Impact Factor ISSN: 1837-9664
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Issue 4; 2026
- Volume 17; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Special Issues
Introduction
Colorectal Cancer
CRC carcinogenesis- molecular...
Colon and the microbiota
Mucosal defense mechanisms and...
How inflammation is linked to...
Evading host immune system
Drivers, passengers and keystone...
Bacteria of special interest
Bacterial metabolites: Short...
Animal studies
Chemotherapy
Obesity, inflammation and cancer
Conclusion
Abbreviations
References

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(17):3378-3395. doi:10.7150/jca.20497 This issue Cite
Review
Linking Gut Microbiota to Colorectal Cancer
Hans Raskov1 ![]() , Jakob Burcharth2, Hans-Christian Pommergaard3
, Jakob Burcharth2, Hans-Christian Pommergaard3
1. Speciallægecentret ved Diakonissestiftelsen, Frederiksberg, Denmark;
2. Department of Surgery, Zealand University Hospital, University of Copenhagen, Denmark;
3. Department of Surgical Gastroenterology, Rigshospitalet, Copenhagen, Denmark.
Received 2017-4-10; Accepted 2017-8-10; Published 2017-9-20
Abstract
Pre-clinical and clinical data produce mounting evidence that the microbiota is strongly associated with colorectal carcinogenesis. Dysbiosis may change the course of carcinogenesis as microbial actions seem to impact genetic and epigenetic alterations leading to dysplasia, clonal expansion and malignant transformation. Initiation and promotion of colorectal cancer may result from direct bacterial actions, bacterial metabolites and inflammatory pathways. Newer aspects of microbiota and colorectal cancer include quorum sensing, biofilm formation, sidedness and effects/countereffects of microbiota and probiotics on chemotherapy. In the future, targeting the microbiota will probably be a powerful weapon in the battle against CRC as gut microbiology, genomics and metabolomics promise to uncover important linkages between microbiota and intestinal health.
Keywords: microbiota, dysbiosis, mucosal defense mechanisms, inflammation, carcinogenesis, colorectal cancer.
Introduction
The general understanding of CRC: colorectal cancer; is that it is basically a genetic disease resulting from a sequence of mutations following well-established molecular pathways and accumulating over many years. Sporadic CRC develops through the CIS: chromosomal instability pathway; while HNPCC: hereditary non-polyposis colon cancer; and CRC in FAP: familial adenomatous polyposis; develop along the MIS: microsatellite instability; pathway and from a germline mutation in the APC: adenomatous polyposis coli; gene respectively - both leading to invasive cancer at younger ages.1,2 More recently an additional pathway - the serrated pathway - displaying both MIS and non-MIS tumors has been defined.3, 4
The molecular biology of colorectal carcinogenesis is mapped in great detail with the majority of CRC demonstrating accumulation of mutations and epigenetic changes according to the adenoma-carcinoma sequence with loss of function of tumour suppressor genes and gain of function in oncogenes.5
However, pathways used by pathogens to establish infections (quorum sensing, invasion, biofilm formation) are able to derail mechanisms controlling cellular proliferation and pre-clinical and clinical research are producing mounting evidence that the gut microbiota is strongly associated with CRC carcinogenesis. Pertubations of the microbiota may effectively change the course of carcinogenesis directly as well as indirectly as microbial actions seem to impact both genetic and epigenetic alterations promoting dysplasia, clonal expansion, tumour growth and invasive cancer.6, 7
Intestinal microbial eubiosis and host-microbiota interactions are extremely important for human health. Dysbiosis and chronic, subclinical, low-grade inflammation in the bowel wall contribute to and may even initiate development of gastrointestinal diseases, such as CRC, IBD: inflammatory bowel disease; and IBS: irritable bowel syndrome. Dysbiosis may also contribute to extra-intestinal, systemic conditions such as atherosclerosis, obesity, type 2 diabetes, metabolic syndrome and mental perturbations.
Only recently the microbiota was integrated in the framework of translational CRC research and it seems clear that dysbiosis and subsequent immunological responses facilitate CRC carcinogenesis and is linked to prognosis as well.8-15 Although the microbiota is stable over long periods of time, a variety of factors such as aging, obesity, western diet, lack of exercise, diseases and antibiotics shifts the microbiota towards a less diverse and pro-inflammatory profile.16
Potential bacterial drivers and carcinogenic metabolites, intraluminal events and inflammatory pathways are subject to intense research15, 17-19 as manipulation of the microbiota (e.g. use of pro-, pre- or antibiotics) changes the course of tumorigenesis, may alter or arrest tumorigenesis.
16S rRNA sequencing and whole genome shotgun sequencing metagenomics studies show lower bacterial diversity and a higher rate of certain pro-tumerogeneic bacteria (e.g. Fusobacteria and Porphyromonas) associated with CRC.6, 13, 14, 17, 18, 20-27
Here, we review recent research linking gut microbiota to CRC and discuss future implications. We also present the latest findings regarding possible implications of gut microbiota on sidedness of CRC and effects on chemotherapy.
Colorectal Cancer
CRC is a common malignancy in Western Europe, North America, Australia and New Zealand affecting 5-7% of the populations. Despite population screening, advances in surgery and oncology, CRC is still among the leading causes of cancer deaths. Due to Western influences on lifestyle and diet, CRC rates are on the rise in many Eastern nations as well and the global burden of CRC is estimated increase by 60% with 2.2 million new cases and 1.1 million deaths annually by 2030.28 Although the 5-year survival rate has improved in the US (from 50 % to 65 % over the last 40 years), CRC remains a heavy burden on health care economics. More than 1.18 million US residents were living with CRC in 2013 and the prevalence is increasing. 29, 30
Recently attention has been drawn to the impact of primary tumor sidedness in terms of reduced progression-free and overall survival for patient with right-sided CRC. 31 The CRYSTAL and FIRE-3 studies looked at 764 KRAS wild-type patients with mCRC: metastatic CRC; receiving conventional chemotherapy combined with bevacuzimab or cetuximab. Patients with right-sided primary tumors had significantly reduced progression-free survival and overall survival compared to patients with a left-sided primary.32
These findings were confirmed in a prospective study (CALGB/SWOG 80405) of 1137 patients with mCRC from right- or left-sided primary tumors and RAS-wild-type, both receiving conventional chemotherapy with bevacuzimab or cetuximab. Patients with left-sided primary CRC had a median overall survival 33.3 months compared to 19.4 months for right-sided primary CRC. The overall conclusion from retrospective analyses of these studies was that patients with right-sided CRC did not benefit from EGFR: epidermal growth factor receptor; antibodies, regardless of their RAS-status.33
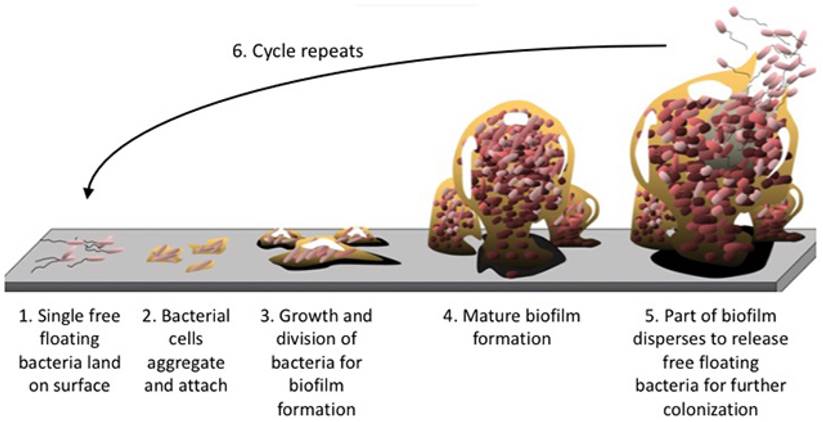
Interestingly, in a recent study substantial biofilm formation was found in the right colon from virtually all right-sided colon adenomas and cancers, while left-sided cancers infrequently were biofilm positive (89% vs. 12%). Importantly, histologically normal colon mucosa, collected from the surgical resection margin, was also biofilm positive or negative, demonstrating 100% concordance with their paired cancer.34
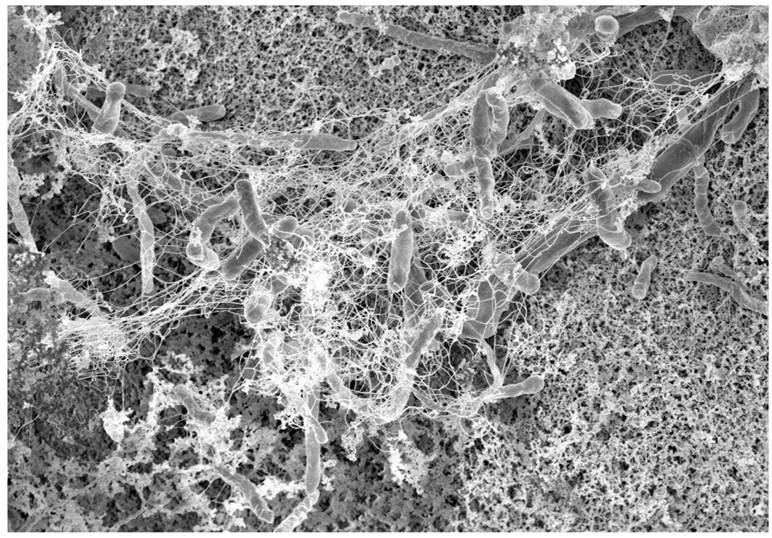
Microbial biofilm (figure 1) may be regarded as an independent driver at an early stage of CRC carcinogenesis, before the malignant transformation from adenoma to carcinoma occurs. Various processes mediating the action of biofilms in driving the CRC process have been proposed, including reduced levels of E-cadherin in colonic crypts, increased intestinal permeability, the production of polyamine metabolites and their subsequent acetylation, and elevated induction of IL-6: Interleukin-6;/STAT3: Signal Transducer and Activator of Transcription 3; signaling. 35
Biofilm formation. The structural components of the extracellular matrix is a highly hydrated, robust structure with high tensile strength and represents up to 90% of the biofilm mass keeping bacteria in close proximity, enabling intimate cell-to-cell interactions and DNA exchange while at the same time protecting the biomass from damaging agents. No copyright.
CRC carcinogenesis- molecular pathways
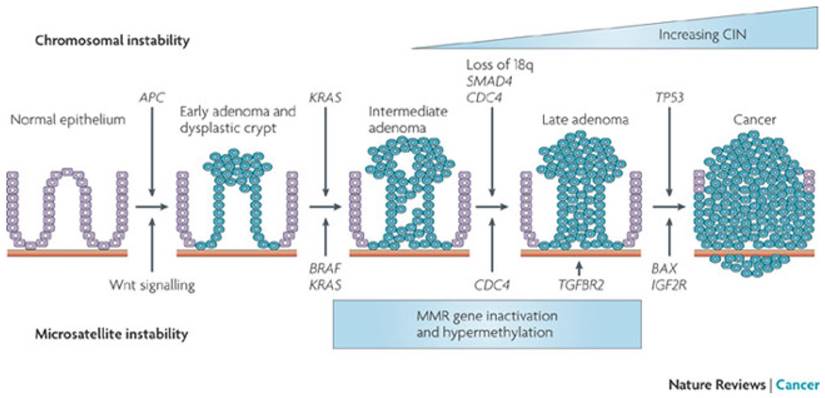
The two major pathways involved in most cases of CRC are the CIS pathway developing through mutations in KRAS, APC, MYC, Wnt, MAPK and P53 along the adenoma - carcinoma sequence (figure 2) and the MSI pathway through mutations in MMR genes. More than 90 % of CRC are sporadic with only a small number caused by germline mutations.
