Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(17):3607-3614. doi:10.7150/jca.21200 This issue Cite
Meeting Report
Scientific Summary from the Morgan Welch MD Anderson Cancer Center Inflammatory Breast Cancer (IBC) Program 10th Anniversary Conference
Wendy A. Woodward1, 2 ![]() , Massimo Cristofanilli3, 4, 5, Sofia D. Merajver6, 7, 8, 9, Steven Van Laere10, Lajos Pusztai11, Francois Bertucci12, Fedor Berditchevski13, Kornelia Polyak14, 15, 16, 17, 18, Beth Overmoyer14, Gayathri R. Devi19, 20, Esta Sterneck21, Robert Schneider22, 23, Bisrat G. Debeb1, 24, Xiaoping Wang1, 24, Kenneth L. van Golen25, Randa El-Zein26, 27, Omar M. Rahal1, 2, Angela Alexander1, 24, James M. Reuben1, 28, Savitri Krishnamurthy1, 29, Anthony Lucci1, 30, Naoto T. Ueno1, 24
, Massimo Cristofanilli3, 4, 5, Sofia D. Merajver6, 7, 8, 9, Steven Van Laere10, Lajos Pusztai11, Francois Bertucci12, Fedor Berditchevski13, Kornelia Polyak14, 15, 16, 17, 18, Beth Overmoyer14, Gayathri R. Devi19, 20, Esta Sterneck21, Robert Schneider22, 23, Bisrat G. Debeb1, 24, Xiaoping Wang1, 24, Kenneth L. van Golen25, Randa El-Zein26, 27, Omar M. Rahal1, 2, Angela Alexander1, 24, James M. Reuben1, 28, Savitri Krishnamurthy1, 29, Anthony Lucci1, 30, Naoto T. Ueno1, 24
1. MD Anderson Morgan Welch Inflammatory Breast Cancer Research Program and Clinic, The University of Texas MD Anderson Cancer Center, Houston, TX;
2. Department of Radiation Oncology, The University of Texas MD Anderson Cancer Center, Houston, TX;
3. Developmental Therapeutics Program of Division of Hematology Oncology, Northwestern University Feinberg School of Medicine, Chicago, IL, USA;
4. Department of Medicine, Northwestern University Feinberg School of Medicine, Chicago, IL, USA;
5. Robert H. Lurie Comprehensive Cancer Center of Northwestern University, Chicago, IL, USA;
6. Program in Cellular and Molecular Biology, University of Michigan Medical School, Ann Arbor, MI;
7. Department of Internal Medicine, University of Michigan, Ann Arbor, MI;
8. University of Michigan Comprehensive Cancer Center, Ann Arbor, MI, 48109, USA. The Office for Health Equity and Inclusion, University of Michigan, Ann Arbor, MI;
9. Program in Cancer Biology, University of Michigan Medical School, Ann Arbor, MI;
10. Center for Oncological Research (CORE), Faculty of Medicine and Health Sciences, University of Antwerp, Antwerp Belgium;
11. Breast Medical Oncology, Yale Cancer Center, Yale School of Medicine, New Haven, CT;
12. Department of Medical Oncology, Institute Paoli-Calmettes, Marseille, France;
13. School of Cancer Sciences of the University of Birmingham, Birmingham, United Kingdom;
14. Department of Medical Oncology, Dana-Farber Cancer Institute, Boston, MA;
15. Department of Medicine, Brigham and Women's Hospital, Boston, MA;
16. Department of Medicine, Harvard Medical School, Boston, MA;
17. BBS Program, Harvard Medical School, Boston, MA;
18. Broad Institute, Cambridge, MA;
19. Department of Surgery, Division of Surgical Sciences, Duke University Medical Sciences, Durham, NC;
20. Women's Cancer Program, Duke Cancer Institute, Durham, NC;
21. Laboratory of Cell and Developmental Signaling, Center for Cancer Research, National Cancer Institute, Frederick, MD;
22. Department of Microbiology, New York University School of Medicine, New York, NY;
23. Perlmutter Cancer Center, New York University School of Medicine, New York, NY;
24. Department of Breast Medical Oncology, Division of Cancer Medicine, The University of Texas MD Anderson Cancer Center, Houston, TX;
25. Department of Biological Sciences and The Center for Translational Cancer Research, The University of Delaware, Newark, DE;
26. Department of Radiology, Houston Methodist Research Institute, Houston, TX;
27. Department of Epidemiology, The University of Texas MD Anderson Cancer Center, Houston, TX;
28. Department of Hematopathology, The University of Texas MD Anderson Cancer Center, Houston, TX;
29. Department of Pathology, The University of Texas MD Anderson Cancer Center, Houston, TX;
30. Department of Surgical Oncology, The University of Texas MD Anderson Cancer Center, Houston, TX.
Received 2017-5-26; Accepted 2017-8-28; Published 2017-10-9
Citation:
Woodward WA, Cristofanilli M, Merajver SD, Van Laere S, Pusztai L, Bertucci F, Berditchevski F, Polyak K, Overmoyer B, Devi GR, Sterneck E, Schneider R, Debeb BG, Wang X, van Golen KL, El-Zein R, Rahal OM, Alexander A, Reuben JM, Krishnamurthy S, Lucci A, Ueno NT. Scientific Summary from the Morgan Welch MD Anderson Cancer Center Inflammatory Breast Cancer (IBC) Program 10th Anniversary Conference. J Cancer 2017; 8(17):3607-3614. doi:10.7150/jca.21200. https://www.jcancer.org/v08p3607.htm
Other stylesAbstract
In 2006, a remarkable collaboration between University of Texas MD Anderson Cancer Center clinicians and Texas and New Mexico State legislators led to the formation of a dedicated IBC Research Program and Clinic at MD Anderson. This initiative provided funding and infrastructure to foster coordination of an IBC World Consortium of national and international experts, and launch the first ever IBC international conference in 2008, which brought together experts from around the world to facilitate collaborations and accelerate progress. Indeed great progress has been made since then. National and international experts in IBC convened at the 10th Anniversary Conference of the MD Anderson IBC Clinic and Research Program and presented the most extensive sequencing analysis to date comparing IBC to non-IBC, gene- and protein-based immunoprofiling of IBC versus non-IBC patients, and converging lines of evidence on the specific role of the microenvironment in IBC. Novel models, unique metabolic mechanisms, and prominent survival pathways have been identified and were presented. Multiple clinical trials based on the work of the last decade are in progress or in development. The important challenges ahead were discussed. This progress and a coordinated summary of these works are presented herein.
