Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(16):3296-3308. doi:10.7150/jca.19810 This issue Cite
Research Paper
Association between Mismatch-repair Genetic variation and the Risk of Multiple Primary Cancers: A Meta-Analysis
Pengfei Kong1, 2*, Ruiyan Wu1*, Yadong Lan3*, Wenzhuo He1, 2*, Chenlu Yang1, 4, Chenxi Yin1, 5, Qiong Yang1, 2, Chang Jiang1, 2, Dazhi Xu1, 6, Liangping Xia1, 2 ![]()
1. State Key Laboratory of Oncology in South China, Collaborative Innovation Centre for Cancer Medicine, Guangzhou, 510000, PR China;
2. Department of the VIP region, Sun Yat-sen University Cancer Centre, Guangzhou, 510000, PR China;
3. Department of Oncological Surgery, the Second People's Hospital of Lu-an City, Lu-an, 237000, PR China;
4. Department of Gynecology, Guangdong Women and Children Hospital, Guangzhou, 510010, PR China;
5. Department of the ICU, Sun Yat-sen University Cancer Centre, Guangzhou, 510000, PR China;
6. Department of Gastric and Pancreatic Surgery, Sun Yat-sen University Cancer Centre, Guangzhou, 510000, PR China.
* Equal contributors.
Received 2017-2-25; Accepted 2017-8-17; Published 2017-9-16
Abstract
Microsatellites instability (MSI) is a risk factor for multiple primary cancers (MPCs). However, a variety of studies focused on the risk in the hereditary non-polyposis colorectal cancer (HNPCC) not the sporadic colorectal cancer (CRC) patients. The aim of this meta-analysis was to comprehensive overview and quantitative summary the association between MSI and risk of MPCs. A comprehensive literature search in MEDLINE, EMBASE, Web of science, ScienceDirect, Weily and OVID was conducted. Up to May 2016, we identified 22 observational studies. We calculated the summary relative risk (RR) for the risk of MPCs in MSI patients compared with microsatellites stability (MSS) patients using fixed- or random-effects models. The RR of the association between mismatch-repair gene (MMR) genotype and MPCs was 2.59 (95% confidence interval [CI], 2.06 to 3.27); the RR was 2.14 (95% CI, 1.78 to 2.57) for sporadic CRC and 5.59 (95% CI, 2.69 to 11.59) for HNPCC for the MSI versus MSS category. The subgroup analyses showed different mutant gene, mutant locus, and mutant level of MMR with different influence on the patients susceptible to MPCs. In addition, MSI genotype increase the risk of MPC was not associated with an apparently specific in regard to site, timing, age and detection method. In conclusion, this meta-analysis indicates that MSI is associated with an increased risk of MPCs both in the HNPCC and sporadic CRC patients. Our findings will form the backbone of the treatment for MSI genotype may be an important valuable strategy for MPCs prevention.
Keywords: microsatellites instability, multiple Primary Cancers, risk factor, meta-analysis.
Introduction
DNA duplication is an accurate process and often generates incorrect base-pairing (base-base mismatches) or unmatched DNA loops (insertion-deletion loops), which needs a DNA repair system to provides a mechanism for removing these changes and maintaining genomic stability [1]. One of the repair system is the mismatch-repair gene (MMR), which is composed of a series of MMR proteins including mutL homolog 1 (MLH1), mutL homolog 3 (MLH3), mutS homolog 2 (MSH2), mutS homolog 3 (MSH3), mutS homolog 6 (MSH6), postmeiotic segregation increased 1 (PMS1), and postmeiotic segregation increased 2 (PMS2) [2, 3]. Microsatellites, short (1-6 base pairs) and tandem repeated sequences that are scattered throughout the genome, are extremely sensitive to the replication errors [4]. Commonly, the alterations in the length of these tandem nucleotide repeats results from deficient-MMR (dMMR) known as microsatellites instability (MSI) [5].As deficiencies occur in these MMR genes, the mistakes generated during the process of DNA synthesis cannot be timely corrected, and will eventually trigger genetic or epigenetic events [6, 7].
Consistent with the current thinking, cancer can be defined as a gene disease, and therefore MSI might be one of the potential mechanism responsible for particularly gene mutation and subsequently cancer development [8]. One of the most famous example is hereditary non-polyposis colorectal cancer (HNPCC),which is collectively known as germ line mutation in mismatch-repair(MMR) gene and preferentially induces early onset of colorectal cancer (CRC) as well as cancer of the endometrium, pancreas stomach, ovary and urogenital synchronously or metachronously [9]. It has generally been reported that MSI is detected in 90% of cancers from patients with HNPCC [10], which accounts for 1~2% of all CRC [11]. Besides, it should be also noted that MSI were account for 12% ~17% of sporadic colorectal cancer [12, 13] as well as in a subset of several other types of sporadic cancer with quite high frequencies, like gastric cancer (22%) [14], ovarian cancer (10%) [15], endometrial cancer (22%-33%) [16, 17], esophageal adenocarcinoma (7%) [18], head and neck (3%) [19], renal cell carcinoma (2%) [20].
Since 2000s, advances in cancer diagnosis and treatment and consequently improved CRC overall five-year survival rate, about 10% of cancer patients, even if completely cured. We noted, however, that approximately 20% or more CRC patients are at high risk of developing a secondary cancer, and the prognostic outcome of multiple primary cancers (MPCs) patients remains ominous [21-23], highlighting intensively prevention and quick diagnosis of secondary cancers are urgently needed. In addition, to date, very little is known about a reliable marker to identify these patients. With these considerations in mind, we systematically synthesized data from all existing studies to evaluate the role of MSI as a marker for CRC patient developing multiple primary cancers, especially for sporadic CRC.
In this meta-analysis, we reviewed 47898 researches from the systematic literature search, analyzed a total of 11399 cancer patients with MSI status. Overall, our study clearly indicates that CRC patients with MSI have an increased risk of MPCs compared with the patients with microsatellites stability (MSS), and this effect is independent of the HNPCC or sporadic CRC. Here, our findings also provide the first evidence that the detailed association between mutant gene, mutant locus, and level of MSI on MMR status and MPCs risk. Further, we explicitly demonstrated that no significance site and timing specific in MPCs resulted from MSI.
Materials and Methods
Identification of studies
Related studies before May 31, 2016 were extensively searched using following data bases: Medline (PubMed), Web of science, ScienceDirect, Weily, OVID, and Embase databases. Using following key words: (“gastric cancer” or “stomach cancer” or “colorectal cancer” or “ovarian cancer”) combined with (“MSI” or “microsatellite instability” or “mismatch repair” or “MMR” or “MLH1” or “MSH2” or “MSH6” or “PMS2”) (for detail search terms see Supplementary Table S1 and Supplementary Table S2). In addition, reference lists of all retrieved articles were searched manually for additional potentially relevant studies. No restriction was applied except for the language in English. Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines was performed in the process of this study.