Adenoma-carcinoma sequence. CIS involves loss of APC function and KRAS, followed by loss of chromosome 18q with SMAD4 and mutation in TP53. MSI CRC is characterized by a deficiency of the MMR leading to slippage in microsatellites. CIN: chromosomal instability. From Walther & al. Nat Rev Cancer 2009;9:489-99. Permission from Macmillan Publishers Limited.
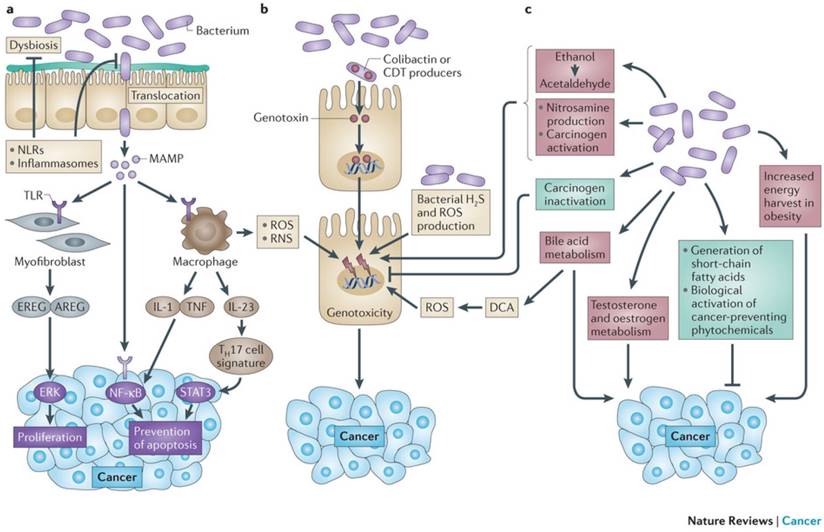
Mechanisms by which the bacterial microbiome modulates carcinogenesis. The microbiota promotes carcinogenesis through different mechanisms. A | Dysbiosis and inflammation induced by MAMP activating TLR and other PPR (e.g. NLR). B Detrimental effects are mediated by bacterial toxins such as colibactin and CDT: Cytolethal Distending Toxin, ROS, Reactive Nitrogen Species and H2S. C Metabolic actions activating toxins such as acetaldehydes and nitrosamines. The microbiota mediates preventive effects (in green) through inactivation of carcinogens and production of SCFA: short chain fatty acids; such as butyrate and propionate. From Schwabe RF, Jobin C. Nature Reviews Cancer 2013;13:800-12. With permission from Macmillan Publishers Ltd.
CRC in FAP is a rare malignancy (<1% of CRC) caused by a frameshift mutation in the APC gene on chromsome 5q22. The mutation simulates a stop codon causing a truncation of the APC protein inhibiting β-catenin from binding to the cytoplasmic part of the e-cadherin complex. The free intra-cytoplasmic β-catenin translocates to the cell nucleus resulting in upregulation of Wnt-signalling. FAP results in multiple adenomas and inevitably CRC before the age of 40 years.2
Chronic inflammation is linked to carcinogenesis as 20 % of patients with ulcerative colitis develop CRC within 30 years of onset.36 IBD (ulcerative colitis and Crohns disease) is widely accepted as one of the important risk factors for CRC. A meta-analysis of 116 studies found the mean age of IBD-CRC diagnosis to be 43 years, which is 10-15 years younger than sporadic CRC but with a similar 5-year survival rate.37 Furthermore, it is well established that long term use of NSAIDs reduce lifetime risk of CRC, again pointing towards a link between inflammation and carcinogenesis.38
The finding that long-term antibiotic use in early adulthood seems to be associated with increased risk of colorectal adenomas later in life also points towards an association between microbiota and neoplasia. 39
In sporadic CRC, the accumulation of multiple mutations in growth promoting oncogenes and growth limiting tumor suppressor genes occurs with no apparent genetic predisposition although familial clustering is seen.
A constant proliferation rate in the normal colorectal mucosa is a result of a delicate equilibrium between proto-oncogenes and tumor-suppressor genes. Proliferation studies show that the rapid turnover and immense number of mitoses in the colon crypts lead to tens of thousands of mutations every day.40-42
Efficient DNA repair mechanisms, among others the MMR, the base excision repair system, the nucleotide excision repair system and the double strand break repair systems continuously scan the genome for replication errors. DNA defects are repaired immediately, the base excision repair system alone accounting for more than 10,000 repairs in the colon per day.43 If time is needed for DNA repair, the G1 cell cycle phase is slowed down or put on stand-by by tumor-suppressor genes, among which the p53 is one of the most important and well known.44, 45 If mutations are too many or too extensive to repair, apoptosis is initiated through a complex signal pathway shutting down mitochondrial function resulting in immediate cell death.46
Although the DNA repair systems are extremely effective, mutations do slip through from time to time explaining both evolution and genetic diseases. Apart from germline/hereditary cancers, the development of CRC usually requires decades to accumulate mutations in key oncogenes and tumor suppressor genes powerful enough to initiate colorectal carcinogenesis.47
Although it is not yet proven that any specific strains of bacteria are able to provide the first hit to initiate carcinogenesis, the current understanding of the adenoma-carcinoma pathway has expanded to include environmental factors and luminal events including perturbations in the microbiota and dysbiosis.48
Colon and the microbiota
Microorganisms have evolved over more than a billion years and the human body is host to trillions (3.5-4 x 1013) of microbes residing in the GI tract. The number of microbes is slightly higher than the number of cells in our body (approx. 3 x 1013/70kg) and they harbor 150 times as many genes (3.3 mill.) compared to the human genome. It is postulated that the human colon is one of the most densely populated microbial ecosystems on the planet.49-52
In the colon, the concentration of bacteria reaches 1011 bacteria pr. gram content, which is a million times higher than in the small bowel. The small bowel contains a very different microbiota much more dynamic and dependent on diet. Despite the harsh chemical environment in the small bowel, the risk of small bowel cancer is 12 times less than the risk of CRC, suggesting a potential role of the microbiota in carcinogenesis.13, 53-55
The gut microbiota is an ecologic system of diverse commensal microorganisms metabolizing food remnants, intestinal secretions, digestive juices and exfoliated colonocytes. The colon produces vitamins (vitamin K, B12, thiamin, riboflavin), absorbs water and electrolytes and transports waste products (feces) to the rectum for defecation. Proteolytic fermentation increases with high protein intake and results in production of phenolic compounds, amines, ammonia, NOC: N-nitroso compounds; and indoles, all considered to have a carcinogenic impact on epithelial cell differentiation and proliferation.56
Colonic metabolism involves primarily extraction of nutrients and energy from otherwise indigestible carbohydrates such as starch and proteins by fermentation. Providing 10% of our daily energy requirements, the colon is a bioreactor also producing heat, which is important for maintaining a stable intraluminal environment for microbes as well as a stable body temperature. Microorganisms produce more heat than any other organism per weight unit, and it is estimated that 70% of the body heat at rest results from bacterial metabolism in the gut.14, 53, 57, 58
The human tissues are separated from the microbiota by a single layer of epithelial cells representing the mucosal surface. Epithelial cells are protected from potential pathogens by a mucus layer and by the immune system. A breach in this otherwise robust barrier may result in a state of chronic inflammation.59
In the human colon, the total proliferation rate is 3-10 billion colonocytes per day, being the highest proliferation rate in any mammalian organ. This constant proliferation requires readily available nutrients and energy, making the process very responsive to dietary changes. In the bottom of each colonic crypt, 4-6 stemcells proliferate into an enormous amount of colonocytes while accumulating genetic and epigenetic changes.60 As a result of the continuous proliferation, new colonocytes move from the bottom of the crypt towards the surface by approximately 1 cell position per hour. Colonocytes reaching the luminal surface are exfoliated. A crypt, and thus the entire colonic mucosa, is fully renewed in only a few days.61-63
The microbiota has a great influence on the expression of a broad array of human genes, e.g. particular strains of Bifidobacteria, Lactobacilli and E. coli influence gene expression of mucins, TLR: Toll-like receptors; on macrophages and dendritic cells as well as caspase expression and thus modulate immunological activity and apoptosis. However, during eubiosis the interaction between commensal bacteria and the immune cells constitute a delicate balance of pro-inflammatory genes and proto-oncogenes on one side and anti-inflammatory genes and tumor-suppressor genes on the other side.64, 65 Although identification of the gut microbiota, consisting of a great variety of microbes with a majority of anaerobic bacteria, has been ongoing for some years, the majority of the microbiota is still not characterized66. Multidisciplinary approaches for characterizing the microbiota show major discrepancies between morphological and molecular studies. It is therefore necessary to perform multiple techniques to assess the overall bacterial diversity in the gut.67-69 Currently, it is estimated that 500-1000 different bacterial species and more than 7000 individual bacterial strains create the delicate community of commensal organisms.50, 70
Besides bacteria the microbiota includes vast numbers of viruses, protozoa, archae and fungi. 99.9 % of colonic bacteria are strict anaerobes (bacteroidetes, bifido-, fuso-, peptostreptococcus, atopobium and eubacterium).71
The gut microbiota is dominated by five bacterial phyla
| Firmicutes (50-60% of the microbiota) including more than 180 species of Lactobacillus, Clostridiales and Streptococcaceae. |
| Bacteroidetes (25-40% of the microbiota) with Bacteroides Fragilis and Porphyromonadaceae both reported upregulated in CRC |
| Actinobacteria (2-5%) including the Bifidobacteriae |
| Proteobacteria (2-5%) E. coli), salmonella, yersinia, shigella, vibrio, haemophilus, |
| Fusobacteria (2-5%) Fusobacterium Nucleatum, faecalibacterium prausnitzii |
Although a state of symbiosis exists between the microbiota and the human body, most microbes are pathogenic if translocated from the gut lumen into surrounding tissues. Hospital infections are most often Gram-negative with E. coli as the most common etiologic organism, followed by Pseudomonas aeruginosa, Klebsiella species and Enterobacter species. Bacteroides are found in most anaerobic infections and carry a high mortality.72-74
In the colon, the fecal (luminal) microbiota differs substantially from the microbiota within the mucine layer of the bowel wall (the juxta- or mucosal microbiota). The fecal microbiota is much richer in diversity than the mucosal microbiota and stool samples are thus poor mirror images of the mucosal microbiota as many microbes in the stool are simply passing through. Therefore, the adherent microbes in the deeper mucus layer with access to the epithelial cells (directly or indirectly) are more likely key players in carcinogenesis.75 In fact, most of the mucosal microbes are residing the in the outer layer of the mucus membrane while the innermost part of the mucus layer contains only small amounts of bacteria.76
The microbiota is in part determined by the genetic profile of the host and the microbiota constantly evolves during the lifespan of the individual. The microbiota is generally stable for long periods of time and in younger adults the microbiota is substantially different from the elderly, especially with regard to the proportion of the firmicutes phylum decreasing with age.77, 78
A core microbiota comprising 66 taxonomic units is shared by approximately 40% of all humans but still we have no clear understanding of what constitutes a healthy microbiota. Conversely, 60% of the microbiota is variable and determined by a combination of host factors.50, 79, 80 Although each individual has a unique microbiota (microbiological fingerprint), the abundance and distribution between the bacterial phylotypes is almost similar among healthy persons.