Introduction
In the last 10 years, coordinated focus on the treatment and biology of IBC has yielded significant progress. Presentations at the first International IBC Conference included important work by those who have been pursuing this disease for decades [1-18], and also identified a long list of unmet needs. Genomic studies had been performed but in many cases on small data sets without opportunity for validation. Important signaling had been identified but clear druggable targets were still lacking. Tissue for more exhaustive studies with state of the art technology was needed, and models facilitating pre-clinical studies were limited. Funds from the State of Texas Rare and Aggressive Breast Cancer Grant provided infrastructure in Texas to build a large sustainable research program and this in part provided the impetus to coordinate national and international collaborations, combine patient data, develop new models, and bring new investigators into the field. While research programs at the University of Michigan, NYU, and the University of Delaware have a long-standing dedication to IBC, new programs were developed at the Dana Farber Cancer Institute, the University of Texas, and Duke University, and long-committed international investigators have expanded genomic and clinical contributions through transatlantic patient data sharing to richly enhance this network. At the MD Anderson Cancer Center Morgan Welch IBC 10th Anniversary Conference, update from these experts was presented, in many cases contributing to the progress of developing clinical trials for IBC patients. The state of the science is summarized herein.
Summary
Big Data
Dr. Cristofanilli presented a sweeping overview of the work his program has done, including Foundation One sequencing of 53 IBC tumors demonstrating 266 genome alterations (GA) with an average of 5.0 GA/tumor (range 1-15). At least one alteration associated with an FDA-approved therapy or clinical trial was identified in 51/53 (96%) of cases with an average of 2.6 CRGA/case. The most frequently altered genes were TP53 (62%), MYC (32%), PIK3CA (28%), ERBB2 (26%), FGFR1 (17%), BRCA2 (15%), and PTEN (15%) [9]. He reviewed as well correlative studies demonstrating viral load in IBC patients as well as bacterial correlations although these have yet to be validated with evidence for causality [19]. Indeed, at the conference Dr. Pusztai presented the first whole genome sequencing (WGS) results of 20 IBC cases compared to a matched cohort of non-IBC on behalf of a team of investigators from Yale and MDACC. The results showed that there is no single genomic abnormality that is shared by all IBC and could therefore define the disease in genomic terms [20]. The overall mutation load, the number of somatic single nucleotide variants (SNV), and distribution of mutational signatures were similar between IBC and non-IBC. Most somatic mutations were in the non-coding region of the genome. There were only a few recurring mutations and the frequency of top 20 recurrent mutations were not different between IBC and non-IBC. TP53 was the top most frequently mutated gene. Considering significantly differently mutated high functional impact SNVs between IBC and non-IBC the team has identified variants in the non-coding regulatory regions of the Microtubule-Associated Serine/Threonine Kinase 2 (MAST2) gene as one of the few mutations significantly more prevalent in IBC (4 vs 0 cases, P = 0.039). At the DNA copy number (CNV) level, there was significantly more CNV losses of chromosomes 7 and 16 and gains of chromosomes 11 and 17 in IBC compared to non-IBC, but these anomalies still affected only a minority of IBC cases. Overall these results show high level of genomic complexity in IBC with each cancer having a unique combination of genomic alterations without any unique abnormality that would genomically define IBC. Interestingly, when germline variants were examined, the overall level of germline disturbance was higher in IBC compared to non-IBC, suggesting that host factors may contribute to the disease biology. The investigators also examined DNA sequences that did not match to the human genome to search for bacteria or viruses that may be pathognomonic for IBC. Instead they found a red herring, more propionic bacterium acne related to contamination with skin cells in samples.
Considering genetics of innate immunity differences, comparing distribution of alleles of HLA-A,-B, -C and -DRB1 loci suggested some differences in innate immunity. Specifically, reviewing 68 IBC patients of European ancestry and 776 ethnically-matched healthy unrelated volunteers, the alleles B*0801, Cw*0701 and DRB1*0301 were found to have significantly lower frequencies, and the alleles Cw*0602 and DRB1*1301 had higher frequencies among IBC patients. Along these lines, examining white cell phenotypes in blood, Mego et al. found metastatic IBC patients have significant reductions in adaptive immunity [21, 22].
Dr. Van Laere presented provocative genomic evidence for the role of the microenvironment in IBC. Using mRNA-based immune profiling he described no overt difference in total TIL counts in IBC versus non-IBC, but major differences in specific white cell populations including B-cells, T-cells, Tregs, and M2 macrophages. He then reviewed immunostaining for CD163, Foxp3, CD8, and CD79a in tissue assessed both by pathologists as well as using image quantitation to distinguish the tumor stroma boundary and provide a read out of stromal TIL. He reported that stromal TIL proportions were similar in IBC and non-BC and in contrast to non-IBC there was no subtype specific TIL variation in IBC. Confirming mRNA data, PDL1 expression on immune cells was increased in IBC independently of molecular subtype. IBC was more characterized by an adaptive immune response although all TME subtypes were observed. TILS in IBC represented CD70a- and FOXP3-positive cells and immune cell PDL1 was related to CD8 positivity. Using the IBC-like signature to review the mutational load of TCGA data, signature 13 appeared increased in IBC-like cases. Of course this group includes non-IBC as well and it was noted that the WGS of 20 cases of IBC revealed no statistically significant mutation signature differences. Lastly, three immune response phenotypes defined based on TIL and PDL1 expression were identified in IBC cases: mixed polarization, type 2 polarization, and ignorant. In interesting related data, Dr. Devi profiled laser micro-dissected epithelium and stroma from 35 IBC patients for immune mRNA based profiles and reports no significant differences between African-American and Caucasian IBC patients.