Eligibility criteria
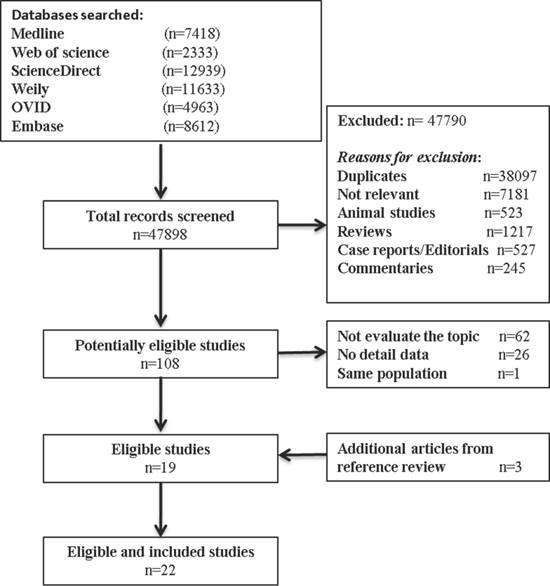
Our inclusion criteria for article were as follows: (1) report the occurrence of MSI (mutant of gene MLH1, MSH2, MSH6, PMS2); (2) measured the occurrence of double/MPCs (exclude metastasis); (3) randomized clinical trials (RCTs), cohort studies, or case-control studies. The exclusion criteria included the following: (1) animal studies, review articles, case reports, editorials, commentaries, and duplicate studies; (2) articles without solitary primary cancer as control groups; and (3) if the study utilized the same population or overlapping database. If necessary, we contacted the authors for the detail or additional unpublished data. The entire process of study of study identification is summarized in Figure 1.
Flow diagram summarizing study identification and selection.
Data extraction and assessment
Two reviewers independently scanned each eligible article to obtain and confirm data using a standardized form. Any inconsistencies between two reviewers were settled by a third investigator. The following information were extracted: first author, publication year, country, study period, number of patients, tumor site, MSI detection method, incidence rate of MSI, incidence rate of double/MPCs, MSS in patients with multiple and solitary cancer, RRs with corresponding 95% CIs for the risk of MPCs MSI group compared with MSS group, and adjusted or matched variables. The Newcastle-Ottawa scale was performed to assess the quality of the included study (Supplementary Table S3) [24].
Statistical analysis
Relative risk (RR) and corresponding 95% confidence interval (CIs) from each study were calculated, and heterogeneity between individual studies was assessed by χ2 test and I2 test; p<0.05 and/or I2>50% indicated significant heterogeneity. The choice of effects model depended on the significant of heterogeneity: the fixed-effect model with Mantel-Haenszel method for insignificant heterogeneity while the random one was applied when the heterogeneity was significant. To identify sources of between-studies heterogeneity, subgroup analyses were further conducted according age, gender, sites of cancer, MSI status (MSI-H and MSI-L), MSI detection method (PCR and IHC), mutant gene (MLH1, MSH2, MSH6 and PMS2), mutant locus (BAT25, BAT26, BAT40, D2S123, D2S136, D3S1067, D5S346, D11S922 and D17S250) and adjustments for covariates. Additionally, sensitivity analysis was also performed to dissect the heterogeneity. As our previous study described, to explore the publication bias risk, funnel plots and Egger's test method were used.[25, 26] Two-sided p values were calculated, with a p value <0.05 defined significant. All statistical analyses were conducted using the Stata software (V.19.0; Stata Corp, College Station, Texas, USA).
Result
Search results and study characteristics
Our search process for eligible studies was shown in Figure 1. We obtained initial 47898 article candidates from the systematic literature search. Of those, 38,097 were duplicates, 9,693 were excluded based on the title and abstract: 7,181 not relevant, 523 animal studies; 1217 reviews; 527 case reports or editorials; and 245 commentaries. The retained 108 articles were further evaluated by reading the full-text, from which 89 were excluded: 62 not focus on our topic; 26 no detail data; and 1 same population. At the end, 19 eligible studies qualified the criterion in the present meta-analysis. In addition, 3 other studies met the inclusion criterion were identified by manual searching the reference lists of articles. Finally, 22 studies including 11,399 cancer patients with MSI status were included in our meta-analysis.
The general characteristics of the enrolled studies are presented in Table 1 and Supplementary Table S4. Twenty of the included studies were hospital case-control studies [21, 22, 27-45] and the rest were cohort studies [23, 46]. Most articles reported the method of MSI detection except two [38, 46], and the percentage of MSI ranging from 7.4% to 36.8% [29, 33]. PCR assay were the major way to detect MSI status, only one study used IHC alone for hMSH2/hMLH1 [34]. In addition, the enrolled studies also reported the percentage of MPCs, ranging from 2.7% to 60.0% [23, 33]. In the whole enrolled studies, except 2 studies only can obtained in abstract form [27, 28] and three other studies [29, 31, 45], all included studies were mostly regarded as high quality (Supplementary Table S3).
Characteristics of included studies.
| Author/Year | MSI detection method | MSI% | Multiple primary cancers% |
|---|---|---|---|
| Horri/1994 | PCR | 25.0 | 17.9 |
| Brown/1998 | PCR | 16.2 | 44.5 |
| Kiriu/1998 | PCR | 7.4 | 29.6 |
| Abe/2000 | PCR | 28.2 | 39.1 |
| Kim HS/2001 | PCR | 12.5 | 14.2 |
| Mark/2002 | PCR | 11.3 | 8.7 |
| Kim YH/2005 | PCR | 16.0 | 9.2 |
| Lawes/2005 | PCR IHC | 36.8 | 60.0 |
| Yamamoto/2006 | IHC | 23.2 | 44.8 |
| Hayashi/2006 | PCR | 20.3 | 49.5 |
| Yun HR/2009 | PCR | 15.2 | 4.0 |
| Nosho/2009 | PCR | 14.5 | 2.7 |
| Kim SH/2010 | PCR | 10.9 | 3.1 |
| Yoon/2010 | Unconfirmed | 9.8 | 3.3 |
| Bae JM/2012 | PCR IHC | 13.2 | 30.5 |
| Hu H/2013 | PCR IHC | 20.4 | 34.7 |
| Huang/2014 | PCR | 19.5 | 21.8 |
| Lee JW/2014 | Unconfirmed | 12.5 | 4.6 |
| Malesci/2014 | PCR | 10.7 | 5.7 |
| Cho I/2014 | PCR | 8.1 | 5.9 |
| Meng/2015 | PCR | 12.8 | 3.9 |
| Kim YB/2016 | PCR IHC | 14.4 | 8.4 |
Abbreviations: MSI: microsatellites instability, PCR: Polymerase chain reaction, IHC: immunohistochemistry.