With increasing age and age-related loss of CD4 T cells, the microbiota shifts towards a more pro-inflammatory and less diverse profile linked to adverse health issues and tumorigenesis. Loss of T cell differentiation diminishes the ability of the immune systems to suppress tumor-promoting inflammation in the colon.16, 79 Also among the age-related changes in the microbiota is a reduction in butyrate producing bacteria (especially the Firmicutes phyla) resulting in a decrease of the primary energy source for colonocytes as well as an increase in intra-colonic pH values. The higher pH values, inflammation and dysbiosis create a hostile environment for colonocytes and contribute to tumorigenesis.48
When looking at individual bacterial strains in relation to colorectal carcinogenesis, there appears to be opposing roles for protective, butyrate-producing Gram-negative populations and pro-inflammatory, mucin-degrading Gram-positive populations. Several papers have looked at the levels of different strains in relation to CRC carcinogenesis in animal settings and in CRC patients. Methods, materials and results are inconsistent but a reproducible pattern in most studies show a reduction in lactic acid bacteria and an increase in fusobacteria, staphylococcaceae and porphyromonadaceae. Fusobacteria and porphyromonadaceae are known to synergistically promote oral cancer.81
The microbiota located in close relation to tumor tissue and polyps did not differ significantly from the normal mucosa within the same individual, suggesting that the microbiota in CRC patients had not changed secondary to the neoplasia per se, suggesting that a CRC distinctive microbiota was already present in early stages of carcinogenesis.14, 36
In general, studies were based on small sample numbers and controls were often not comparable and at this point in time, it is not possible to characterize a specific cancer associated gut microbiome. Rather than a single carcinogenic bacterial strain, it seems more likely that certain bacterial compositions and their metabolites create an environment involved in carcinogenesis mediated through continuous/chronic inflammatory processes and changes in the immune system.7
Mucosal defense mechanisms and inflammation
In healthy individuals, the structure and function of the colorectal epithelium together with the immune system preserve a mutually beneficial relationship between the microbiota and the host. The preservation of eubiosis is important for maintaining the integrity of the intestinal gut barrier as well as the participation in the uptake of nutrients and production of vitamins. The healthy microbiota is able to sort food ingredients and other intraluminal substances into useful, useless and pathogenic and to handle them accordingly.82
The single-layer of epithelial cells between the microbiota and the host is not only a physical and chemical barrier counteracting microbial invasion. It also balances the communication between the host, the microbiota and the intraluminal environment. During eubiosis, a physiologic and controlled state of low-grade inflammation is present in the bowel wall, supervised by the innate immune system. The healthy microbiota does not cause significant inflammatory responses in the host mucosa as the immune system is trained to recognize and accept the normal and naturally occurring microorganisms.
Predominant anti-inflammatory cytokines such as TGF-β: transforming growth factor-β; and IL-10 are produced by epihelial cells, T regs: regulatory T cells; and stromal cells in order to keep colonic inflammation in check. Even commensal bacteria induce IL to maintain a basal level of Th17: T helper cells; protecting against mucosal pathogens in the bowel wall.83-85
The immune system contains an immunologic archive, based on PRR: pattern recognition receptors; able to distinguish potentially pathogenic microorganisms from harmless commensals. PPR primarily recognizes surface molecules derived from microbes, especially bacterial lipopolysccarides, lipoproteins, prokaryotic DNA and foreign nucleic acids, so-called MAMP: microorganism-associated molecular patterns; or PAMP: pathogen-associated molecular patterns.
TLR belong to a major class of PRR expressed on membranes of macrophages and dendritic cells (fig. 1). TLR signaling trigger immune defense mechanisms, primarily through production of proinflammatory cytokines, and enhances barrier function by reinforcing tight junctions and zonula occludens thereby obstructing paracellular microbial invasion.86 CpG (cytidine-phospate-guanosine) motifs of damaged human DNA or apoptotic debris (damage-associated molecular pattern - DAMP) may activate TLR9, an intracellular DNA sensor, and trigger a self-destructive, chronic B-cell immune response. Chronic TLR9 triggering may accelerate growth and dissemination of tumors in the GI tract.87, 88
Another group of PRR is NLR: NOD-like receptors; a big family of closely related intracellular sensors belonging to a super-family of glycoprotein receptors, among those the NOD1 - 14, Ipaf and Naip detecting PAMPs entering cells via phagocytosis or pores in the cell membrane. NLR are located in the cytoplasm of both immune cells and non-immune cells such as colonocytes. NLR not only recognize bacterial wall fragments being the typical NLR activators, but also react to damaged host cell membranes exposing glycans from the outer leaflets of membrane to the cytoplasmic NLR receptors. NLR activation triggers structural rearrangement of the receptor to conduct signal spread activating multiple signal pathways to induce production of pro-inflammatory cytokines and autophagy activity.89
Disruption or mutations in PPR result is a dramatic thinning of the mucus layer and a sustained inflammatory reaction. Especially NOD2 mutations are associated with Crohns disease and increased risk of CRC as well as worsened prognosis in CRC.90-95
Dysbiosis favors invasion and growth of pathogenic species and disrupt homeostasis of the immune system and mucosal barrier. The subsequent inflammatory process results in increased permeability allowing gut microbes to drive a continuous state of inflammation.66, 86, 96-98
How inflammation is linked to microbiota and carcinogenesis
When pathogens invade the well-calibrated functions of microbiota and innate immune system, the normal, anti-inflammatory environment driving anti-tumor immunity is transformed by the prompt activation of immune cells releasing cytokines and growth factors such as interleukins, TGF-β, TNF: tumor necrosis factor and VEGF: vascular endothelial growth factor.99
While PPR try to maintain epithelial barrier function by reinforcement of cellular junctions, an opposite effect is produced by the pro-inflammatory cytokines causing increased mucosal permeability because of cytoskeletal contraction of the peri-junctional ring providing paracellular passageways for microbes. Both cytokines and growth factors drive the inflammatory process negatively influencing cell differentiation and thereby supporting the growth and survival of dysplastic cells. In a self-sustaining process, tumor cells and even bacteria may produce increasing amounts cytokines themselves.100, 101
If a state of persistent inflammation develops, the ongoing cascades of inflammatory signals result in proliferation, angiogenesis, inhibition of apoptosis and increased release of growth factors - all paving the way for cancer.19
Several cytokines may be used as biomarkers and treatment targets. A classification system (Immunoscore®) may improve prognostication of CRC patients. A multivariate analysis revealed that Immunoscore was superior to testing of microsatellite instability in predicting recurrence-free survival and overall survival.102-105
When eubiosis is impaired TGF-β, IL-6 and TNF produced by macrophages and T cells trigger the differentiation of naive CD4 T cells into pro-inflammatory Th17 cells leading the adaptive immune response against pathogens. In case of sustained mucosal inflammation, the prolonged presence of Th17 has been linked to development of CRC. Infiltration of Th17 cells in CRC tissue and elevated levels of Th17-mitogenic cytokines, especially IL-17 and IL-22, are associated to poor prognosis and reduced survival.75, 99
Interestingly, certain types of commensal bacteria, especially Clostridia species, as well as dysplasia promote the accumulation of Th17 in the intestinal mucosa and drive IL-17 production in epithelial cells and Th17 has been shown to be a key driver of CRC in Min-mouse models exposing the animals to ETBF: Enterotoxigenic Bacteroides Fragilis. In experimental settings, limiting Th17 by depletion of Tregs reduces formation of microadenoma.75, 91, 106-109 On the other hand, tumours with marked infiltration of CD3+ (and maybe of CD8+) are reported to be associated with good clinical outcome in CRC in terms of longer disease-free survival and overall survival.110
IL-6 is a powerful pro-inflammatory cytokine clearly associated to CRC development and important for angiogenesis in cancer tissue. IL-6 produced by macrophages, T cells and fibroblasts of the tumor-supporting stroma leads to activation of STAT3 and tumor progression. An elevated level of serum IL6 is an independent prognostic marker for tumor size, liver metastases and poor survival in CRC patients.111, 112
Another multifunctional cytokine, TGF-β, produced by inflammatory cells and macrophages, plays an important role in cell growth, differentiation and apoptosis. Under normal circumstances TGF-β activation in epithelial cells inhibits abnormal cell growth by inducing cell-cycle checkpoints and growth arrest, but inactivating mutations causing loss of TGF-β signaling molecules and alterations in TGF-β receptors are frequent in CRC.113, 114 TGF-β produced by tumor stromal fibroblasts has the opposite effect and drives proliferation and metastatic potential of CRC cells. Tumors characterized by elevated TGF-β production are associated with a high-risk of recurrence upon treatment because of a survival advantage to metastatic cells.115
Macrophages also produce TNF, a powerful pro-inflammatory cytokine, recruiting inflammatory cells, increasing vascular permeability and mitogenic signaling.
TNF activates oncogenic signaling pathways in epithelial cells, including the Wnt and NF-κB: Nuclear Factor κ-B; a transcription factor, thereby regulating growth and survival. Exposure of epithelial cells to TNF increases CIS and mutation rates, suggesting a direct mechanism by TNF on cancer development.116-119 TNF serum levels positively correlate with disease progression and poor survival in CRC patients.120, 121
Possible pathophysiological mechanisms of inflammation also include the oxidative stress caused by an imbalance between the production of ROS: Reactive Oxygen Species; and antioxidant defenses resulting in DNA damage, dysplasia and thus a tumor-supporting environment. Oxidative stress induces NFκB linking chronic inflammation to cancer through the ability of up-regulation of pro-inflammatory and tumor promoting cytokines, such as TNFα, IL-1 and -6 as well as mitogenic and anti-apoptotic signaling. NF-κB directly regulates a complex of genes (including bcl-x and survivin) increasing proliferation and protecting tumor cells from apoptosis.100, 122 Interestingly, obesity increases NFκB expression in most tissues also increasing the risk of type-2 diabetes. NFκB could be a factor linking obesity, type 2 diabetes, cardio-vascular disease, IBD and cancer.13, 123
Evading host immune system
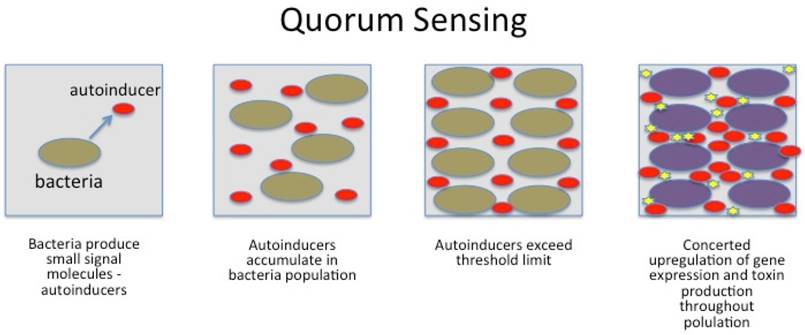
Gut bacteria are able to invade host tissues, evade host immune system and maintain inflammation. Bacteria communicate internally using chemical molecules in a process called QS: quorum sensing; (figure 4) and by nanotubes, both of great importance for bacterial virulence. Ways to interrupt QS (quorum quenching) and the formation of biofilms are believed to be next generation of antibiotics and may impact tumorigenesis and is therefore focus intense research. QS involves production, release, detection and response to chemical signalling by small hormone-like molecules called autoinducers.
Schematic presentation of bacterial gene regulation by Quorum sensing. No copyright.
Early stage of bacterial biofilm with visible intercellular polymeric matrix.
