224-32
29. Lohse I, Reilly P, Zaugg K. The CPT1C 5'UTR contains a repressing upstream open reading frame that is regulated by cellular energy availability and AMPK. PLoS One. 2011;6:e21486
30. Sanchez-Macedo N, Feng J, Faubert B. et al. Depletion of the novelp53-target gene carnitine palmitoyltransferase 1C delays tumor growth in theneurofibromatosis type I tumor model. Cell Death Differ. 2013;20(4):659-68
Author contact
![]() Corresponding author: Department of Biochemistry and Molecular Biology, Second Military Medical University, No.800 Xiangyin Road, Shanghai 200433, China.Tel:+86-21-81870969,Fax:+86-21-65334333.Email:yaozhenzhen1128com.
Corresponding author: Department of Biochemistry and Molecular Biology, Second Military Medical University, No.800 Xiangyin Road, Shanghai 200433, China.Tel:+86-21-81870969,Fax:+86-21-65334333.Email:yaozhenzhen1128com.
Citation styles
APA
Wang, R., Cheng, Y., Su, D., Gong, B., He, X., Zhou, X., Pang, Z., Cheng, L., Chen, Y., Yao, Z. (2017). Cpt1c regulated by AMPK promotes papillary thyroid carcinomas cells survival under metabolic stress conditions. Journal of Cancer, 8(18), 3675-3681. https://doi.org/10.7150/jca.21148.
ACS
Wang, R.; Cheng, Y.; Su, D.; Gong, B.; He, X.; Zhou, X.; Pang, Z.; Cheng, L.; Chen, Y.; Yao, Z. Cpt1c regulated by AMPK promotes papillary thyroid carcinomas cells survival under metabolic stress conditions. J. Cancer 2017, 8 (18), 3675-3681. DOI: 10.7150/jca.21148.
NLM
Wang R, Cheng Y, Su D, Gong B, He X, Zhou X, Pang Z, Cheng L, Chen Y, Yao Z. Cpt1c regulated by AMPK promotes papillary thyroid carcinomas cells survival under metabolic stress conditions. J Cancer 2017; 8(18):3675-3681. doi:10.7150/jca.21148. https://www.jcancer.org/v08p3675.htm
CSE
Wang R, Cheng Y, Su D, Gong B, He X, Zhou X, Pang Z, Cheng L, Chen Y, Yao Z. 2017. Cpt1c regulated by AMPK promotes papillary thyroid carcinomas cells survival under metabolic stress conditions. J Cancer. 8(18):3675-3681.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.