Genomic studies of pathological complete response (pCR) after primary chemotherapy in IBC are rare. Dr. Bertucci presented consortium data including 137 IBC and 252 non-IBC, in which a gene expression signature (GES) associated with pCR was identified in IBC, mainly based on immunity genes. Interestingly, this signature also worked in non-IBC, and reciprocally, published GES signatures that predict pCR in non-IBC also worked in IBC [23]. He additionally reported that PDL1 mRNA expression is higher in IBC than in non-IBC and associated with pCR in IBC [7]. Interestingly, Dr. Berditchevski presented novel work on a smaller series of IBC patients suggesting the IBC over-expresses CD151, a tetraspanin that is associated with aggressive breast cancer features and regulates functions of integrins and matrix metalloproteases (MMPs). It was associated with worse outcome when present, and correlated to more extensive infiltrate of CD68 positive cells (P = 0.005). CD151 depletion didn't affect the growth of IBC cells but regulated the chemoattractant potential of IBC cells via mechanisms involving exosomes.
Microenvironment and novel models
Dr. Woodward presented four pieces of evidence suggesting the breast tissues themselves may be complicit in the phenotype of IBC. Examination of breast tissue remote from the tumor in IBC has more CD68+ cells and more CD44CD49CD133+ stem cells than breast tissue from non-IBC tumors. This was predicted by epidemiology observations of excess obesity and early pregnancy in IBC reviewed by Dr. El-Zein from the > 400 patients in the IBC registry. Dr. Woodward showed that adding mesenchymal stem cells (MSC) to SUM149 animal models and PDX models increases the incidence of skin invasion similar to IBC patient symptoms in an IL6, macrophage-dependent manner [24, 25]. In an early promotion model of SUM149 cells injected into intact mammary glands with or without gland priming by macrophages and MSCs, priming promotes the diffuse clustered pathologic pattern described early in the meeting by IBC pathologist Dr. Krishnamurthy. Lastly, co-culture of polarized macrophages and MSC promoted the intrinsic radiation resistance of IBC tumor cells in vitro. Concordant data described above from Dr. Van Laere highlight the prevalence of M2 macrophages in IBC versus non-IBC based on mRNA profiling and IHC. Dr. Merajver has also recently published the role of macrophage conditioned media in promoting migration of IBC cells. Dr. Berditchevski later showed that CD68-positive cells in IBC and CD3+ TIL were associated with worse survival. In total, a compelling argument was made for a role for the microenvironment in the clinical presentation of IBC, and consideration was given to the possibility that modifiable epidemiology factors may prime the breast prior to the development of a sufficiently aggressive genetic mutation that initiated breast cancer in a breast that promotes migration over proliferation and mass development. Dr. Devi demonstrated an elegant window-based model of lymphatic assessment in live animals (Arora et al. in press). Dr. Debeb reported that IBC cell lines generate brain metastases under tail vein injection, a simple technique that incorporates the biology of traversing the lung prior to brain colonization. Colonization was blocked by mIR-141 knock-down in IBC cells [26]. MDA-231 cells that do not express mIR-141 were promoted to colonize the brain with miR-141 overexpression. In all cases strong epithelial phenotype was demonstrated in brain metastases [26].
Survival signaling in IBC
Potentially bridging the gap between host factors and intrinsic tumor issues, Dr. Van Golen described the role of lipid rafts in IBC tumor cells. Caveolin, a protein expressed in lipid rafts is associated with RhoC, a known IBC signaling factor in IBC tissues. Further, Caveolin is expressed in IBC cell lines and knockdown reduces RhoC and invasion of IBC cells without affecting RhoC regulatory functions like GDI, GAP, and hydrolysis [27]. Akt is also inhibited [27].
Dr. Merajver made complex work simple demonstrating that IBC cells unexpectedly enter the TCA cycle in reverse and make citrate Acetyl-CoA from glutamine. They have low basal oxidative capacity, and make use of glutamine in a RhoC not HIF1a-dependent mechanism.[28] Looking for stores from which acetate may be derived, an unbiased screen pulled out N-acetyl aspartate (NAA). Further, glycogen may be an important store in hypoxic conditions. These stores may help IBC cells become highly plastic under a broad range of conditions in different tissues.
Dr. Overmoyer showed Dana-Farber Cancer Institute data from Dr. Polyak's lab demonstrating efficacy of JAK1/2 inhibition with Ruxolitinib. Significant efficacy was demonstrated in two IBC breast cancer cell lines SUM149 and SUM190 in cell culture and in xenografts in combination with paclitaxel. This has formed the basis of a soon to enroll Translational Breast Cancer Research Consortium sponsored neoadjuvant trial including Ruxolitinib discussed below. She also presented the pre-clinical rational for targeting angiogenesis in IBC. There is clear evidence of increased angiogenesis in tumors, and pre-clinical work suggests that targets in this space are viable, however the addition of anti-angiogenic agents to neoadjuvant chemotherapy were discouraging [29].
Dr. Robert Schneider suggested a follow up trial combining mTOR and JAK inhibition may be even more efficacious based on pre-clinical studies. In IBC tissues, very strong activation of mTORC1 & JAK/STAT pathways, IL6, and TAMs specifically in tumors compared to stroma suggest a therapeutic window for targeting this pathway in IBC.[30] 95% of IBC tumors showed strong dual activation of mTORC1 (pS6) and JAK2 (pJAK2) in tumor and not stroma highlighting a potential role for dual targeted strategies. Supporting this, in SUM149 xenografts optimal tumor growth delay was observed using dual inhibitors, WP1066 and INK128, targeting JAK2/STAT3 and mTOR, respectively. Further, data were presented demonstrating that the genotoxic DNA damage response includes a coordinated transcriptional and translational response involving mTORC1/2 together but not mTORC1 alone. mTORC1 inhibition in cancer cells down regulates very few mRNAs and their translation, while dual inhibition selectively block many important survival pathways (Silvera et al. MCB in press). Interestingly, many of the critical transcripts in cancer cell survival have such significant tertiary structure that only minimal blockade of translation is required to selectively inhibit them. Thus one would expect the dual blockade of mTORC1/2 to inhibit recovery after genotoxic stress like radiation, and indeed in SUM149 xenografts almost complete inhibition of tumor regrowth was seen with radiation (6Gy x 3) and INK128 compared to controls of RT alone, drug alone, Rad001 alone or Rad001+radiation. The effect is also seen in an ovarian cancer model in combination with platinum.[31] Explicitly comparing the genome wide mRNA translation in this model clearly illustrates the hypothesis that mild inhibition of translation selectively targets critical apoptosis transcripts including survivin, MCL1, BIRC3 and BIRC5.