Bacteria also use nanotubes (small bridging tunnels of 30-130 nm in diameter), through which cargo can be exchanged in the form of cytoplasmic components (e.g. resistance). Furthermore, nanotubes are not restricted to interaction between the same species.9 For bacteria to launch a successful attack on the host, timing and communication is the key. If not executed precisely, bacteria are immediately wiped out by the host's immune system. By the use of quorum sensing, a finely tuned and powerful attack may overcome the defense mechanisms and even kill the host. Bacteria use quorum sensing to count themselves, synchronize their behavior and to express specific genes at the same time. They wait until their numbers exceed a critical mass (threshold for signal molecules) for a successful invasion (swarming) and secrete their toxins simultaneously. Once gene expression is upregulated, a number of bacteria produce exopolysaccarides to glue themselves together in a sticky, protective layer (biofilm) having significant importance for virulence and resistance.124
Bacteria have evolved ways to perceive environmental changes and gauge whether it is beneficial to remain as planktonic (free-flowing) organisms or whether it is time to resume a safer life within a biofilm. When bacteria switch from planktonic to community mode, they undergo a shift in behavior that involves alterations in the activity of numerous genes. Once colonization of the biofilm has begun, it grows through a combination of cell division and recruitment.125
Biofilm formation enables bacterial pathogens to colonize a wide variety of host niches and to persist in harsh environments making their eradication by the innate immune system particularly difficult. Because biofilms often develop their own metabolism, they are sometimes compared to tissues of higher organisms, in which closely packed cells benefit from the collective gene-pool for the greater good and safety of the community.126
Biofilm formation is not a random process but have distinct architectures developing into 3-dimentional structures housing millions or billions of individual bacteria. Attached to the surface by pili and fimbriae and with the help of the self-produced intercellular polymeric matrix the bacteria glue themselves together and form colonies resembling small cities with towers, roads, bridges and channels for the flow of nutrients and at the same time allowing residents to enter and exit the biofilm. Living inside biofilms, bacteria increase their chance of survival by increased persistence and resistance to hostile environmental factors.124, 127
If overcrowding in the biofilm occurs, communication molecules prompt bacteria to leave the biofilm (figure 1). In the colon, biofilms may be found in the inner protective mucus layer allowing for direct interaction between bacteria and colonocytes thereby triggering inflammation. Approximately 15% of healthy individuals harbour thin bacterial biofilms predominantly in the right side of the colon whereas nearly 100% of right-sided CRC were found to have substantial biofilms. In addition, biofilms proximal to the hepatic flexure constitutes a higher frequency of epithelial KRAS and BRAF mutations and are linked to a worse prognosis for right-sided cancers.9, 12, 34, 75, 128-130
Once an attack is launched, bacteria have developed highly effective mechanisms to evade the immune system. Wearing coats of carbohydrates camouflaging their surface antigens (lipopolysaccarides and peptidoglycans) bacteria are able to disguise themselves and evade immune surveillance and PPR recognition. They can invade their new territory without detection and may even circulate systematically and unnoticed within the body.129
Inside the host, bacteria disguise themselves by changing expression of surface molecules by switching simple structural molecules. Some bacteria even possess a genetic switch to turn gene expression on and off. As the acquired immunity of the host relies on memory of previous exposure to antigens, variation or covering of surface antigens is very effective in circumventing both humoral and cellular responses.129, 131
Bacteria may alter host immune response or even avoid it by affecting downstream inflammatory cytokines and by secretion of enzymes such as IgA proteases degrading immunoglobulins. Bacteria anchor to host cell surfaces by adhesins or receptor ligands and by the use of invasins or fusion proteins they force their way into host cells. Inside infected cells, bacteria may block normal apoptosis mechanisms, and using the cell like a Trojan horse, bacteria multiply within the infected cell.132
If discovered by the immune system, several bacteria are able to defend themselves by injecting cytotoxic agents directly into immune cells through pili and nanotubes. Due to their size, bacteria are perfect phagocytic targets but many bacteria have developed ways of avoiding phagocytosis by injection of effector mediators neutralizing phagocytic activity. The ability to avoid internalization and killing plays a central role in bacterial virulence.133
When caught and trapped inside a macrophage, bacteria can use several strategies to avoid killing. Escaping the phagosomes, most commonly by blockage of phagosome-lysosome fusion or by utilization of mechanisms to allow survival in phagolysosomes. Species of Shigella and Listeria and some Rickettsia species secrete lysins that are highly effective at lysing the vacuolar membrane that engulfs internalized organisms.129
Drivers, passengers and keystone pathogens
The mechanisms by which the microbiota potentiate carcinogenesis range from production of carcinogenic toxins to manipulation of inflammatory pathways. The driver-passenger model describes the existence of certain bacteria in the microbiota possessing special virulence traits (e.g ETBF, Fusobacteria spp. and Porphyromonadaceae) creating DNA damage and driving genomic instability. Drivers are subsequently replaced by commensals (passengers) with tumor-promoting or tumor-suppressing properties such as the pro-inflammatory Streptococcaceae, Klebsiella and Proteus. During bacterial invasion, passengers or “back-seat drivers” do not play any active role. They are excluded from the communication network and are not impacted by quorum sensing but can be activated at a later stage. By the use of nanotubes, passengers may bypass any diffusion barrier or inhibitory systems put up by the immune system.9
According to the keystone pathogen (or α-bug) hypothesis, the effects of some bacterial species are disproportionately large relative to their abundance. Thus, small-abundance pathogens like ETBF, Fusobacteria and colibactin-producing E.coli can cause dysbiosis, increase permeability, translocate through the mucosal barrier and with their toxins cause chronic inflammation, inflammatory diseases and contribute to carcinogenesis.18, 49, 134
Bacteria of special interest
Although dysbiosis may take many forms and no consistent cancer-related dysbiotic signature is likely to be discovered, carcinogenesis may depend on structure and metabolic activities of communities of bacteria (polymicrobial synergy), i.e. no single virulence factor, disruption of mucosal barrier or long lasting inflammation, but rather all these components in concert.
Although conflicting data exist, there are a number of reports of significant differences between the mucosal and fecal microbiota from CRC patients and controls.75, 135
Overall, the common findings in CRC patients are loss of bacterial diversity and dysbiosis. Particularly there is often a marked decrease in firmicutes, bacteroides and lactic acid bacteria together with an increase in fusobacterium, and porphyromonas.
E. faecalis, E. coli and Fusobacterium nucleatum are capable of triggering Wnt mitogenic signalling, DNA damage, interference with DNA repair processes and to increase mucosal permeability by rendering adhesion molecules (e-cadherins) dysfunctional - all of which are critical to CRC carcinogenesis.49, 55, 75
In experimental settings, proteobacteria, especially a relative abundance of Lactobacillus, was inversely proportional to tumour burden, whereas Bacteroidetes and proteobacteria consistently were among the phylae increasing tumour counts and tumour burden. Indeed, the balance between the predominant Bacteroidetes and Firmicutes phyla seems to be critical in relation to disease progression.7, 8, 75
In 2014, European Molecular Biology Laboratory performed metagenomic sequencing of stool samples from 156 individuals with CRC and from controls. Metagenomics predicted the presence or absence of CRC using the relative abundance of 22 bacterial species, including Porphyromonas and Fusobacterium. It was possible to predict CRC with about the same accuracy as fecal occult blood testing commonly used as a screening tool. Fifty percent of cancers were correctly identified with less than 10 percent of false positives. When metagenomics and fecal occult blood testing were combined, they found more than 70 percent of cancers. The accuracy of metagenomic CRC detection did not differ between early- and late-stage cancers. The method was later validated in independent patient and control populations from several countries.136
Fusobacterium Nucleatum
FN: Fusobacterium Nucleatum; is an anaerobic Gram-negative bacteria linked to CRC. 16S rRNA and whole genome sequencing has shown enrichment of FN in CRC associated mucosa and in the mucosa of adenoma patients when compared to healthy individuals. FISH: fluorescence in situ hybridization; technique also show excess FN in tumor tissue as well as invasiveness of the bacteria has been demonstrated.137, 138
FN is able to directly promote carcinogenesis when the secreted anchoring adhesin FadA binds to the extracellular domain of E-cadherin on epithelial cells rendering the adhesive complex dysfunctional. The result is openings of paracellular passageways for pathogens to the submucous tissues. The malfunctioning intercellular part of the adhesion complexes cannot bind free cytoplasmic beta-catenin, which instead translocates to the cell nucleus. Here β-catenin upregulates mitogenic signaling through the Wnt pathway, leading to increased expression of transcription factors, Wnt genes, inflammatory genes and growth stimulation of CRC cells.139 FadA may even compromise DNA repair as high-level FN colonization has been significantly associated with microsatelite instability.55 A 3-fold increase in risk of adenomas among individuals with the highest tertile of FN counts has been demonstrated. Presence and counts increased with malignant transformation from adenomatous polyp to CRC.7 FadA levels in the colon tissue from patients with adenomas and adenocarcinomas is >10-100 times higher compared to normal individuals and the potential as a diagnostic marker is being investigated.139 Examination of adenomatous and carcinomatous tissues demonstrated an elevated number of FadA gene copies and high levels of FN-DNA in tumor tissue was associated with increased lymph node metastases and poorer outcome in terms of shorter survival for CRC patients.140, 141
FN is often found in tumor microenvironment, and it seems that the function of infiltrating natural killer cells is inhibited in the presence of FN. The cytotoxicity of NK: natural killer; cells appear to be inhibited through a direct interaction between the Fap2 protein produced by FN and the inhibitory immune receptor TIGIT: T cell immunoreceptor with Ig and immunoreceptor tyrosine-based inhibition motifs; present on all human NK cells and various T cells. A recent study identifies how the tumors exploit the Fap2 protein of FN to evade the immune system and inhibit immune cell activity via TIGIT.