MSI status and MPC risk
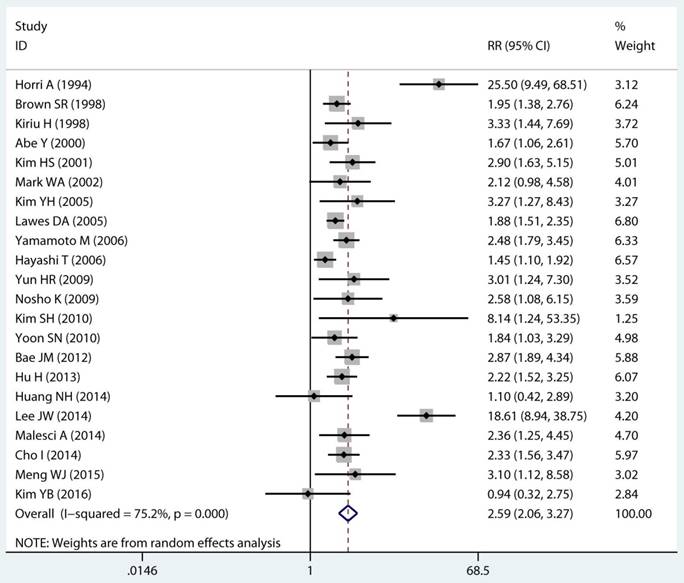
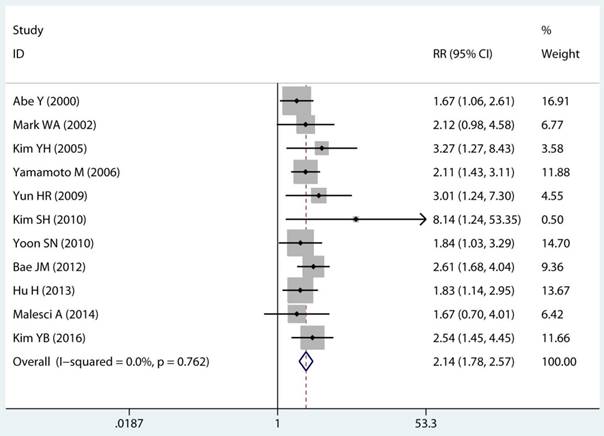
Twenty-two studies investigated the association between MSI status and MPCs risk. The multivariable-adjusted RRs for each study and the combined RR for MSI status and the risk of MPCs are delineated in Figure 2A and Supplementary Figure S1. Among all included studies, 19 showed a statistically significant direct association between MSI status and MPCs risk [21-23, 27-31, 33-40, 43, 45, 46]. Above all, the pooled analysis represented a summary RR of 2.59 (95% CI 2.06 to 3.27) with significant heterogeneity (I2=75.2%, p<0.0001) (Figure 2A). In addition, there were five studies in a relative low quality when using the Newcastle-Ottawa scale system to evaluate or the outcomes lack of effective adjust [27-29, 31, 45]. After excluding the study with relative low quality and unreliable results, the heterogeneity decreased across all studies (I2=70.9%, p<0.0001) and the adjusted RRs were 2.37 (95% CI 1.85 to 3.04) (Supplement Figure S1). Among all high quality study, eleven studies reported the data explore the association between MSI status and MPCs risk in non-HNPCC [21, 22, 30, 32, 34, 36-40, 44]. Meta-analysis of all non-HNPCC related studies elucidated that, among non-HNPCC patients, the MSI status was associated with a statistically significant more than two-fold increment in MPCs incidence (adjusted RR 2.14, 95 % CI 1.78 to 2.57) (Figure 2B). Accordingly, MSI status can significantly augment the risk of MPCs in CRC and non-HNPCC (adjusted RR 5.59, 95 % CI 2.69 to 11.59) (Supplementary Figure S2).
Forest plot of MMR status and risk of MPCs (MSI vs MSS). (A) Overall and (B) Non-HNPCC. MMR: mismatch-repair, MPCs: multiple primary cancers, MSI, microsatellites instability, MSS: microsatellite stable, HNPCC: hereditary non-polyposis colorectal cancer, CI, confidence interval.
Mutant gene, mutant locus, and level of MSI on MSI status and MPC risk
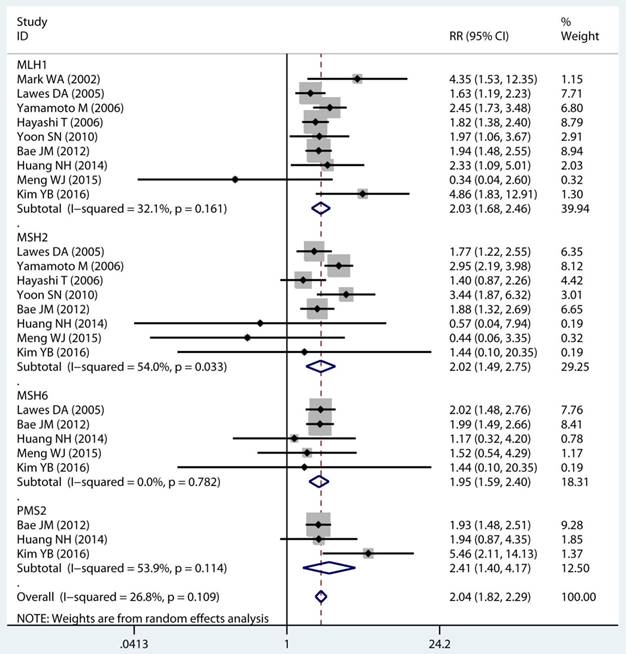
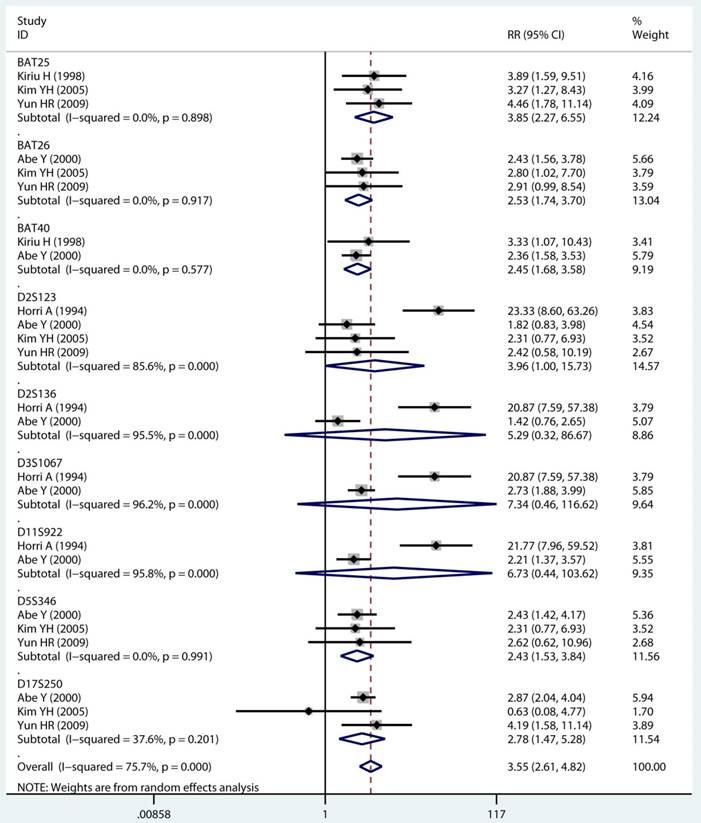
As displayed in Table 2, the details of mutant gene, mutant locus, and level of MSI was divided into 4, 9, and 2 subgroups, respectively. Firstly, among subgroup analyses stratified by mutant gene types, studies on MLH1 (adjusted RR 1.97, 95 % CI 1.69 to 2.30) [32-35, 38, 39, 41, 43, 44], studies on MSH2 (adjusted RR 2.02, 95 % CI 1.49 to 2.75) [33-35, 38, 39, 41, 43, 44], studies on MSH6 (adjusted RR 1.78, 95 % CI 1.33 to 2.38) [30, 33, 39, 41, 44], and studies on PMS2 (adjusted RR 2.41, 95 % CI 1.40 to 4.17) [39, 41, 44]. Secondly, in mutant locus subgroup, adjusted RR= 3.81 (95% CI 2.20 to 6.62) for BAT25 mutant [21, 29, 36], adjusted RR= 2.63 (95% CI 1.71 to 4.06) for BAT26 mutant [21, 30, 36], adjusted RR= 2.44 (95% CI 1.67 to 