Dr. Devi reported on the role of a key regulator of programmed cell death, X-linked inhibitor of apoptosis protein (XIAP), specifically in suppressing immune mediated anti-tumor cell kill. CTLs and NK cells release Caspase-8 and Granzyme B leading to apoptosis in a process regulated by XIAP, a potent caspase inhibitor. They have identified how high expression of XIAP suppresses granzyme activity, reactive oxygen accumulation, and activates NFkB target genes in suppression of antibody-mediated cytotoxicity- (Evans, 2016 Cell Death & Disease).[32] In clonal isolates from SUM149 and SUM190 cells that exhibit resistance to cetuximab- or transtuzumab-mediated ADCC (rSUM149) and in SUM190 cells resistant to XIAP expression was increased and Caspase 3/7 activity was decreased. Silencing XIAP and expression of mutant XIAP that cannot bind caspases demonstrated direct inhibition of the caspase activity and failed immune-mediated lysis. Dr. Devi also demonstrated XIAP suppresses granzyme mediated cell lysis in a caspase independent manner [32]. Importantly, XIAP depletion in SUM149 and SUM190 overcame resistance to immune-mediated apoptosis, demonstrated reduction in transcripts related to inflammation and tumor promotion (Devi, 2016 AACR Abstract: http://cebp.aacrjournals.org/content/26/2_Supplement/B44). Their earlier studies have shown a unique translational mechanism that allows for XIAP overexpression during cellular stress [33], which then contributes to therapeutic resistance to tyrosine kinase inhibitors and chemotherapeutics in IBC cells and tumors [4, 6, 34]. Furthermore, recent work from their laboratory has demonstrated expression of XIAP and NFkB in IBC patient samples and tumor emboli, revealing a new druggable pathway for IBC therapy (Arora, 2017 in press Oncotarget online doi: 10.18632/oncotarget.15667).
Dr. Balamurugan et al. made a compelling case for the similarity between IBC features and activity of the transcription factor C/EBPδ. For example, C/EBPδ promotes hypoxia adaption, lymphangiogenesis, and pro-inflammatory signaling pathways as well as expression of VEGF and CXCR4 [35]. Indeed, C/EBPδ was found to be expressed in IBC cell lines (SUM190, KPL4, SUM149, and MDA-IBC3 when cultured as emboli) and further induced in emboli cultures. Convincing staining in patient emboli was shown as well. Silencing of C/EBPδ in SUM149 or IBC3 cells reduced the expression of key inflammation and stemness genes, and inhibited cell invasion and emboli formation in culture. Importantly, treatment with clinically relevant HDAC inhibitors such as LBH589, MS275, SAHA, and curcumin inhibited C/EBPδ expression and emboli formation and attenuated SUM149 tumor growth in mice.
Dr. Lewis-Wambi highlighted published work by Dr. Bertucci et al. demonstrating the increased expression of inflammatory pathways in IBC including INFα, INFγ, and TNFα [36]. These signals can converge on signaling through Interferon induced-transmembrane protein - 1 (IFITM1), a protein with an unknown role in IBC. It is over-expressed in SUM149 but not SUM190 or IBC3 cells [37]. Ogony et al. demonstrated IFITM1 knockdown reduces the aggressiveness of IBC cells and that STAT2 is a critical regulator. In the MIND model in vivo, knockdown of IFITM1 inhibits tumor growth. Consistent with the body of work showing the role of JAK/STAT inhibition, IFITM1 is blocked by Ruxolitinib and modest effect on SUM149 cells with this drug was again demonstrated. Provocatively, IFITM1 is overexpressed in IBC and TNBC tumors, but notable mostly from AA patients. It was pointed out the SUM149 cells are from an AA IBC patient and this may explain the dramatic difference in these cells.
Dr. Wang re-capped the well-demonstrated role of EGFR in IBC noting it is highly expressed in 30% of patients and correlated to worse overall survival [38]. In IBC cell lines, knockdown inhibits the stem-like population using multiple surrogate assays of stemness and demonstrates an interesting correlation with COX2 expression. COX2 expression in IBC patients is also correlated with worse outcome and she demonstrated that EGFR targets stemness via a COX2-mediated mechanism. Like previously reported EGFR inhibition [39], COX2 inhibition also reversed EMT features of in vivo IBC tumors. Interestingly, COX-2 functions through a previously unreported and potentially targetable connection to Nodal. These targets will be explored in an accruing EGFR targeted clinical trial discussed below.
Epidemiology of IBC
Dr. El-Zein presented a summary of the epidemiologic profiles associated with the development of IBC among different ethnicities and proposed scenarios that were not ethnically exclusive but seemed to be ethnically predominant. Among the African American (AA) patients, younger age at first pregnancy and multiparity were significantly presented. Breastfeeding was over-represented in Caucasians and the duration between the age at menarche and age at first pregnancy was significantly longer among the Caucasians patients. In case of the AA patients, the younger age at first pregnancy together with shorter reproductive/childbearing period and the lack of breastfeeding, generate a cancer prone microenvironment in the breasts. Lack of breastfeeding leads to accumulation of the pro-inflammatory microenvironment and with consequent pregnancies, further enrichment of the tumorigenic microenvironment and hormonal induced cell proliferation and clonal expansion occurs [40]. The scenario seems to be different for Caucasians, where a longer period between age at menarche and age at first child birth leads to breast tissue aging, with accumulation of molecular damage due to high susceptibility of undifferentiated breast tissues to carcinogens [41]. During pregnancy, hormonal induction of cell proliferation, which in the presence of existing molecular damage, leads to clonal expansion and tumorigenesis [40]. Therefore, there may be several modifiable lifestyle and reproductive factors among AA and Caucasians that could be the subject of targeted preventive efforts.