FN potentiates CRC tumorigenesis in APCmin mice and represents a typical driver bacterium. FN cannot colonize the colon on its own but needs the interaction of several other species to establish colonies, which in turn support the growth of peptostreptococcus and porphyromonas. Furthermore, FN is implicated in other diseases like rheumatoid arthritis and liver cirrhosis.81, 138, 142
Enterotoxigenic Bacteroides fragilis
BF: Bacteroides Fragilis; is a common anaerobic commensal in most humans representing approx. 1 percent of the microbiota. A subgroup of BF is the ETBF, a common cause of diarrhea in children. ETBF has only one recognized virulence factor, the ETBF-toxin or fragilysin being a 20kDa zink-dependant metalloproteinase toxin binding to epithelial receptors on colonocytes.7
Fragilysin rapidly alters structure and function of epithelial cells including cleavage of E-cadherin thereby increasing mucosal permeability and cytokine secretion, upregulation of Wnt signaling, NF-κB activation, cell proliferation and DNA damage. In vivo and in vitro experimental models together with early human data support that ETBF may promote colon carcinogenesis.6, 49, 143 Small amounts of Fragilysin can be demonstrated in up to 40% of healthy adults. Persistent colonization with ETBF may result in a subclinical IL-17 dominant colitis with concommitant STAT3 activation. Increased amounts of ETBF and Fragilysin have been demonstrated in 100% of advanced CRC cases 144, 145. In Min mice (Apc min/+), colonizaton with ETBF rapidly resulted in adenoma formation and visible colon tumors already after only one month. Thus, ETBF may represent the optimal bacterial driver for CRC.49
Escherischia coli
E. coli is a facultative anaerobic commensal bacteria belonging to the proteobacteria, not extremely abundant in the colon but easily cultivable. There are several phylogroups of which the B2 strain is pathogenic and often involved in IBD and CRC. The B2 strain harbors a genomic island called “pks” codings for the production of a polyketide-peptide genotoxin, Colibactin.146
Colibactin is able to penetrate the cell membrane of colonocytes and translocate to the cell nucleus and function as a DNAse causing double strand DNA breaks, cell cycle arrest and incomplete DNA repair, all of which will lead to chromosome aberrations. Micronuclei, aneuploidy, ring chromosomes, and anaphase bridges persisted in dividing cells up to 21 days after infection, indicating occurrence of DNA breaks and chromosomal instability. The infected cells showed significant mutation frequency demonstrating the mutagenic and transforming potential of E. coli type B2.147, 148
Germfree mice have very few IgA producing cells in their intestinal mucosa. IgA is important for the exclusion of pathogens and neutralization of toxins. If these mice are colonized with either E. coli type B2 or E. faecalis, they both develop inflammation, but only those colonized with E. coli type B2 developed colon tumors characterizing colibactin as a carcinogenetic bacterial metabolite.143
E. coli phylotype B2 harboring the pks island represents another perfect driver bacteria and colonization with the B2 strain may contribute to the development of sporadic CRC. The B2 strain and colibactin represents the overall current strongest candidate for bacterial contribution to CRC carcinogenesis.49, 149
Porphyromonas
PM: Porphyromonas; compromises the integrity of the epithelial cell layer, allowing for invasion and colonization. Once inside the host cell, PM inhibit cell apoptosis by inhibiting cytochrome C release from mitochondria and downregulating of caspase 3 activity. Also, there is upregulation of anti-apoptosis genes encoding Bcl-2 and survivin as well as changes in the level and phosphorylation status of cyclins, p53 and PI3K controlling the cell cycle. Furthermore, it appears that PM may increase TNFα and nitric oxide synthetase expression. Taken together, the actions of PM lead to a pro-tumorigenic inflammatory environment.150, 151
Bacterial metabolites: Short chain fatty acids and secondary bile acids
Probiotics produce SCFA such as butyrate, acetate, propionate and valerate. SCFA provide colonocytes with fuel and suppress cancer and inflammation through several pathways: depressing anti-apoptotic Bcl-2, converting procaspase to active caspase, irreversible binding to mutagens such as nitrosamines and hydrogen peroxide, cell cycle regulation and induction of autophagy. Inflammation is primarily suppressed by inhibiting IL-2,-6,-10,-12, TNFα and NFκB. Probiotics contribute to the barrier function of the mucosa by maintenance and assembly of tight junctions and increasing mucin production.152
Other microbial metabolites, e.g. SBA promote carcinogenesis. Butyrate and SCFA are both thoroughly investigated in relation to inflammation and CRC carcinogenesis. Beneficial effects of SCFAs are listed in table 2. From bacterial breakdown of dietary fiber SCFA has the capability of influencing host gene expression by a number of different routes and also modulate host inflammatory response, among others by histone-deactylase inhibitor affecting gene expression. The Bacteroidetes and Firmicutes phylae produce large amounts of acetate and propionate, whereas Bifidobacteriae are the main source of butyrate production.153
Beneficial effects of SCFA produced by probiotics
| Predominant energy source for colonocytes |
| Induction of mucin synthesis |
| Augmentation of tight junction assembly |
| Mediation of cross-talk between commensals and host immune system for maintenance of gut homeostasis |
| Conditioning gut epithelial cells to mount protective immunity through MAP kinase signaling |
| Inhibiting pro-inflammatory cytokines, NFκB and TNFα |
| Inactivation of mutagens |
Besides liberating beneficial SCFA, fermentation of amino acids produces several harmful compounds potentially playing roles in CRC and IBD. Compounds like ammonia, phenols, p-cresol, certain amines and H2S, may take part in the initiation or progression of cancer through chronic inflammation and DNA damage.53 Butyrate possesses anti-tumorigenic properties; among others the induction of IL-8 in the colonic epithelium, driving T cell differentiation of naive T-naive cells to regulatory T cells and reduction of tumorigenic ILs and metalloproteinase activity.36, 152, 154
A reduction of butyrate producing bacteria results in an increase in intra-colonic pH values creating a hostile environment for colonocytes. From the cecum to the rectum the pH increases contributing to increased susceptibility for tumorigenesis in the left colon and rectum.48
The PBA: primary bile acids; cholic acid and chenodeoxycholic acid, produced from cholesterol in hepatocytes, are conjugated with taurine or glycine to increase solubility and impermeability critical for the formation of micelles. SBA: secondary bile acids; primarily lithocholic acid and DOC: deoxycholic acid; are a very potent bacterial metabolites produced by commensal bacteria by deconjugation (dehydroxylation) of PBA.155, 156
High-fat diets increase formation of SBA and may induce significant changes in the composition and function of the microbiota together with changes in the gut immune system. Repeatedly exposure of colnocytes to increased levels of DOC induced resistance to apoptosis.157 Exposure of colonocytes to high concentrations of DOC induces formation of ROS and RNS and the primary carcinogenic actions of SBA are through ROS, NF-kappa-B activation and direct DNA damage.158
Ridlon et al. propose mechanisms by which microbial metabolism of taurocholate (whose formation is favored by diets high in animal protein) may promote CRC through sulfide formation from taurine and via the tumor-promoting activity of DOC.159
Animal studies
Based on the initial composition of the microbiota in an animal study using the inflammation based model (AOM/DSS) and a pattern recognition algorithm, it was possible to predict subsequent tumor development and even time antibiotic treatment to arrest/revert carcinogenesis. Eight different bacterial community structures were generated by different antibiotic treatments prior to AOM injection and despite a non-significant reduction in the bacterial load due to the antibiotics they varied in the ability to drive tumorigenesis.6, 36
When gnotobiotic mice are colonized with microbiota from mice with CRC of chronic inflammation the number of tumors increases.36 However, IL-10 and p53 knockout mice do not develop CRC under germfree conditions. If colonized with feces of healthy mice tumorigenesis is significantly reduced.6 In the AOM model using DSS as inflammatory agent, mice do not develop tumors if they receive antibiotics as well as antibody blockade of IL-17. 106
In conclusion, animal studies have shown that perturbating the microbiota may halt carcinogenesis and that the microbiota interacts with the host to impact tumor susceptibility. A tumor-associated microbiota transferred to germ-free mice resulted in increased tumor development when compared to healthy mice.
Chemotherapy
Microbiota may also influence tumorigenesis in a positive manner providing a critical boost to antitumoral T cell responses. Some chemotherapeutics (e.g. Oxaliplatin) even rely on the differentiation gene MyD88: Myeloid Differentiation Primary Response 88 gene; signalling triggered by microbes. The MyD88 encodes a cytosolic adapter protein playing a central role in the innate and adaptive immune response. The gene product functions as a signal transducer in the interleukin-1 and Toll-like receptor signaling pathways. Both pathways regulate the activation of numerous proinflammatory genes. The protein consists of an N-terminal death domain and a C-terminal Toll-interleukin1 receptor domain.
Efficacy of immunotherapy is found to rely on commensal bacteria such as bifidobacteria and bacteroides providing augmented dendritic cell functions, enhanced CD8+ T cell priming and accumulation in the tumour microenvironment.75
Clinical trials have demonstrated lack of effect of EGFR antibodies in patients with mCRC from right-sided primaries irrespective of their RAS-status and one could speculate these results are related to the biofilm formation found in these patients.32, 34
Animal trials have shown that a normal functioning microbiota is required for optimal effect of both oxaliplatin and cyclophosphamide160, 161 and in vitro studies showed prolonged and increased response to 5-fluorouracil, a fluoropyrimidine, in the presence of a healthy microbiota composition. Capecitabine and TAS-102 (tipiracil hydrochloride) are both fluoropyrimidines used for systemic treatment in colorectal cancer patients.162
In theory, this could in part explain some cases of lack of effect of chemotherapy (e.g. Oxaliplatin in rectal cancer) due to preoperative radiotherapy followed by a quite extensive surgical trauma and frequent use of antibiotics postoperatively. Consequently, the use of antibiotics in cancer patients should be administered with care as several chemotherapeutics may depend on a functional microbiota. Increasing production of SCFA and modulating the microbiota towards a health-promoting profile by increasing lactic acid bacteria may prove beneficial during recovery from surgery and chemotherapy.75, 163, 164
Targeting the microbiota may also improve the effect on certain chemotherapeutics, e.g. inhibiting β-glucoronidase produced by gut bacteria prevents the metabolism of the topoisomerase-inhibitor Irinotecan and the administration of Bifidobacteria enhances anti-tumor immunity partly by increasing anti-PD-L1 (check-point blockade) efficacy.164, 165
Obesity, inflammation and cancer
Obesity rates are on the rise in most parts of the world and increase with age and lower educational attainment. In Europe, 16% of adults are obese and 11% of CRC cases have been attributed to obesity. In the US, obesity constitutes the fifth leading risk for overall mortality in the US and is associated with a number of diseases.164, 166
Obesity (BMI > 30) is often combined with widespread low-grade inflammation in many organs and tissues, especially the adipose tissue. Obese individuals are known to harbor different types of microbiota in terms of phylum-level changes, reduced bacterial diversity and altered representation of bacterial genes and metabolic pathways. Although an obesity-related decrease in the ratio of Bacteroidetes to Firmicutes has been demonstrated, it remains controversial if this is a general obesity trait. Epidemiological data associate obesity to a 30-70 percent increased risk of CRC.160
Obesity may also be associated with worse outcome, e.g. recurrence and mortality. Several factors, including reduced sensitivity to antiangiogenic-therapeutic regimens, might explain these differences. Except for wound infection, obesity has no significant impact on surgical procedures. The underlying mechanisms linking obesity to CRC are still a matter of debate, but metabolic syndrome, insulin resistance and modifications in levels of adipocytokines seem to be of great importance. NFκB could be a factor linking obesity, type-2 diabetes and cancer.53, 167, 168
Conclusion
Dysbiosis generates a proinflammatory response and a self-reinforcing hostile environment in the colonic mucosa. It is unlikely that dysplasia and malignant transformation is related to only a few microbes but rather to a polymicrobial change in microbiota. Convincing data show that microbiota is deeply involved in colorectal carcinogenesis and the understanding of the microbial processes, especially with regard to interpreting the crosstalk between the microbes and the host, will help us explore the manipulation of the microbiota in the prevention and treatment of CRC through diets, pre-, pro- and antibiotics including next-generation antibiotics of quorum sensing inhibitors and biofilm inhibitors.
Biofilms are advanced microbial arrangements or higher-order structures that may enhance or accelerate dysplasia and malignant tissue transformation. A biofilm invading the colonic mucus layer indicates a pathologic state. The recent discovery of colonic biofilms in patients with right-sided colon cancer adds to the concept that colorectal cancer may arise as a result of the actions of microorganisms and therefore may, at least in theory, be regarded as an infectious disease. Data on biofilm formation in patients with CRC may change screening practice to also include biopsies from the right colon and examination (FISH, electron microscopy) for biofilm if results are confirmed in larger trials. Biofilm-positive individuals maybe should be offered the same attention and follow-up as adenoma patients. New biofilm inhibitors may change the course for CRC the same way antibiotics did for Helicobacter Pylori and gastric cancer.