3.56) for BAT40 mutant [29, 30], adjusted RR= 3.96 (95% CI 1.00 to 15.73) for D2S123 mutant [21, 27, 30, 36], adjusted RR= 5.29 (95% CI 0.32 to 86.67) for D2S136 mutant [27, 30], adjusted RR= 7.34 (95% CI 0.46 to 116.62) for D3S1067 mutant [27, 30], adjusted RR= 2.42 (95% CI 1.38 to 4.23) for D5S346 mutant [21, 30, 36], adjusted RR= 6.73 (95% CI 0.44 to 103.62) for D11S922 mutant [27, 30], and adjusted RR= 2.57 (95% CI 1.71 to 3.87) for D17S250 mutant [21, 30, 36]. Lastly, we also divided the level of MSI status into MSI-L and MSI-H group. Notably, we observed a clearly trend of increasing risk of MPCs highly correlated with MMR status. For the detail, in the MSI-H group [21, 23, 27, 29, 30, 33, 35, 44], adjusted RR= 2.19 (95% CI 1.36 to 3.52). Similarly, in the MSI-L group [21, 27, 29, 35, 44], adjusted RR= 2.91 (95% CI 1.06 to 7.98). Furthermore, we summarized the above-mentioned results and presented with intact forest plots in Figure 3 A, Figure 3B, and Supplementary Figure S3. In our study, because of significant heterogeneity in several subgroup, we all used the random-effect model in the total landscape figure. Together, it was unraveled a different MPCs risk increment for different mutant gene, mutant locus, and MSI scale.
Mutant gene, mutant locus, and level of MSI on MSI status and multiple primary cancer risk
| Group | NO. of reports | RR (95%) | Heterogeneity test | ||
|---|---|---|---|---|---|
| χ2 | p | I2(%) | |||
| Mutant gene | |||||
| MLH1 | 9 | 1.97 (1.69, 2.30) | 11.78 | 0.161 | 32.10 |
| MSH2 | 8 | 2.02 (1.49, 2.75) | 15.23 | 0.033 | 54.00 |
| MSH6 | 5 | 1.78 (1.33, 2.38) | 1.75 | 0.782 | 0.00 |
| PMS2 | 3 | 2.41 (1.40, 4.17) | 4.34 | 0.114 | 53.90 |
| Mutant locus | |||||
| BAT25 | 3 | 3.81 (2.20, 6.62) | 0.22 | 0.898 | 0.00 |
| BAT26 | 3 | 2.63 (1.71, 4.06) | 0.17 | 0.917 | 0.00 |
| BAT40 | 2 | 2.44 (1.67, 3.56) | 0.31 | 0.577 | 0.00 |
| D2S123 | 4 | 3.96 (1.00, 15.73) | 20.90 | <0.0001 | 85.60 |
| D2S136 | 2 | 5.29 (0.32, 86.67) | 22.10 | <0.0001 | 95.50 |
| D3S1067 | 2 | 7.34 (0.46, 116.62) | 26.29 | <0.0001 | 96.20 |
| D5S346 | 3 | 2.42 (1.38, 4.23) | 0.02 | 0.991 | 0.00 |
| D11S922 | 2 | 6.73 (0.44, 103.62) | 24.09 | <0.0001 | 95.80 |
| D17S250 | 3 | 2.57 (1.71, 3.87) | 3.20 | 0.201 | 37.60 |
| MSI level | |||||
| MSI-H | 8 | 2.19 (1.36, 3.52) | 27.75 | <0.0001 | 74.80 |
| MSI-L | 5 | 2.91 (1.06, 7.98) | 22.93 | <0.0001 | 82.60 |
Abbreviations: NO: number, RR: relative risk, MSI: microsatellites instability, MSI-H: microsatellites instability high, MSI-L: microsatellites instability low.
Forest plot of MMR detailed status and risk of MPCs (MSI vs MSS). (A) Mutant gene and (B) Mutant locus. MMR: mismatch-repair, MPCs: multiple primary cancers, MSI, microsatellites instability, MSS: microsatellite stable, CI, confidence interval.