Trials from Pre-clinical IBC studies
Dr. Ueno's group presented high PCR rates among triple negative IBC patients in a phase II study of Panitumumab, Nab-paclitaxel, Carboplatin followed by FEC q3w x 4 pre-operatively. Including RCB1 patients who have been shown to have similar outcome to pCR, 67% of patients achieved excellent response. Dr. Ueno has previously shown that EGFR targeted therapy reverses EMT in IBC and presented that EGFR can mediate the crosstalk between IBC tumor cells and macrophages [42]. A randomized trial is accruing comparing Panitumumab, Paclitaxel, Carboplatin followed by AC q3w x 4 pre-operatively.
Dr. Overmoyer presented the pre-clinical work from the Dana Farber Cancer Institute that led to TBCRC 039, an Incyte sponsored trial of pre-operative Ruxolitinib +/- paclitaxel in a 7 day run in follow by paclitaxel alone (N = 16) versus Ruxolitinib and paclitaxel (N = 48), all followed by ddAC x r, MRM, and PMRT. A current clinical trial related to the pre-clinical work discussed above targets expression of angiogenesis and EMT-related genes (NCT02623972), and an anti-HER2 preoperative study is soon to achieve accrual goals, and limits toxic chemotherapy while optimizing HER2-directed treatment (NCT01796197).
Dr. Cristofanilli presented the developing clinical trial QUILT-IBC combining three novel immune-agents. ALT-803 is an IL15 super-agonist that potently activates NK cells, T cell, and facilitated memory. Nivolumab is a PD-1 inhibitor thought to release the brakes on the immune cells that are suppressed by the tumor [43]. Activated natural killer cells (aNK) are modified cells that have had the inhibitory aspects of the NK cell targeted to create a permanently “on” NK cell. These have been tested in 40 patients to date with metastatic breast cancers. Combining all three, aNK cells are expected to compensate for reduced frequency and impaired function of endogenous NK cells, ALT-803 is expected to improve NK cell number and facilitate T cell immunity and memory, and Nivolumab is expected to release the brakes on the adaptive immune system to enhance T cell function in the microenvironment and restore NK function. In the trial design still under development, aNK and ALT-803 with chemotherapy would be given as an induction phase, followed by consolidation with all three novel drugs and Nab-paclitaxel. Subsequent continuation is dependent on response after 8 weeks of consolidation.
Conclusion
Global efforts from dedicated and committed physicians, scientists, patient advocates, and health care providers working together and sharing intellectual and material resources are aligned to achieve one common goal: improve survival and find cures for patients with IBC. Significant progress has been made towards achieving that goal. For instance, after the refinement of multimodality clinical management of IBC, the 5-year overall survival has significantly increased to up to 62% from a very low (< 5%) historical survival rate. However, IBC still remains the most lethal entity of breast cancer, with a very poor prognosis (i.e. higher incidence of micrometastatic disease) compared to locally advanced non-IBC.
Dr. Bertucci's results showing that same gene expression signatures equally predict pCR in both IBC and non-IBC are particularly intriguing, especially in the context of the recent WGS studies by Dr. Pusztai's group, results of which, conclude that there is no single genomic abnormality that was shared by all IBC cases that could define the disease in genomic terms. These significant molecular profiling studies encourage to explore a potential role for non-genetic heterogeneity playing a key role in IBC progression. Recent studies have highlighted the role of non-genetic heterogeneity in driving aggressive behavior in melanoma from a rare population of drug resistant cells, and in EGFR inhibitor-resistant lung cancers via genetic evolution of EGFRT790M-negative pre-existing drug-tolerant cells [44, 45]. A similar non-genetic mechanism is suggested to be at play in a rare population of IBC cells displaying a hybrid epithelial and mesenchymal phenotype (hybrid E/M phenotype) with expression of epithelial markers (E-cadherin) and mesenchymal characteristics of high invasion and migration.[46] The dual nature of hybrid E/M IBC cells would allow them to strongly adhere to each other while at same time migrate collectively to form higher number of clusters of circulating tumor cells (CTCs). Relative to single CTCs, cancer cells within cluster CTCs have synergistic ability to form metastasis and are more resistant to apoptosis in metastatic sites in mice.[47] Clinically, breast cancer patients with high circulating cluster CTCs have a poor overall and progression-free survival compared to those with single CTCs.[47] Higher number of circulating cluster CTCs in IBC patients, relative to non-IBC, have been associated with survival, possible due to treatment resistance.[48, 49] More studies comparing single vs cluster CTCs in IBC patients and their correlation with clinical hallmarks of IBC diagnosis and treatment prognosis are encouraged and needed.
Despite low availability of tissue samples, several elegant and often independent molecular profiling studies have shed light on IBC-specific alterations at the RNA and DNA level related to druggable oncogenic pathways such as those of mTOR/PI3K, MYC, VEGF, JAK/STAT, and EGFR among others. Interestingly, at the same time, these studies across multiple labs and approaches have suggested a key role for the tumor microenvironment in the etiology and/or aggressive nature of IBC. In this regard, IBC tumors appear to be enriched in M2-macrophage signature and stromal MSCs that home to the tumor from the bone marrow and can promote clinical hallmarks and metastasis of IBC. It was recognized that a deeper understanding of the non-genomic alterations in IBC tumors, such as proteomic, metabolomic, epigenetic, immunologic, and tumor micro-environment studies are urgently needed to better understand IBC and support the development of novel treatment strategies that will complement the current standard of care for IBC patients. A summary of the state of the art as presented at the conference is depicted in Figure 1. Importantly, multiple clinical trials from this and other work on IBC specific biology are now ongoing or promising to emerge in the future.