Probiotics and their SCFA seem to be a safe tool in conjunction with conventional treatment of CRC and already one clinical trial has demonstrated beneficial effects of probiotics for CRC patients including shorter hospital stay and lower infection rate.89
A fecal or microbiota transplant (MT) to restore a healthy microbiota is an interesting concept in CRC but has still not been elucidated in CRC. MT is a simple, inexpensive and presumably safe way to manipulate the microbiota. MT is effective in recurrent or chronic clostridial infections.169 Effective components of MT still need to be identified and there is no consensus as to the optimal route of MT (naso-duodenal or rectal), composition or amount of the optimal MT.An optimal donor profile also has to be defined and the donor screened for contagious diseases. To our knowledge, there are no reported adverse effects of MT, but until we know more about benefits and potential harms of MT authorities are reluctant to approve it. With the current speed of pharmacological and microbiological research, these issues are likely to be solved in the near future. An ongoing study aims to detect a bacterial profile that predicts if patients with mCRC will respond to systemic treatment and/or experience chemotoxicity. If a favorable microbiota composition can be identified, MT is a better alternative to halt/reduce or switch chemotherapy.162
In the future, targeting the microbiota will probably be a powerful weapon in the battle against CRC. The combination of gut microbiology with new genomic and metabolomics data promises to uncover more important linkages between microbial metabolites and health in the future.
In affluent and modern societies, the microbiota and the relation between microbiota and host is dramatically changing as a consequence of lifestyle, environment and diet, especially processed food, increase in carbohydrate intake, lack of exercise and obesity.
Research and increasing knowledge will provide better understanding of the impacts of diet, obesity and inter-individual variation and should reveal new avenues for disease prevention. In addition, prebiotics, probiotics and microbiota transplantation may restore microbial homeostasis, reduce the toxic effects of certain microbes, inflammation, mitogenic and antiapoptotic pathways and even antibiotics may contribute to the efficacy of chemotherapeutics in cancer patients.
Lastly, new classes of antibiotics like quorum sensing inhibitors and biofilm inhibitors may also become prominent tools in terms of changing the virulence and resistance of certain bacterial drivers and the course of bacterial driven carcinogenesis.
Abbreviations
AOM: azoxymethane; APC: adenomatous polyposis coli; AREG: amphiregulin; BA: bile acids; BF: bacteroides fragilis; CDT: cytolethal distending toxin; CIS: chromosomal instability; CRC: colorectal cancer; DOC: deoxycholic acid; DSS: dextran sodium sulphate; EREG: epiregulin; ETBF: Enterotoxic bacteroides fragilis; ETBFT: Enterotoxic bacteroides fragilis toxin; FAP: familial adenomatous polyposis; FISH: fluorescence in situ hybridization; FOBT: fecal occult blood test; FN: fusobacterium nucleatum; HNPCC: Hereditary non-polyposis colorectal cancer; IBD: inflammatory bowel disease; IBS: irritable bowel syndrome; IL: interleukin; MAMP: microorganism-associated molecular pattern; MAPK: mitogen-activated protein kinase; MIS: microsatellite instability; MMR: mismatch repair; NF-kB: nuclear factor kappa-light-chain-enhancer of activated B cells; NLR: NOD-like receptor; NOC: N-nitroso compounds; NOD: nucleotide-binding oligomerization domain-like receptor; NSAID: non-steroidal anti-inflammatory drugs; PAMP: pathogen-associated molecular pattern; PBA: primary bile acids; PM: porphyromonas; PRR: pattern recognition receptor; RNS: reactive nitrogen species; ROS: reactive oxygen species; SBA: secondary bile acids; SCFA: short chain fatty acids; STAT3: signal transducer and activator of transcription 3; TGF: transforming growth factor; Th cells: T helper cells; TIGIT: T cell immunoreceptor with Ig and immunoreceptor tyrosine-based inhibition motifs; TLR: toll-like receptor; TNF: tumor necrosis factor; UC: ulcerative colitis; VEGF: vascular endothelial growth factor; Wnt: wingless-related integration site.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Carethers JM, Stoffel EM. Lynch syndrome and Lynch syndrome mimics: The growing complex landscape of hereditary colon cancer. World J Gastroenterol. 2015;21:9253-61
2. Waller A, Findeis S, Lee MJ. Familial Adenomatous Polyposis. Journal of pediatric genetics. 2016;5:78-83
3. Snover DC. Update on the serrated pathway to colorectal carcinoma. Hum Pathol. 2011;42:1-10
4. Kedrin D, Gala MK. Genetics of the serrated pathway to colorectal cancer. Clinical and translational gastroenterology. 2015;6:e84
5. Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759-67
6. Zackular JP, Baxter NT, Chen GY, Schloss PD. Manipulation of the Gut Microbiota Reveals Role in Colon Tumorigenesis. mSphere. 2016:1
7. Sun J, Kato I. Gut microbiota, inflammation and colorectal cancer. Genes & Diseases. 2016;3:130e43
8. Sinha R, Ahn J, Sampson JN, Shi J, Yu G, Xiong X. et al. Fecal Microbiota, Fecal Metabolome, and Colorectal Cancer Interrelations. PLoS One. 2016;11:e0152126
9. Schertzer JW, Whiteley M. Microbial communication superhighways. Cell. 2011;144:469-70
10. Russo E, Taddei A, Ringressi MN, Ricci F, Amedei A. The interplay between the microbiome and the adaptive immune response in cancer development. Therap Adv Gastroenterol. 2016;9:594-605
11. Raskov H, Burcharth J, Pommergaard HC, Rosenberg J. Irritable bowel syndrome, the microbiota and the gut-brain axis. Gut microbes. 2016;7:365-83
12. Marx V. Cell communication: stop the microbial chatter. Nature. 2014;511:493-7
13. Jurjus A, Eid A, Al Kattar S, Zeenny MN, Gerges-Geagea A, Haydar H. et al. Inflammatory bowel disease, colorectal cancer and type 2 diabetes mellitus: The links. BBA clinical. 2016;5:16-24
14. Flemer B, Lynch DB, Brown JM, Jeffery IB, Ryan FJ, Claesson MJ. et al. Tumour-associated and non-tumour-associated microbiota in colorectal cancer. Gut. 2016
15. Borges-Canha M, Portela-Cidade JP, Dinis-Ribeiro M, Leite-Moreira AF, Pimentel-Nunes P. Role of colonic microbiota in colorectal carcinogenesis: a systematic review. Rev Esp Enferm Dig. 2015;107:659-71
16. Collins SM, Bercik P. The relationship between intestinal microbiota and the central nervous system in normal gastrointestinal function and disease. Gastroenterology. 2009;136:2003-14
17. Vogtmann E, Hua X, Zeller G, Sunagawa S, Voigt AY, Hercog R. et al. Colorectal Cancer and the Human Gut Microbiome: Reproducibility with Whole-Genome Shotgun Sequencing. PLoS One. 2016;11:e0155362
18. Tjalsma H, Boleij A, Marchesi JR, Dutilh BE. A bacterial driver-passenger model for colorectal cancer: beyond the usual suspects. Nature reviews Microbiology. 2012;10:575-82
19. Bultman SJ. Emerging roles of the microbiome in cancer. Carcinogenesis. 2014;35:249-55
20. Wang T, Cai G, Qiu Y, Fei N, Zhang M, Pang X. et al. Structural segregation of gut microbiota between colorectal cancer patients and healthy volunteers. The ISME journal. 2012;6:320-9
21. Sobhani I, Tap J, Roudot-Thoraval F, Roperch JP, Letulle S, Langella P. et al. Microbial dysbiosis in colorectal cancer (CRC) patients. PLoS One. 2011;6:e16393
22. Shen XJ, Rawls JF, Randall T, Burcal L, Mpande CN, Jenkins N. et al. Molecular characterization of mucosal adherent bacteria and associations with colorectal adenomas. Gut microbes. 2010;1:138-47
23. Kostic AD, Gevers D, Pedamallu CS, Michaud M, Duke F, Earl AM. et al. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. 2012;22:292-8
24. Geng J, Fan H, Tang X, Zhai H, Zhang Z. Diversified pattern of the human colorectal cancer microbiome. Gut Pathog. 2013;5:2
25. Chen W, Liu F, Ling Z, Tong X, Xiang C. Human intestinal lumen and mucosa-associated microbiota in patients with colorectal cancer. PLoS One. 2012;7:e39743
26. Chen HM, Yu YN, Wang JL, Lin YW, Kong X, Yang CQ. et al. Decreased dietary fiber intake and structural alteration of gut microbiota in patients with advanced colorectal adenoma. Am J Clin Nutr. 2013;97:1044-52
27. Ahn J, Sinha R, Pei Z, Dominianni C, Wu J, Shi J. et al. Human gut microbiome and risk for colorectal cancer. J Natl Cancer Inst. 2013;105:1907-11
28. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87-108
29. Marley AR, Nan H. Epidemiology of colorectal cancer. International journal of molecular epidemiology and genetics. 2016;7:105-14
30. Howlader N, Noone AM, Krapcho M, Miller D, Bishop K, Kosary CL. et al. SEER Cancer Statistics Review, 1975-2014, National Cancer Institute. Bethesda, MD, https://seer.cancer.gov/csr/1975_2014/, based on November 2016 SEER data submission, posted to the SEER website, April. 2017
31. Karakas Y, Dizdar O. Tumor Sidedness and Prognosis in Colorectal Cancer: Is Microbiome the Missing Link?. JAMA oncology. 2017
32. Tejpar S, Stintzing S, Ciardiello F, Tabernero J, Van Cutsem E, Beier F. et al. Prognostic and Predictive Relevance of Primary Tumor Location in Patients With RAS Wild-Type Metastatic Colorectal Cancer: Retrospective Analyses of the CRYSTAL and FIRE-3 Trials. JAMA oncology. 2016
33. Venook AP. Right-sided vs left-sided colorectal cancer. Clin Adv Hematol Oncol. 2017;15:22-4
34. Dejea CM, Wick EC, Hechenbleikner EM, White JR, Mark Welch JL, Rossetti BJ. et al. Microbiota organization is a distinct feature of proximal colorectal cancers. Proc Natl Acad Sci U S A. 2014;111:18321-6
35. Li S, Konstantinov SR, Smits R, Peppelenbosch MP. Bacterial Biofilms in Colorectal Cancer Initiation and Progression. Trends Mol Med. 2017;23:18-30
36. Yamamoto M, Matsumoto S. Gut microbiota and colorectal cancer. Genes and environment: the official journal of the Japanese Environmental Mutagen Society. 2016;38:11
37. Dyson JK, Rutter MD. Colorectal cancer in inflammatory bowel disease: what is the real magnitude of the risk? World J Gastroenterol. 2012;18:3839-48
38. Hawk ET, Levin B. Colorectal cancer prevention. J Clin Oncol. 2005;23:378-91
39. Cao Y, Wu K, Mehta R, Drew DA, Song M, Lochhead P. et al. Long-term use of antibiotics and risk of colorectal adenoma. Gut. 2017
40. Barnes DE, Lindahl T. Repair and genetic consequences of endogenous DNA base damage in mammalian cells. Annu Rev Genet. 2004;38:445-76
41. Pancione M, Remo A, Colantuoni V. Genetic and epigenetic events generate multiple pathways in colorectal cancer progression. Patholog Res Int. 2012;2012:509348