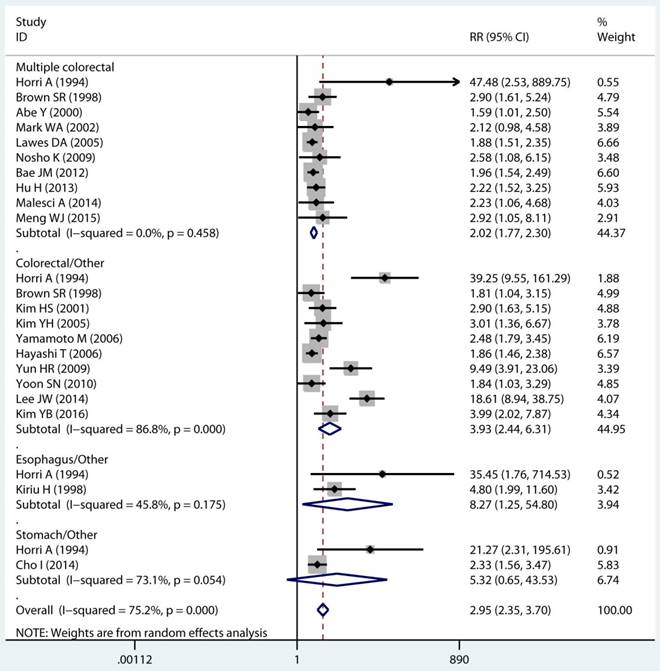
Site and timing specific and MPC risk
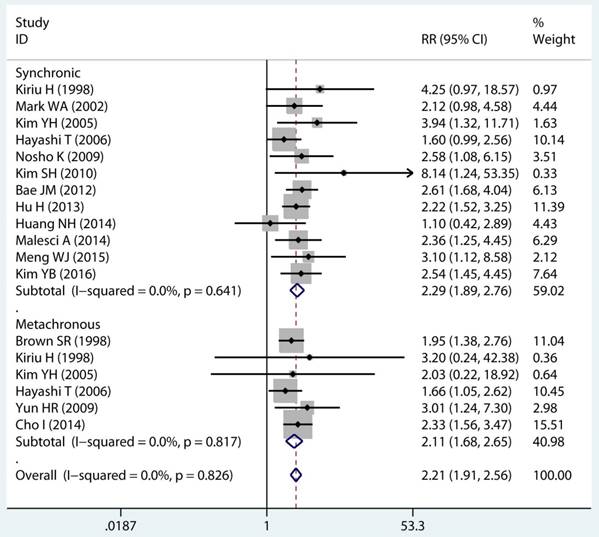
In twenty studies, 1,070 site specific MPCs were identified, which were classified as multiple colorectal [22, 23, 27, 28, 30, 32, 33, 39, 40, 43], colorectal/Other [21, 27, 28, 31, 34-36, 38, 44, 46], esophagus/Other [27, 29], and stomach/Other [27, 45] group. On pooled analysis, it was found MSI genotype was associated with a statistically significant increase in the risk of multiple colorectal cancer (adjusted RR 2.08, 95 % CI 1.82 to 2.39), colorectal-based MPCs (adjusted RR 3.93, 95 % CI 2.44 to 6.31), and esophagus-based MPCs (adjusted RR 7.06, 95 % CI 3.06 to 16.31), though the results with considerable heterogeneity among studies in two subgroup (p heterogeneity < 0.01, I2 >50%) (Table 3) (Figure 4A). Additionally, fifteen studies reporting on the timing type of MPCs were included in the analysis. When all the estimates were pooled, the summary RR in timing type of synchronic [21-23, 29, 32, 35, 37, 39-41, 43, 44] and metachronous [21, 28, 29, 35, 36, 45] from the fix-effects models were 2.29 (95% CI 1.89 to 2.76) and 2.11 (95% CI 1.68 to 2.65), respectively. Significant heterogeneity was not present across studies (p heterogeneity > 0.05, I2 <50%) in two groups (Table 3).
Site and timing specific and multiple primary cancer risk.
| Group | NO. of reports | RR (95%) | Heterogeneity test | |||
|---|---|---|---|---|---|---|
| χ2 | p | I2 (%) | ||||
| Site of cancer | ||||||
| Multiple colorectal | 10 | 2.08 (1.82, 2.39) | 8.77 | 0.458 | 0.00 | |
| Colorectal/Other | 10 | 3.93 (2.44, 6.31) | 68.01 | <0.0001 | 86.80 | |
| Esophagus/Other | 2 | 7.06 (3.06, 16.31) | 1.84 | 0.175 | 45.80 | |
| Stomach/Other | 2 | 5.32 (0.65, 43.53) | 3.71 | 0.054 | 73.10 | |
| Timing of MPC | ||||||
| Synchronic | 12 | 2.29 (1.89, 2.76) | 8.79 | 0.641 | 0.00 | |
| Metachronous | 6 | 2.11 (1.68, 2.65) | 2.22 | 0.817 | 0.00 | |
| Hereditary | ||||||
| HNPCC | 4 | 5.54 (2.42, 12.64) | 5.77 | 0.056 | 65.40 | |
| Non-HNPCC | 13 | 2.13 (1.81, 2.51) | 8.17 | 0.772 | 0.00 | |
Abbreviations: NO: number, RR: relative risk, MPC: multiple primary cancer, HNPCC: hereditary non-polyposis colorectal cancer.
Forest plot of site and timing specific and risk of MPCs (MSI vs MSS). (A) Site specific and (B) Timing specific. MPCs: multiple primary cancers, MSI, microsatellites instability, MSS: microsatellite stable, CI, confidence interval.
Subgroup analysis
Age
We performed preplanned stratified analyses of studies based on patient's age (Table 4). In eight studies reported data of relative young age group (<65 years) [21, 29, 31, 32, 34, 35, 39, 41], MSI was associated with a significant risk increment in incidence of MPCs (adjusted RR 2.15, 95 % CI 1.26 to 3.68), although there was obvious heterogeneity within the group. In addition, pooled analysis of relative old age (≥65 years) subgroup also showed a significant increase in MPCs to be associated with MSI status (adjusted RR 2.06, 95 % CI 1.52 to 2.79) and the outcomes without heterogeneity.
Subgroup analyses of MSI status and multiple primary cancer risk
| Group | NO. of reports | RR (95%) | Heterogeneity test | |||
|---|---|---|---|---|---|---|
| χ2 | p | I2(%) | ||||
| Total | 22 | 2.59 (2.06, 3.27) | 81.70 | <0.0001 | 75.20 | |
| Adjusted | 17 | 2.37 (1.85, 3.04) | 54.96 | <0.0001 | 70.90 | |
| Age | ||||||
| <65 | 8 | 2.15 (1.26, 3.68) | 24.87 | 0.001 | 71.90 | |
| ≥65 | 7 | 2.06 (1.52, 2.79) | 6.60 | 0.360 | 9.10 | |
| Gender | ||||||
| Male | 8 | 2.70 (1.36, 5.34) | 34.30 | <0.0001 | 79.60 | |
| Female | 7 | 3.80 (2.11, 6.84) | 14.17 | 0.028 | 57.70 | |
| Detection method | ||||||
| PCR | 19 | 2.36 (1.93, 2.89) | 46.19 | <0.0001 | 61.00 | |
| IHC | 5 | 2.19 (1.88, 2.55) | 3.22 | 0.521 | 0.00 | |
Abbreviations: MSI: microsatellites instability, NO: number, RR: relative risk, PCR: Polymerase chain reaction, IHC: immunohistochemistry.
Gender
Studies were stratified by gender, The RRs were 2.70 (95% CI 1.36 to 5.34) for studies conducted in male cohorts [21, 29, 31, 32, 34, 37, 39, 40], 3.80 (95% CI 2.11 to 6.84) for studies in female cohorts [21, 31, 32, 34, 37, 39, 40]. These results indicate, in contrast to male, female had a more apparent affinity between MSI and MPCs risk.
Detection method
The possible association between the detection method was reported by twenty studies, five in IHC [33, 34, 39, 40, 44], and nineteen in PCR [21-23, 27-33, 35-37, 39-41, 43-45]. In this subgroup pooled analyses, IHC and PCR group both were yielded statistically significant RRs. The method of IHC for MSI detection, with a pooled RR of 2.19 (95% CI 1.88 to 2.55), was concordance with that for PCR method, with a summary RR of 2.36 (95% CI 1.93 to 2.89).