Figure 1
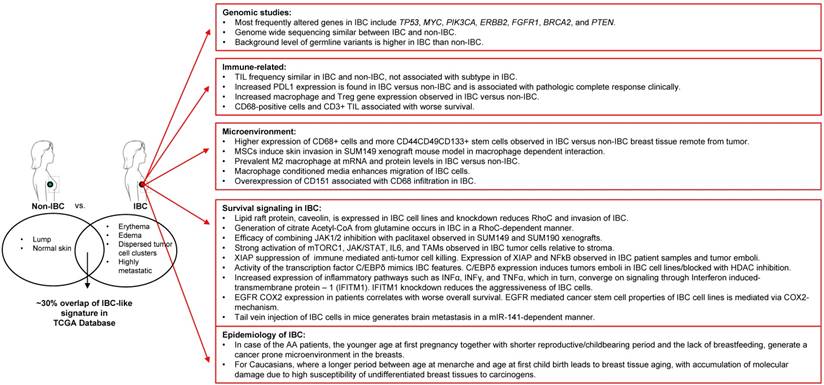
Summary of novel IBC research and findings.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Costa R, Santa-Maria CA, Rossi G, Carneiro BA, Chae YK, Gradishar WJ, Giles FJ, Cristofanilli M. Developmental therapeutics for inflammatory breast cancer: Biology and translational directions. Oncotarget. 2016
2. Yamauchi H, Woodward WA, Valero V, Alvarez RH, Lucci A, Buchholz TA, Iwamoto T, Krishnamurthy S, Yang W, Reuben JM. et al. Inflammatory breast cancer: what we know and what we need to learn. Oncologist. 2012;17(7):891-899
3. van Golen KL, Cristofanilli M. The Third International Inflammatory Breast Cancer Conference. Breast Cancer Res. 2013;15(6):318
4. Williams KP, Allensworth JL, Ingram SM, Smith GR, Aldrich AJ, Sexton JZ, Devi GR. Quantitative high-throughput efficacy profiling of approved oncology drugs in inflammatory breast cancer models of acquired drug resistance and re-sensitization. Cancer Lett. 2013;337(1):77-89
5. Levine PH, Hashmi S, Minaei AA, Veneroso C. Inflammatory breast cancer clusters: A hypothesis. World J Clin Oncol. 2014;5(3):539-545
6. Allensworth JL, Evans MK, Bertucci F, Aldrich AJ, Festa RA, Finetti P, Ueno NT, Safi R, McDonnell DP, Thiele DJ. et al. Disulfiram (DSF) acts as a copper ionophore to induce copper-dependent oxidative stress and mediate anti-tumor efficacy in inflammatory breast cancer. Mol Oncol. 2015;9(6):1155-1168
7. Bertucci F, Finetti P, Colpaert C, Mamessier E, Parizel M, Dirix L, Viens P, Birnbaum D, van Laere S. PDL1 expression in inflammatory breast cancer is frequent and predicts for the pathological response to chemotherapy. Oncotarget. 2015;6(15):13506-13519
8. Pierga JY, Petit T, Levy C, Ferrero JM, Campone M, Gligorov J, Lerebours F, Roche H, Bachelot T, Charafe-Jauffret E. et al. Pathological response and circulating tumor cell count identifies treated HER2+ inflammatory breast cancer patients with excellent prognosis: BEVERLY-2 survival data. Clin Cancer Res. 2015;21(6):1298-1304
9. Ross JS, Ali SM, Wang K, Khaira D, Palma NA, Chmielecki J, Palmer GA, Morosini D, Elvin JA, Fernandez SV. et al. Comprehensive genomic profiling of inflammatory breast cancer cases reveals a high frequency of clinically relevant genomic alterations. Breast Cancer Res Treat. 2015;154(1):155-162
10. Tsai CJ, Li J, Gonzalez-Angulo AM, Allen PK, Woodward WA, Ueno NT, Lucci A, Krishnamurthy S, Gong Y, Yang W. et al. Outcomes After Multidisciplinary Treatment of Inflammatory Breast Cancer in the Era of Neoadjuvant HER2-directed Therapy. Am J Clin Oncol. 2015;38(3):242-247
11. Bertucci F, Goncalves A, Viens P. Bevacizumab in HER2-negative inflammatory breast cancer. Oncoscience. 2016;3(11-12):297-298
12. Buchheit CL, Schafer ZT. BIM-EL localization: The key to understanding anoikis resistance in inflammatory breast cancer cells. Mol Cell Oncol. 2016;3(1):e1011474
13. Do Nascimento VC, Rajan R, Redfern A, Saunders C. Inflammatory breast cancer: A decade of experience. Asia Pac J Clin Oncol. 2016;12(3):242-247
14. Goh G, Schmid R, Guiver K, Arpornwirat W, Chitapanarux I, Ganju V, Im SA, Kim SB, Dechaphunkul A, Maneechavakajorn J. et al. Clonal Evolutionary Analysis during HER2 Blockade in HER2-Positive Inflammatory Breast Cancer: A Phase II Open-Label Clinical Trial of Afatinib +/- Vinorelbine. PLoS Med. 2016;13(12):e1002136
15. Nahleh ZA, Barlow WE, Hayes DF, Schott AF, Gralow JR, Sikov WM, Perez EA, Chennuru S, Mirshahidi HR, Corso SW. et al. SWOG S0800 (NCI CDR0000636131): addition of bevacizumab to neoadjuvant nab-paclitaxel with dose-dense doxorubicin and cyclophosphamide improves pathologic complete response (pCR) rates in inflammatory or locally advanced breast cancer. Breast Cancer Res Treat. 2016;158(3):485-495
16. Ross NL, Sullivan MO. Overexpression of caveolin-1 in inflammatory breast cancer cells enables IBC-specific gene delivery and prodrug conversion using histone-targeted polyplexes. Biotechnol Bioeng. 2016;113(12):2686-2697
17. Alexander A, Karakas C, Chen X, Carey JP, Yi M, Bondy M, Thompson P, Cheung KL, Ellis IO, Gong Y. et al. Cyclin E overexpression as a biomarker for combination treatment strategies in inflammatory breast cancer. Oncotarget. 2017
18. Manai M, Thomassin-Piana J, Gamoudi A, Finetti P, Lopez M, Eghozzi R, Ayadi S, Lamine OB, Manai M, Rahal K. et al. MARCKS protein overexpression in inflammatory breast cancer. Oncotarget. 2017;8(4):6246-6257
19. El-Shinawi M, Mohamed HT, El-Ghonaimy EA, Tantawy M, Younis A, Schneider RJ, Mohamed MM. Human cytomegalovirus infection enhances NF-kappaB/p65 signaling in inflammatory breast cancer patients. PLoS One. 2013;8(2):e55755
20. Li X. KS, Kumar S, Harmonic A, Kitchen R, Wali V.B, Reddy S, Woodward, W, Reuben J, Chuang J, Hatzis C, Ueno N.T, Gerstein M, Pusztai L: Landscape of Somatic Mutations in Inflammatory Breast Cancer by Whole-Genome Sequencing. In: San Antonio Breast Cancer Symposium: 2016. 2016