42. Khare S, Verma M. Epigenetics of colon cancer. Methods Mol Biol. 2012;863:177-85
43. Lang T, Maitra M, Starcevic D, Li SX, Sweasy JB. A DNA polymerase beta mutant from colon cancer cells induces mutations. Proc Natl Acad Sci U S A. 2004;101:6074-9
44. Richardson RB. p53 mutations associated with aging-related rise in cancer incidence rates. Cell cycle. 2013;12:2468-78
45. Shin YJ, Chen KY, Sayed AH, Hencey B, Shen X. Post-translational regulation enables robust p53 regulation. BMC Syst Biol. 2013;7:83
46. Wang S, El-Deiry WS. Requirement of p53 targets in chemosensitization of colonic carcinoma to death ligand therapy. Proc Natl Acad Sci U S A. 2003;100:15095-100
47. Blanpain C. Tracing the cellular origin of cancer. Nat Cell Biol. 2013;15:126-34
48. Raskov H, Pommergaard HC, Burcharth J, Rosenberg J. Colorectal carcinogenesis-update and perspectives. World J Gastroenterol. 2014;20:18151-64
49. Sears CL, Garrett WS. Microbes, microbiota, and colon cancer. Cell host & microbe. 2014;15:317-28
50. Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C. et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59-65
51. Kostic AD, Howitt MR, Garrett WS. Exploring host-microbiota interactions in animal models and humans. Genes Dev. 2013;27:701-18
52. Sender R, Fuchs S, Milo R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLoS Biol. 2016;14:e1002533
53. Marchesi JR, Adams DH, Fava F, Hermes GD, Hirschfield GM, Hold G. et al. The gut microbiota and host health: a new clinical frontier. Gut. 2016;65:330-9
54. Koropatkin NM, Cameron EA, Martens EC. How glycan metabolism shapes the human gut microbiota. Nature reviews Microbiology. 2012;10:323-35
55. Gagniere J, Raisch J, Veziant J, Barnich N, Bonnet R, Buc E. et al. Gut microbiota imbalance and colorectal cancer. World J Gastroenterol. 2016;22:501-18
56. Rowland IR. The role of the gastrointestinal microbiota in colorectal cancer. Curr Pharm Des. 2009;15:1524-7
57. Rosenberg E, Zilber-Rosenberg I. Do microbiotas warm their hosts? Gut microbes. 2016;7:283-5
58. Flint HJ, Bayer EA, Rincon MT, Lamed R, White BA. Polysaccharide utilization by gut bacteria: potential for new insights from genomic analysis. Nature reviews Microbiology. 2008;6:121-31
59. Peterson LW, Artis D. Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nat Rev Immunol. 2014;14:141-53
60. Zhao R, Michor F. Patterns of proliferative activity in the colonic crypt determine crypt stability and rates of somatic evolution. PLoS Comput Biol. 2013;9:e1003082
61. Wilson TJ, Ponder BA, Wright NA. Use of a mouse chimaeric model to study cell migration patterns in the small intestinal epithelium. Cell Tissue Kinet. 1985;18:333-44
62. Potten CS, Kellett M, Roberts SA, Rew DA, Wilson GD. Measurement of in vivo proliferation in human colorectal mucosa using bromodeoxyuridine. Gut. 1992;33:71-8
63. David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE. et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014;505:559-63
64. Plaza-Diaz J, Gomez-Llorente C, Fontana L, Gil A. Modulation of immunity and inflammatory gene expression in the gut, in inflammatory diseases of the gut and in the liver by probiotics. World J Gastroenterol. 2014;20:15632-49
65. Maukonen J, Saarela M. Human gut microbiota: does diet matter? Proc Nutr Soc. 2015;74:23-36
66. Gritz EC, Bhandari V. The human neonatal gut microbiome: a brief review. Frontiers in pediatrics. 2015;3:17
67. Hugon P, Lagier JC, Robert C, Lepolard C, Papazian L, Musso D. et al. Molecular studies neglect apparently gram-negative populations in the human gut microbiota. J Clin Microbiol. 2013;51:3286-93
68. Rajilic-Stojanovic M, Smidt H, de Vos WM. Diversity of the human gastrointestinal tract microbiota revisited. Environmental microbiology. 2007;9:2125-36
69. Zoetendal EG, Rajilic-Stojanovic M, de Vos WM. High-throughput diversity and functionality analysis of the gastrointestinal tract microbiota. Gut. 2008;57:1605-15
70. Ley RE, Peterson DA, Gordon JI. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell. 2006;124:837-48
71. Clemente JC, Ursell LK, Parfrey LW, Knight R. The impact of the gut microbiota on human health: an integrative view. Cell. 2012;148:1258-70
72. Peleg AY, Hooper DC. Hospital-acquired infections due to gram-negative bacteria. N Engl J Med. 2010;362:1804-13
73. Abdallah Ismail N, Ragab SH, Abd Elbaky A, Shoeib AR, Alhosary Y, Fekry D. Frequency of Firmicutes and Bacteroidetes in gut microbiota in obese and normal weight Egyptian children and adults. Arch Med Sci. 2011;7:501-7
74. Dominguez-Bello MG, Blaser MJ, Ley RE, Knight R. Development of the human gastrointestinal microbiota and insights from high-throughput sequencing. Gastroenterology. 2011;140:1713-9
75. Drewes JL, Housseau F, Sears CL. Sporadic colorectal cancer: microbial contributors to disease prevention, development and therapy. Br J Cancer. 2016;115:273-80
76. Johansson ME, Phillipson M, Petersson J, Velcich A, Holm L, Hansson GC. The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc Natl Acad Sci U S A. 2008;105:15064-9
77. Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M. et al. Diversity of the human intestinal microbial flora. Science. 2005;308:1635-8
78. Mira-Pascual L, Cabrera-Rubio R, Ocon S, Costales P, Parra A, Suarez A. et al. Microbial mucosal colonic shifts associated with the development of colorectal cancer reveal the presence of different bacterial and archaeal biomarkers. J Gastroenterol. 2015;50:167-79
79. Claesson MJ, Cusack S, O'Sullivan O, Greene-Diniz R, de Weerd H, Flannery E. et al. Composition, variability, and temporal stability of the intestinal microbiota of the elderly. Proc Natl Acad Sci U S A. 2011;108(Suppl 1):4586-91
80. Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI. The human microbiome project. Nature. 2007;449:804-10
81. Flynn KJ, Baxter NT, Schloss PD. Metabolic and Community Synergy of Oral Bacteria in Colorectal Cancer. mSphere. 2016:1
82. Grenham S, Clarke G, Cryan JF, Dinan TG. Brain-gut-microbe communication in health and disease. Front Physiol. 2011;2:94
83. Hong SN, Rhee PL. Unraveling the ties between irritable bowel syndrome and intestinal microbiota. World J Gastroenterol. 2014;20:2470-81
84. Smythies LE, Maheshwari A, Clements R, Eckhoff D, Novak L, Vu HL. et al. Mucosal IL-8 and TGF-beta recruit blood monocytes: evidence for cross-talk between the lamina propria stroma and myeloid cells. J Leukoc Biol. 2006;80:492-9
85. Winter SE, Baumler AJ. Dysbiosis in the inflamed intestine: chance favors the prepared microbe. Gut microbes. 2014;5:71-3
86. Bischoff SC, Barbara G, Buurman W, Ockhuizen T, Schulzke JD, Serino M. et al. Intestinal permeability-a new target for disease prevention and therapy. BMC Gastroenterol. 2014;14:189
87. Lin L, Zhang J. Role of intestinal microbiota and metabolites on gut homeostasis and human diseases. BMC Immunol. 2017;18:2
88. Kauppila JH, Karttunen TJ, Saarnio J, Nyberg P, Salo T, Graves DE. et al. Short DNA sequences and bacterial DNA induce esophageal, gastric, and colorectal cancer cell invasion. APMIS. 2013;121:511-22
89. Gao R, Gao Z, Huang L, Qin H. Gut microbiota and colorectal cancer. Eur J Clin Microbiol Infect Dis. 2017;36:757-69
90. Philpott DJ, Sorbara MT, Robertson SJ, Croitoru K, Girardin SE. NOD proteins: regulators of inflammation in health and disease. Nat Rev Immunol. 2014;14:9-23
91. Song X, Gao H, Lin Y, Yao Y, Zhu S, Wang J. et al. Alterations in the microbiota drive interleukin-17C production from intestinal epithelial cells to promote tumorigenesis. Immunity. 2014;40:140-52
92. Tian Y, Li Y, Hu Z, Wang D, Sun X, Ren C. Differential effects of NOD2 polymorphisms on colorectal cancer risk: a meta-analysis. Int J Colorectal Dis. 2010;25:161-8
93. Wlodarska M, Thaiss CA, Nowarski R, Henao-Mejia J, Zhang JP, Brown EM. et al. NLRP6 inflammasome orchestrates the colonic host-microbial interface by regulating goblet cell mucus secretion. Cell. 2014;156:1045-59
94. Nabatov AA. The vesicle-associated function of NOD2 as a link between Crohn's disease and mycobacterial infection. Gut Pathog. 2015;7:1
95. Caruso R, Warner N, Inohara N, Nunez G. NOD1 and NOD2: signaling, host defense, and inflammatory disease. Immunity. 2014;41:898-908
96. Piche T. Tight junctions and IBS-the link between epithelial permeability, low-grade inflammation, and symptom generation? Neurogastroenterol Motil. 2014;26:296-302
97. Kennedy PJ, Cryan JF, Dinan TG, Clarke G. Irritable bowel syndrome: a microbiome-gut-brain axis disorder? World J Gastroenterol. 2014;20:14105-25
98. Dantzer R, O'Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9:46-56
99. Mager LF, Wasmer MH, Rau TT, Krebs P. Cytokine-Induced Modulation of Colorectal Cancer. Front Oncol. 2016;6:96
100. Klampfer L. Cytokines, inflammation and colon cancer. Curr Cancer Drug Targets. 2011;11:451-64
101. AS K, GV M, SS K, DH W, M Y, DB K. Intercellular signaling and the multiplication of prokaryotes: bacterial cytokines. Symp Soc Gen Microbiol. 1999;57:33-69
102. Mlecnik B, Bindea G, Angell HK, Maby P, Angelova M, Tougeron D. et al. Integrative Analyses of Colorectal Cancer Show Immunoscore Is a Stronger Predictor of Patient Survival Than Microsatellite Instability. Immunity. 2016;44:698-711
103. Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T. et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature. 2014;515:577-81
104. Galon J, Mlecnik B, Bindea G, Angell HK, Berger A, Lagorce C. et al. Towards the introduction of the 'Immunoscore' in the classification of malignant tumours. J Pathol. 2014;232:199-209
105. Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C. et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313:1960-4
106. Wu S, Rhee KJ, Albesiano E, Rabizadeh S, Wu X, Yen HR. et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat Med. 2009;15:1016-22
107. de Kivit S, Tobin MC, Forsyth CB, Keshavarzian A, Landay AL. Regulation of Intestinal Immune Responses through TLR Activation: Implications for Pro- and Prebiotics. Front Immunol. 2014;5:60
108. Chae WJ, Gibson TF, Zelterman D, Hao L, Henegariu O, Bothwell AL. Ablation of IL-17A abrogates progression of spontaneous intestinal tumorigenesis. Proc Natl Acad Sci U S A. 2010;107:5540-4
109. Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y. et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science. 2011;331:337-41