Sensitivity Analyses and Evaluation of Heterogeneity
Sensitivity analyses were performed to explore possible causes of heterogeneity and the effect of various exclusion criteria on the overall result were examined. Four studies that were not adjusted for age, gender, and location of tumor were omitted [27, 28, 35, 41]. The remaining studies produced an RR of 2.60 (95% CI, 2.11 to3.20), with substantial evidence of decreasing heterogeneity (P=0.001, I2= 59.4%). Restricting analysis to the five studies that were adjusted for family history produced similar results (RR: 1.78, 95% CI: 1.45 to 2.19), and the heterogeneity was significantly decreasing (I2= 28.1%). Further exclusion of any single study did not change the overall outcomes, which ranged from 2.32 (95% CI: 1.94 to 2.77) to 2.70 (95% CI: 2.11-3.46).
Meta-regression analysis demonstrated that study design (P=0.025) and study quality (P=0.005) were significant sources of heterogeneity, but the outcomes indicate that MSI detection method, publication year, and country area were not the main origin of heterogeneity. Study design alone explained 50.16% of the τ2 in the meta-regression analyses and study quality explained 26.94% of the τ2 (Supplementary Table S5).
Publication Bias
The funnel plot showed a detectable asymmetry (Supplementary Figure S4). Additionally, publication bias was detected using the Begg's test (P = 0.037) and Egger's test (P = 0.015) for the overall studies.
Discussion
This meta-analysis is the first to evaluate the association between MMR genotype and the risk of MPCs in CRC patients and in different clinical scenarios. Our results support the concept that patients with MSI have an increased risk of MPCs compared with the patients with MSS, and this effect is independent of the HNPCC or sporadic CRC.
First, our work firstly provides convincing evidence that, compared to the patients with MSS, patients with MSI have an increased risk of MPCs in the sporadic CRC. So far, an increasing body of research has revealed that a diverse tumor types have MSI, and it is estimated that 15% of CRC, which in turn accounts for 180,000 new CRC cases each year worldwide, are closely correlated to MSI [47, 48]. Additionally, MSI is significantly associated with an increased incidence of MPCs. In proven CRC, MMR status is a very important risk and prognostic measurement [49-52]. Despite MSI is a crucial risk factor for MPCs in the sporadic CRC [30, 32], the majority of studies were to determine the association between MSI and MPCs in HNPCC [53-57]. Thus, no reliable systematic data to unravel the mystery that whether MSI can predicts the risk of MPC in sporadic CRC patient. Surprisingly, in our study, we clearly proved MSI can increase the risk of MPCs both in sporadic CRC and HNPCC. Collectively, contrary to the previous studies, our work is unique and have several overt advantages. Firstly, we comprehensively summarized the existing twenty two high quality published studies and rigorously analysed the data with validated robust and powerful performance methods. Therefore, the evidence base in our meta-analysis is relatively credible. Secondly, since the HNPCC patients contribute only a minority of CRC patients, we focused on sporadic CRC, which may offer the greatest potential benefit for cancer prevention. The risk of MPCs in sporadic CRC patients with MSI (2.14-fold) is higher than that of patients with MSS. As a consequence, our results first reveal the need for special screening and surveillance strategies for these patients. Thirdly, our finding suggested immunotherapeutic interventions involving checkpoint blockade might be selectively effective in CRC related MPCs. Recently, pioneering studies by D.T. etal demonstrated that tumors with a large number of somatic mutations due to mismatch-repair defects may be susceptible to immune checkpoint blockade [58, 59]. For CRC, anti-PD-1 therapies revealed an interesting objective response rates results that 40% in dMMR cohorts and 0% in pMMR cohorts. Thus, as the close association between MSI and MPCs was found in our study, the checkpoint blockade immunotherapy might be a useful therapeutic strategy for advanced MPC. Additionally, a previous work have identified advanced MPCs with MSI-H may be associated with better chemo-sensitivity [60].
Second, our data pointed out that different mutant gene, mutant locus, and mutant level of MMR with different influence on the patients susceptible to MPC. Firstly, our pooled analysis result clearly showed that, contrast with other mutant genes, the PMS2 gene mutant more prone to increase risk (2.41-fold) of MPC, and the result with a low heterogeneity (I2=26.8%, p heterogeneity=0.109; Figure 3A). PMS2 mutation, due to its structure and repetitive nature, is difficult to sequence and clinical testing for PMS2 mutations was not available until very recently [61]. In one side, because of uncertainty of previous testing approach, our result may be missed or miscalled due to the presence of pseudogenes. In the other side, currently, several studies showed PMS2 inactive may be more common than the other MMR genes [62, 63]. In addition, McKinsey L and coworkers reported that PMS2 germline mutations were tightly associated with the early-onset of CRC [64]. Combined with our novel findings, suggesting that PMS2 might be a more powerful marker compared with other MMR markers. Secondly, in the mutant locus analysis, mono-nucleotide microsatellite locus BAT25 and BAT26 showed an obviously low adjusted RR compared with other locus (Figure 3B). It has been reported that the rate of spontaneous mutation in microsatellites with multiple-nucleotide repeat units is more frequent than microsatellites with mono-nucleotide repeat units [12, 65]. Taken together, it implies that microsatellite markers have di-, tri-and tetra-nucleotide repeat units may be more effective for indicating MPC. Thirdly, to our surprise, MSI-L group are more sensitive to develop MPC than MSI-H group (adjusted RR: 2.91 vs 2.19). To our knowledge, MSI-L was associated with lower risk of cancer compare with MSI-H. The discrepancy may be result from the significant heterogeneity and system error (I2=78.2%, p heterogeneity<0.001; Figure S2).
Third, in this study, we found that MSI genotype increase the risk of MPC was not associated with an apparently specific in regard to site, timing, age and detection method (Figure 4). For example, in the subgroup analysis of timing specific, we found similar RRs among synchronic and metachronous patients (adjusted RR: 2.29 vs 2.11). Consistently, limited indications for a potential role of site, age and detection method on MPCs were also provided from our subgroup analysis. Given the modest, borderline significant, association and the still limited data, the issue remains therefore open to explore.
Lastly, several limitations of this meta-analysis need to be addressed. First, an obvious publication bias was detected for the overall studies (Begg's test: P = 0.037), which led to inaccuracy of results to some extent. Nevertheless, there was no evidence of publication bias for non-HNPCC studies, which enrolled most of relative new studies (Begg's test: P = 0.100) (Supplementary Figure S5). Moreover, there was also no evidence of publication bias for non-HNPCC studies (Begg's test: P = 0.150) (Supplementary Figure S6). Hence, publication bias may be at least in part explained by publication year and study quality. Second, the overall summary RR with some heterogeneity (I2=75.2%, p heterogeneity<0.001; Figure 2A). To minimize heterogeneity, we kicked out five studies with relative low quality instead of incorporating all studies into meta-analysis. Interestingly, the heterogeneity of our subset analysis declined to 70.9% (p heterogeneity<0.001; Figure S1). Consequently, heterogeneity may partially due to the relative low quality studies enrolled in the meta-analysis. In addition, the included studies varied with different study design, patients and other confounding factors, seemed to provide one explanation for the main source of heterogeneity between studies. Finally, this meta-analysis was conducted only using published data rather than individual data. Therefore, there might be some covariates that could influence the incidence of MPCs or the status of MMR, but were not reported and thus could not be adjusted.