21. Mego M, De Giorgi U, Hsu L, Ueno NT, Valero V, Jackson S, Andreopoulou E, Kau SW, Reuben JM, Cristofanilli M. Circulating tumor cells in metastatic inflammatory breast cancer. Ann Oncol. 2009;20(11):1824-1828
22. Mego M, Giordano A, De Giorgi U, Masuda H, Hsu L, Giuliano M, Fouad TM, Dawood S, Ueno NT, Valero V. et al. Circulating tumor cells in newly diagnosed inflammatory breast cancer. Breast Cancer Res. 2015;17:2
23. Bertucci F, Ueno NT, Finetti P, Vermeulen P, Lucci A, Robertson FM, Marsan M, Iwamoto T, Krishnamurthy S, Masuda H. et al. Gene expression profiles of inflammatory breast cancer: correlation with response to neoadjuvant chemotherapy and metastasis-free survival. Ann Oncol. 2014;25(2):358-365
24. Lacerda L, Debeb BG, Smith D, Larson R, Solley T, Xu W, Krishnamurthy S, Gong Y, Levy LB, Buchholz T. et al. Mesenchymal stem cells mediate the clinical phenotype of inflammatory breast cancer in a preclinical model. Breast Cancer Res. 2015;17:42
25. Wolfe AR, Trenton NJ, Debeb BG, Larson R, Ruffell B, Chu K, Hittelman W, Diehl M, Reuben JM, Ueno NT. et al. Mesenchymal stem cells and macrophages interact through IL-6 to promote inflammatory breast cancer in pre-clinical models. Oncotarget. 2016;7(50):82482-82492
26. Debeb BG, Lacerda L, Anfossi S, Diagaradjane P, Chu K, Bambhroliya A, Huo L, Wei C, Larson RA, Wolfe AR. et al. miR-141-Mediated Regulation of Brain Metastasis From Breast Cancer. J Natl Cancer Inst. 2016:108 (8)
27. Joglekar M, Elbazanti WO, Weitzman MD, Lehman HL, van Golen KL. Caveolin-1 mediates inflammatory breast cancer cell invasion via the Akt1 pathway and RhoC GTPase. J Cell Biochem. 2015;116(6):923-933
28. Wynn ML, Yates JA, Evans CR, Van Wassenhove LD, Wu ZF, Bridges S, Bao L, Fournier C, Ashrafzadeh S, Merrins MJ. et al. RhoC GTPase Is a Potent Regulator of Glutamine Metabolism and N-Acetylaspartate Production in Inflammatory Breast Cancer Cells. J Biol Chem. 2016;291(26):13715-13729
29. Bertucci F, Fekih M, Autret A, Petit T, Dalenc F, Levy C, Romieu G, Bonneterre J, Ferrero JM, Kerbrat P. et al. Bevacizumab plus neoadjuvant chemotherapy in patients with HER2-negative inflammatory breast cancer (BEVERLY-1): a multicentre, single-arm, phase 2 study. Lancet Oncol. 2016;17(5):600-611
30. Jhaveri K, Teplinsky E, Silvera D, Valeta-Magara A, Arju R, Giashuddin S, Sarfraz Y, Alexander M, Darvishian F, Levine PH. et al. Hyperactivated mTOR and JAK2/STAT3 Pathways: Molecular Drivers and Potential Therapeutic Targets of Inflammatory and Invasive Ductal Breast Cancers After Neoadjuvant Chemotherapy. Clin Breast Cancer. 2016;16(2):113-122 e111
31. Musa F, Alard A, David-West G, Curtin JP, Blank SV, Schneider RJ. Dual mTORC1/2 Inhibition as a Novel Strategy for the Resensitization and Treatment of Platinum-Resistant Ovarian Cancer. Mol Cancer Ther. 2016;15(7):1557-1567
32. Evans MK, Sauer SJ, Nath S, Robinson TJ, Morse MA, Devi GR. X-linked inhibitor of apoptosis protein mediates tumor cell resistance to antibody-dependent cellular cytotoxicity. Cell Death Dis. 2016;7:e2073
33. Aird KM, Ghanayem RB, Peplinski S, Lyerly HK, Devi GR. X-linked inhibitor of apoptosis protein inhibits apoptosis in inflammatory breast cancer cells with acquired resistance to an ErbB1/2 tyrosine kinase inhibitor. Mol Cancer Ther. 2010;9(5):1432-1442
34. Allensworth JL, Sauer SJ, Lyerly HK, Morse MA, Devi GR. Smac mimetic Birinapant induces apoptosis and enhances TRAIL potency in inflammatory breast cancer cells in an IAP-dependent and TNF-alpha-independent mechanism. Breast Cancer Res Treat. 2013;137(2):359-371
35. Balamurugan K, Sterneck E. The many faces of C/EBPdelta and their relevance for inflammation and cancer. Int J Biol Sci. 2013;9(9):917-933
36. Bertucci F, Finetti P, Vermeulen P, Van Dam P, Dirix L, Birnbaum D, Viens P, Van Laere S. Genomic profiling of inflammatory breast cancer: a review. Breast. 2014;23(5):538-545
37. Ogony J, Choi HJ, Lui A, Cristofanilli M, Lewis-Wambi J. Interferon-induced transmembrane protein 1 (IFITM1) overexpression enhances the aggressive phenotype of SUM149 inflammatory breast cancer cells in a signal transducer and activator of transcription 2 (STAT2)-dependent manner. Breast Cancer Res. 2016;18(1):25
38. Cabioglu N, Gong Y, Islam R, Broglio KR, Sneige N, Sahin A, Gonzalez-Angulo AM, Morandi P, Bucana C, Hortobagyi GN. et al. Expression of growth factor and chemokine receptors: new insights in the biology of inflammatory breast cancer. Ann Oncol. 2007;18(6):1021-1029
39. Zhang D, Tari AM, Akar U, Arun BK, LaFortune TA, Nieves-Alicea R, Hortobagyi GN, Ueno NT. Silencing kinase-interacting stathmin gene enhances erlotinib sensitivity by inhibiting Ser(1)(0) p27 phosphorylation in epidermal growth factor receptor-expressing breast cancer. Mol Cancer Ther. 2010;9(11):3090-3099