110. Mei Z, Liu Y, Liu C, Cui A, Liang Z, Wang G. et al. Tumour-infiltrating inflammation and prognosis in colorectal cancer: systematic review and meta-analysis. Br J Cancer. 2014;110:1595-605
111. Morikawa T, Baba Y, Yamauchi M, Kuchiba A, Nosho K, Shima K. et al. STAT3 expression, molecular features, inflammation patterns, and prognosis in a database of 724 colorectal cancers. Clin Cancer Res. 2011;17:1452-62
112. Belluco C, Nitti D, Frantz M, Toppan P, Basso D, Plebani M. et al. Interleukin-6 blood level is associated with circulating carcinoembryonic antigen and prognosis in patients with colorectal cancer. Ann Surg Oncol. 2000;7:133-8
113. Fleming NI, Jorissen RN, Mouradov D, Christie M, Sakthianandeswaren A, Palmieri M. et al. SMAD2, SMAD3 and SMAD4 mutations in colorectal cancer. Cancer Res. 2013;73:725-35
114. Biswas S, Chytil A, Washington K, Romero-Gallo J, Gorska AE, Wirth PS. et al. Transforming growth factor beta receptor type II inactivation promotes the establishment and progression of colon cancer. Cancer Res. 2004;64:4687-92
115. Calon A, Espinet E, Palomo-Ponce S, Tauriello DV, Iglesias M, Cespedes MV. et al. Dependency of colorectal cancer on a TGF-beta-driven program in stromal cells for metastasis initiation. Cancer Cell. 2012;22:571-84
116. Wouters MM, Vicario M, Santos J. The role of mast cells in functional GI disorders. Gut. 2016;65:155-68
117. Martinez C, Gonzalez-Castro A, Vicario M, Santos J. Cellular and molecular basis of intestinal barrier dysfunction in the irritable bowel syndrome. Gut and liver. 2012;6:305-15
118. Elsenbruch S. Abdominal pain in Irritable Bowel Syndrome: a review of putative psychological, neural and neuro-immune mechanisms. Brain Behav Immun. 2011;25:386-94
119. Balkwill F. Tumour necrosis factor and cancer. Nat Rev Cancer. 2009;9:361-71
120. Stanilov N, Miteva L, Dobreva Z, Stanilova S. Colorectal cancer severity and survival in correlation with tumour necrosis factor-alpha. Biotechnology, biotechnological equipment. 2014;28:911-7
121. Al Obeed OA, Alkhayal KA, Al Sheikh A, Zubaidi AM, Vaali-Mohammed MA, Boushey R. et al. Increased expression of tumor necrosis factor-alpha is associated with advanced colorectal cancer stages. World J Gastroenterol. 2014;20:18390-6
122. Gordon JW, Shaw JA, Kirshenbaum LA. Multiple facets of NF-kappaB in the heart: to be or not to NF-kappaB. Circ Res. 2011;108:1122-32
123. Hawinkels LJ, Paauwe M, Verspaget HW, Wiercinska E, van der Zon JM, van der Ploeg K. et al. Interaction with colon cancer cells hyperactivates TGF-beta signaling in cancer-associated fibroblasts. Oncogene. 2014;33:97-107
124. Singh S, Singh SK, Chowdhury I, Singh R. Understanding the Mechanism of Bacterial Biofilms Resistance to Antimicrobial Agents. The open microbiology journal. 2017;11:53-62
125. Kostakioti M, Hadjifrangiskou M, Hultgren SJ. Bacterial biofilms: development, dispersal, and therapeutic strategies in the dawn of the postantibiotic era. Cold Spring Harb Perspect Med. 2013;3:a010306
126. Omar A, Wright JB, Schultz G, Burrell R, Nadworny P. Microbial Biofilms and Chronic Wounds. Microorganisms. 2017:5
127. Guilhen C, Forestier C, Balestrino D. Biofilm dispersal: multiple elaborate strategies for dissemination of bacteria with unique properties. Mol Microbiol. 2017
128. Okuda T, Kokubu E, Kawana T, Saito A, Okuda K, Ishihara K. Synergy in biofilm formation between Fusobacterium nucleatum and Prevotella species. Anaerobe. 2012;18:110-6
129. Finlay BB, McFadden G. Anti-immunology: evasion of the host immune system by bacterial and viral pathogens. Cell. 2006;124:767-82
130. Swidsinski A, Loening-Baucke V, Herber A. Mucosal flora in Crohn's disease and ulcerative colitis - an overview. J Physiol Pharmacol. 2009;60(Suppl 6):61-71
131. Brannon JR, Hadjifrangiskou M. The arsenal of pathogens and antivirulence therapeutic strategies for disarming them. Drug Des Devel Ther. 2016;10:1795-806
132. Doran KS, Banerjee A, Disson O, Lecuit M. Concepts and mechanisms: crossing host barriers. Cold Spring Harb Perspect Med. 2013:3
133. Weiss G, Schaible UE. Macrophage defense mechanisms against intracellular bacteria. Immunol Rev. 2015;264:182-203
134. Geng J, Song Q, Tang X, Liang X, Fan H, Peng H. et al. Co-occurrence of driver and passenger bacteria in human colorectal cancer. Gut Pathog. 2014;6:26
135. Vogtmann E, Goedert JJ. Epidemiologic studies of the human microbiome and cancer. Br J Cancer. 2016;114:237-42
136. Zeller G, Tap J, Voigt AY, Sunagawa S, Kultima JR, Costea PI. et al. Potential of fecal microbiota for early-stage detection of colorectal cancer. Mol Syst Biol. 2014;10:766
137. McCoy AN, Araujo-Perez F, Azcarate-Peril A, Yeh JJ, Sandler RS, Keku TO. Fusobacterium is associated with colorectal adenomas. PLoS One. 2013;8:e53653
138. Kostic AD, Chun E, Robertson L, Glickman JN, Gallini CA, Michaud M. et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell host & microbe. 2013;14:207-15
139. Rubinstein MR, Wang X, Liu W, Hao Y, Cai G, Han YW. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/beta-catenin signaling via its FadA adhesin. Cell host & microbe. 2013;14:195-206
140. Mima K, Nishihara R, Qian ZR, Cao Y, Sukawa Y, Nowak JA. et al. Fusobacterium nucleatum in colorectal carcinoma tissue and patient prognosis. Gut. 2015
141. Castellarin M, Warren RL, Freeman JD, Dreolini L, Krzywinski M, Strauss J. et al. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 2012;22:299-306
142. Gur C, Ibrahim Y, Isaacson B, Yamin R, Abed J, Gamliel M. et al. Binding of the Fap2 protein of Fusobacterium nucleatum to human inhibitory receptor TIGIT protects tumors from immune cell attack. Immunity. 2015;42:344-55
143. Arthur JC, Perez-Chanona E, Muhlbauer M, Tomkovich S, Uronis JM, Fan TJ. et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science. 2012;338:120-3
144. Sears CL, Islam S, Saha A, Arjumand M, Alam NH, Faruque AS. et al. Association of enterotoxigenic Bacteroides fragilis infection with inflammatory diarrhea. Clin Infect Dis. 2008;47:797-803
145. Boleij A, Hechenbleikner EM, Goodwin AC, Badani R, Stein EM, Lazarev MG. et al. The Bacteroides fragilis toxin gene is prevalent in the colon mucosa of colorectal cancer patients. Clin Infect Dis. 2015;60:208-15
146. Veziant J, Gagniere J, Jouberton E, Bonnin V, Sauvanet P, Pezet D. et al. Association of colorectal cancer with pathogenic Escherichia coli: Focus on mechanisms using optical imaging. World J Clin Oncol. 2016;7:293-301
147. Cuevas-Ramos G, Petit CR, Marcq I, Boury M, Oswald E, Nougayrede JP. Escherichia coli induces DNA damage in vivo and triggers genomic instability in mammalian cells. Proc Natl Acad Sci U S A. 2010;107:11537-42
148. Cougnoux A, Dalmasso G, Martinez R, Buc E, Delmas J, Gibold L. et al. Bacterial genotoxin colibactin promotes colon tumour growth by inducing a senescence-associated secretory phenotype. Gut. 2014;63:1932-42
149. Secher T, Samba-Louaka A, Oswald E, Nougayrede JP. Escherichia coli producing colibactin triggers premature and transmissible senescence in mammalian cells. PLoS One. 2013;8:e77157
150. Kuboniwa M, Hasegawa Y, Mao S, Shizukuishi S, Amano A, Lamont RJ. et al. P. gingivalis accelerates gingival epithelial cell progression through the cell cycle. Microbes and infection. 2008;10:122-8
151. Posocco D, Dmitrieva O, Grivennikov SI. Microbiome Implications in Intestinal Tumorigenesis. In: Yang VW, Bialkowska AB (Eds.). Intestinal Tumorigenesis. Springer International Publishing. 2015 eBook ISBN 978-3-319-19986-3 DOI 10.1007/978-3-319-19986-3. Hardcover ISBN 978-3-319-19985-6
152. Sharma M, Shukla G. Metabiotics: One Step ahead of Probiotics; an Insight into Mechanisms Involved in Anticancerous Effect in Colorectal Cancer. Frontiers in microbiology. 2016;7:1940
153. Artis D. Epithelial-cell recognition of commensal bacteria and maintenance of immune homeostasis in the gut. Nat Rev Immunol. 2008;8:411-20
154. Singh N, Gurav A, Sivaprakasam S, Brady E, Padia R, Shi H. et al. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity. 2014;40:128-39
155. Camilleri M. Bile Acid diarrhea: prevalence, pathogenesis, and therapy. Gut and liver. 2015;9:332-9
156. Appleby RN, Walters JR. The role of bile acids in functional GI disorders. Neurogastroenterol Motil. 2014;26:1057-69
157. Bernstein C, Holubec H, Bhattacharyya AK, Nguyen H, Payne CM, Zaitlin B. et al. Carcinogenicity of deoxycholate, a secondary bile acid. Arch Toxicol. 2011;85:863-71
158. Ajouz H, Mukherji D, Shamseddine A. Secondary bile acids: an underrecognized cause of colon cancer. World J Surg Oncol. 2014;12:164
159. Ridlon JM, Wolf PG, Gaskins HR. Taurocholic acid metabolism by gut microbes and colon cancer. Gut microbes. 2016;7:201-15
160. Viaud S, Saccheri F, Mignot G, Yamazaki T, Daillere R, Hannani D. et al. The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science. 2013;342:971-6
161. Iida N, Dzutsev A, Stewart CA, Smith L, Bouladoux N, Weingarten RA. et al. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science. 2013;342:967-70
162. Aarnoutse R, de Vos-Geelen J, Penders J, Boerma EG, Warmerdam F, Goorts B. et al. Study protocol on the role of intestinal microbiota in colorectal cancer treatment: a pathway to personalized medicine 2.0. Int J Colorectal Dis. 2017;32:1077-84
163. Vetizou M, Pitt JM, Daillere R, Lepage P, Waldschmitt N, Flament C. et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science. 2015;350:1079-84
164. Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM. et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science. 2015;350:1084-9
165. Pennisi E. Biomedicine. Cancer therapies use a little help from microbial friends. Science. 2013;342:921
166. Eurostat Press Office. Almost 1 adult in 6 in the EU is considered obese. Eurostat Newsrelease 203/2016. 20 October 2016. http://ec.europa.eu/eurostat/documents/2995521/7700898/3-20102016-BP-EN.pdf/
167. Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE. et al. A core gut microbiome in obese and lean twins. Nature. 2009;457:480-4
168. Bardou M, Barkun AN, Martel M. Obesity and colorectal cancer. Gut. 2013;62:933-47
169. van Nood E, Vrieze A, Nieuwdorp M, Fuentes S, Zoetendal EG, de Vos WM. et al. Duodenal infusion of donor feces for recurrent Clostridium difficile. N Engl J Med. 2013;368:407-15
Author contact
![]() Corresponding author: Hans Raskov, MD., Lundevangsvej 23, DK-2900 Hellerup, Denmark Phone: +45 2441 4031 Email: raskovdk
Corresponding author: Hans Raskov, MD., Lundevangsvej 23, DK-2900 Hellerup, Denmark Phone: +45 2441 4031 Email: raskovdk