Conclusions
In conclusion, unlike early studies, this meta-analysis is a more comprehensive and better designed study mainly focused on MMR genotype of sporadic CRC patients rather than HNPCC patients. There is clear evidence that MSI is associated with an increased risk of MPCs in the sporadic CRC patients. Further, our study highlighted that different mutant gene, mutant locus, and mutant level of MMR with different influence on the patients susceptible to MPCs. The treatment for MSI genotype may be an important valuable strategy for MPCs prevention.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This study was supported by grants from Natural Science Foundation of Guangdong, China (No. 2015A030313010), Science and Technology Program of Guangzhou, China (No. 1563000305) and National Natural Science Foundation of China (No. 81272641and No. 81572409). The authors thank Dr. Lin Yang and Dr. Qiankun Xie for statistical advising and review of the manuscript.
Authors' Contributions
Conception and design: LPX, DZX, PFK.
Development of methodology: PFK, RYW, YDL, WZH.
Acquisition of data: PFK, RYW, YDL, WZH, CXY, QY, CJ.
Analysis and interpretation of data: PFK, RYW, YDL, WZH, CLY.
Writing, review, and/or revision of the manuscript: PFK, RYW, YDL, WZH.
Administrative, technical, or material support: PFK, RYW, YDL, WZH.
Study supervision: LPX, DZX.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Lord CJ, Ashworth A. The DNA damage response and cancer therapy. Nature. 2012;481:287-94
2. Chung DC, Rustgi AK. DNA mismatch repair and cancer. Gastroenterology. 1995;109:1685-99
3. Hewish M, Lord CJ, Martin SA, Cunningham D, Ashworth A. Mismatch repair deficient colorectal cancer in the era of personalized treatment. Nature reviews Clinical oncology. 2010;7:197-208
4. Li GM. Mechanisms and functions of DNA mismatch repair. Cell research. 2008;18:85-98
5. Chao EC, Lipkin SM. Molecular models for the tissue specificity of DNA mismatch repair-deficient carcinogenesis. Nucleic acids research. 2006;34:840-52
6. Wheeler JM, Bodmer WF, Mortensen NJ. DNA mismatch repair genes and colorectal cancer. Gut. 2000;47:148-53
7. Li SK, Martin A. Mismatch Repair and Colon Cancer: Mechanisms and Therapies Explored. Trends in molecular medicine. 2016;22:274-89
8. Prolla TA. DNA mismatch repair and cancer. Current opinion in cell biology. 1998;10:311-6
9. Lynch HT, Smyrk T. Hereditary nonpolyposis colorectal cancer (Lynch syndrome). An updated review. Cancer. 1996;78:1149-67
10. Percesepe A, Borghi F, Menigatti M, Losi L, Foroni M, Di Gregorio C. et al. Molecular screening for hereditary nonpolyposis colorectal cancer: a prospective, population-based study. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2001;19:3944-50
11. Samowitz WS, Curtin K, Lin HH, Robertson MA, Schaffer D, Nichols M. et al. The colon cancer burden of genetically defined hereditary nonpolyposis colon cancer. Gastroenterology. 2001;121:830-8
12. Boland CR, Goel A. Microsatellite instability in colorectal cancer. Gastroenterology. 2010;138:2073-87
13. Carethers JM, Jung BH. Genetics and Genetic Biomarkers in Sporadic Colorectal Cancer. Gastroenterology. 2015;149:1177-90
14. Cancer Genome Atlas Research N. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202-9
15. Murphy MA, Wentzensen N. Frequency of mismatch repair deficiency in ovarian cancer: a systematic review This article is a US Government work and, as such, is in the public domain of the United States of America. International journal of cancer Journal international du cancer. 2011;129:1914-22
16. Zighelboim I, Goodfellow PJ, Gao F, Gibb RK, Powell MA, Rader JS. et al. Microsatellite instability and epigenetic inactivation of MLH1 and outcome of patients with endometrial carcinomas of the endometrioid type. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2007;25:2042-8
17. Hampel H, Frankel W, Panescu J, Lockman J, Sotamaa K, Fix D. et al. Screening for Lynch syndrome (hereditary nonpolyposis colorectal cancer) among endometrial cancer patients. Cancer research. 2006;66:7810-7
18. Farris AB 3rd, Demicco EG, Le LP, Finberg KE, Miller J, Mandal R. et al. Clinicopathologic and molecular profiles of microsatellite unstable Barrett Esophagus-associated adenocarcinoma. The American journal of surgical pathology. 2011;35:647-55
19. Glavac D, Volavsek M, Potocnik U, Ravnik-Glavac M, Gale N. Low microsatellite instability and high loss of heterozygosity rates indicate dominant role of the suppressor pathway in squamous cell carcinoma of head and neck and loss of heterozygosity of 11q14.3 correlates with tumor grade. Cancer genetics and cytogenetics. 2003;146:27-32
20. Thrash-Bingham CA, Salazar H, Freed JJ, Greenberg RE, Tartof KD. Genomic alterations and instabilities in renal cell carcinomas and their relationship to tumor pathology. Cancer research. 1995;55:6189-95
21. Kim YH, Song SY, Kwon YD, Kim DS, Chun HK, Rhee JC. Microsatellite instable double primary cancers of the colorectum and stomach exhibit less favorable outcome. World journal of gastroenterology. 2005;11:3998-4002
22. Malesci A, Basso G, Bianchi P, Fini L, Grizzi F, Celesti G. et al. Molecular heterogeneity and prognostic implications of synchronous advanced colorectal neoplasia. British journal of cancer. 2014;110:1228-35
23. Nosho K, Kure S, Irahara N, Shima K, Baba Y, Spiegelman D. et al. A prospective cohort study shows unique epigenetic, genetic, and prognostic features of synchronous colorectal cancers. Gastroenterology. 2009;137:1609-20
24. Stang A. Critical evaluation of the Newcastle-Ottawa scale for the assessment of the quality of nonrandomized studies in meta-analyses. European journal of epidemiology. 2010;25:603-5
25. Kong P, Cai Q, Geng Q, Wang J, Lan Y, Zhan Y. et al. Vitamin intake reduce the risk of gastric cancer: meta-analysis and systematic review of randomized and observational studies. PloS one. 2014;9:e116060
26. Kong P, Wu R, Liu X, Liu J, Chen S, Ye M. et al. The Effects of Anti-inflammatory Drug Treatment in Gastric Cancer Prevention: an Update of a Meta-analysis. Journal of Cancer. 2016;7:2247-57