40. Yasuda MT, Sakakibara H, Shimoi K. Estrogen- and stress-induced DNA damage in breast cancer and chemoprevention with dietary flavonoid. Genes Environ. 2017;39:10
41. Colditz GA, Frazier AL. Models of breast cancer show that risk is set by events of early life: prevention efforts must shift focus. Cancer Epidemiol Biomarkers Prev. 1995;4(5):567-571
42. Zhang D, LaFortune TA, Krishnamurthy S, Esteva FJ, Cristofanilli M, Liu P, Lucci A, Singh B, Hung MC, Hortobagyi GN. et al. Epidermal growth factor receptor tyrosine kinase inhibitor reverses mesenchymal to epithelial phenotype and inhibits metastasis in inflammatory breast cancer. Clin Cancer Res. 2009;15(21):6639-6648
43. Wang C, Thudium KB, Han M, Wang XT, Huang H, Feingersh D, Garcia C, Wu Y, Kuhne M, Srinivasan M. et al. In vitro characterization of the anti-PD-1 antibody nivolumab, BMS-936558, and in vivo toxicology in non-human primates. Cancer Immunol Res. 2014;2(9):846-856
44. Shaffer SM, Dunagin MC, Torborg SR, Torre EA, Emert B, Krepler C, Beqiri M, Sproesser K, Brafford PA, Xiao M. et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature. 2017;546(7658):431-435
45. Hata AN, Niederst MJ, Archibald HL, Gomez-Caraballo M, Siddiqui FM, Mulvey HE, Maruvka YE, Ji F, Bhang HE, Krishnamurthy Radhakrishna V. et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat Med. 2016;22(3):262-269
46. Jolly MK, Boareto M, Debeb BG, Aceto N, Farach-Carson MC, Woodward WA, Levine H. Inflammatory breast cancer: a model for investigating cluster-based dissemination. NPJ Breast Cancer. 2017;3:21
47. Aceto N, Bardia A, Miyamoto DT, Donaldson MC, Wittner BS, Spencer JA, Yu M, Pely A, Engstrom A, Zhu H. et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell. 2014;158(5):1110-1122
48. Mu Z, Wang C, Ye Z, Austin L, Civan J, Hyslop T, Palazzo JP, Jaslow R, Li B, Myers RE. et al. Prospective assessment of the prognostic value of circulating tumor cells and their clusters in patients with advanced-stage breast cancer. Breast Cancer Res Treat. 2015;154(3):563-571
49. Wang C, Mu Z, Chervoneva I, Austin L, Ye Z, Rossi G, Palazzo JP, Sun C, Abu-Khalaf M, Myers RE. et al. Longitudinally collected CTCs and CTC-clusters and clinical outcomes of metastatic breast cancer. Breast Cancer Res Treat. 2017;161(1):83-94
Author contact
![]() Corresponding author: Wendy A. Woodward, MD, PhD, Associate Professor, Department of Radiation Oncology, Unit 1202, The University of Texas MD Anderson Cancer Center, 1515 Holcombe Blvd., Houston, TX 77030, USA; Tel 713-745-8250; email wwoodwardorg
Corresponding author: Wendy A. Woodward, MD, PhD, Associate Professor, Department of Radiation Oncology, Unit 1202, The University of Texas MD Anderson Cancer Center, 1515 Holcombe Blvd., Houston, TX 77030, USA; Tel 713-745-8250; email wwoodwardorg
Citation styles
APA
Woodward, W.A., Cristofanilli, M., Merajver, S.D., Van Laere, S., Pusztai, L., Bertucci, F., Berditchevski, F., Polyak, K., Overmoyer, B., Devi, G.R., Sterneck, E., Schneider, R., Debeb, B.G., Wang, X., van Golen, K.L., El-Zein, R., Rahal, O.M., Alexander, A., Reuben, J.M., Krishnamurthy, S., Lucci, A., Ueno, N.T. (2017). Scientific Summary from the Morgan Welch MD Anderson Cancer Center Inflammatory Breast Cancer (IBC) Program 10th Anniversary Conference. Journal of Cancer, 8(17), 3607-3614. https://doi.org/10.7150/jca.21200.
ACS
Woodward, W.A.; Cristofanilli, M.; Merajver, S.D.; Van Laere, S.; Pusztai, L.; Bertucci, F.; Berditchevski, F.; Polyak, K.; Overmoyer, B.; Devi, G.R.; Sterneck, E.; Schneider, R.; Debeb, B.G.; Wang, X.; van Golen, K.L.; El-Zein, R.; Rahal, O.M.; Alexander, A.; Reuben, J.M.; Krishnamurthy, S.; Lucci, A.; Ueno, N.T. Scientific Summary from the Morgan Welch MD Anderson Cancer Center Inflammatory Breast Cancer (IBC) Program 10th Anniversary Conference. J. Cancer 2017, 8 (17), 3607-3614. DOI: 10.7150/jca.21200.
NLM
Woodward WA, Cristofanilli M, Merajver SD, Van Laere S, Pusztai L, Bertucci F, Berditchevski F, Polyak K, Overmoyer B, Devi GR, Sterneck E, Schneider R, Debeb BG, Wang X, van Golen KL, El-Zein R, Rahal OM, Alexander A, Reuben JM, Krishnamurthy S, Lucci A, Ueno NT. Scientific Summary from the Morgan Welch MD Anderson Cancer Center Inflammatory Breast Cancer (IBC) Program 10th Anniversary Conference. J Cancer 2017; 8(17):3607-3614. doi:10.7150/jca.21200. https://www.jcancer.org/v08p3607.htm
CSE
Woodward WA, Cristofanilli M, Merajver SD, Van Laere S, Pusztai L, Bertucci F, Berditchevski F, Polyak K, Overmoyer B, Devi GR, Sterneck E, Schneider R, Debeb BG, Wang X, van Golen KL, El-Zein R, Rahal OM, Alexander A, Reuben JM, Krishnamurthy S, Lucci A, Ueno NT. 2017. Scientific Summary from the Morgan Welch MD Anderson Cancer Center Inflammatory Breast Cancer (IBC) Program 10th Anniversary Conference. J Cancer. 8(17):3607-3614.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.