27. Horii A, Han HJ, Shimada M, Yanagisawa A, Kato Y, Ohta H. et al. Frequent replication errors at microsatellite loci in tumors of patients with multiple primary cancers. Cancer research. 1994;54:3373-5
28. Brown SR, Finan PJ, Hall NR, Bishop DT. Incidence of DNA replication errors in patients with multiple primary cancers. Diseases of the colon and rectum. 1998;41:765-9
29. Kiriu H, Yokozaki H, Yasui W, Ito K, Tahara E. Microsatellite instability associated with primary head and neck cancers and secondary esophageal cancers. Japanese journal of clinical oncology. 1998;28:733-9
30. Abe Y, Masuda H. Genetic alterations of sporadic colorectal cancer with microsatellite instability, especially characteristics of primary multiple colorectal cancers. Journal of surgical oncology. 2000;74:249-56
31. Kim HS, Cho NB, Yoo JH, Shin KH, Park JG, Kim YI. et al. Microsatellite instability in double primary cancers of the colorectum and stomach. Modern pathology: an official journal of the United States and Canadian Academy of Pathology, Inc. 2001;14:543-8
32. Norrie MW, Hawkins NJ, Todd AV, Meagher AP, O'Connor TW, Ward RL. The role of hMLH1 methylation in the development of synchronous sporadic colorectal carcinomas. Diseases of the colon and rectum. 2002;45:674-80
33. Lawes DA, Pearson T, Sengupta S, Boulos PB. The role of MLH1, MSH2 and MSH6 in the development of multiple colorectal cancers. British journal of cancer. 2005;93:472-7
34. Yamamoto M, Taguchi K, Baba H, Endo K, Kohnoe S, Okamura T. et al. Loss of protein expression of hMLH1 and hMSH2 with double primary carcinomas of the stomach and colorectum. Oncology reports. 2006;16:41-7
35. Hayashi T, Arai M, Ueno M, Kinoshita H, Tada Y, Koizumi K. et al. Frequency of immunohistochemical loss of mismatch repair protein in double primary cancers of the colorectum and stomach in Japan. Diseases of the colon and rectum. 2006;49:S23-9
36. Yun HR, Yi LJ, Cho YK, Park JH, Cho YB, Yun SH. et al. Double primary malignancy in colorectal cancer patients-MSI is the useful marker for predicting double primary tumors. International journal of colorectal disease. 2009;24:369-75
37. Kim SH, Ahn BK, Nam YS, Pyo JY, Oh YH, Lee KH. Microsatellite instability is associated with the clinicopathologic features of gastric cancer in sporadic gastric cancer patients. Journal of gastric cancer. 2010;10:149-54
38. Yoon SN, Oh ST, Lim SB, Kim TW, Kim JH, Yu CS. et al. Clinicopathologic characteristics of colorectal cancer patients with synchronous and metachronous gastric cancer. World journal of surgery. 2010;34:2168-76
39. Bae JM, Cho NY, Kim TY, Kang GH. Clinicopathologic and molecular characteristics of synchronous colorectal cancers: heterogeneity of clinical outcome depending on microsatellite instability status of individual tumors. Diseases of the colon and rectum. 2012;55:181-90
40. Hu H, Chang DT, Nikiforova MN, Kuan SF, Pai RK. Clinicopathologic features of synchronous colorectal carcinoma: A distinct subset arising from multiple sessile serrated adenomas and associated with high levels of microsatellite instability and favorable prognosis. The American journal of surgical pathology. 2013;37:1660-70
41. Huang HN, Lin MC, Tseng LH, Chiang YC, Lin LI, Lin YF. et al. Ovarian and endometrial endometrioid adenocarcinomas have distinct profiles of microsatellite instability, PTEN expression, and ARID1A expression. Histopathology. 2015;66:517-28
42. Verbeek M, van Bekkum PJ, Dullemans AM, van der Vlugt RA. Torradoviruses are transmitted in a semi-persistent and stylet-borne manner by three whitefly vectors. Virus research. 2014;186:55-60
43. Meng WJ, Yang L, Ma Q, Zhang H, Adell G, Arbman G. et al. MicroRNA Expression Profile Reveals miR-17-92 and miR-143-145 Cluster in Synchronous Colorectal Cancer. Medicine. 2015;94:e1297
44. Kim YB, Lee SY, Kim JH, Sung IK, Park HS, Shim CS. et al. Microsatellite Instability of Gastric and Colorectal Cancers as a Predictor of Synchronous Gastric or Colorectal Neoplasms. Gut and liver. 2016;10:220-7
45. Weinberg DS, Schoen RE. In the clinic. Screening for colorectal cancer. Annals of internal medicine. 2014:160
46. Lee JW, Kim JW, Kim NK. Clinical characteristics of colorectal cancer patients with a second primary cancer. Annals of coloproctology. 2014;30:18-22
47. Peltomaki P. Role of DNA mismatch repair defects in the pathogenesis of human cancer. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2003;21:1174-9
48. Siegel R, DeSantis C, Virgo K, Stein K, Mariotto A, Smith T. et al. Cancer treatment and survivorship statistics, 2012. CA: a cancer journal for clinicians. 2012;62:220-41
49. Lipkin SM, Rozek LS, Rennert G, Yang W, Chen PC, Hacia J. et al. The MLH1 D132H variant is associated with susceptibility to sporadic colorectal cancer. Nature genetics. 2004;36:694-9
50. Hendriks YM, Wagner A, Morreau H, Menko F, Stormorken A, Quehenberger F. et al. Cancer risk in hereditary nonpolyposis colorectal cancer due to MSH6 mutations: impact on counseling and surveillance. Gastroenterology. 2004;127:17-25
51. Koessler T, Oestergaard MZ, Song H, Tyrer J, Perkins B, Dunning AM. et al. Common variants in mismatch repair genes and risk of colorectal cancer. Gut. 2008;57:1097-101
52. Popat S, Hubner R, Houlston RS. Systematic review of microsatellite instability and colorectal cancer prognosis. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2005;23:609-18
53. Park JG, Kim DW, Hong CW, Nam BH, Shin YK, Hong SH. et al. Germ line mutations of mismatch repair genes in hereditary nonpolyposis colorectal cancer patients with small bowel cancer: International Society for Gastrointestinal Hereditary Tumours Collaborative Study. Clinical cancer research: an official journal of the American Association for Cancer Research. 2006;12:3389-93
54. ten Kate GL, Kleibeuker JH, Nagengast FM, Craanen M, Cats A, Menko FH. et al. Is surveillance of the small bowel indicated for Lynch syndrome families? Gut. 2007;56:1198-201
55. Hirai Y, Banno K, Suzuki M, Ichikawa Y, Udagawa Y, Sugano K. et al. Molecular epidemiological and mutational analysis of DNA mismatch repair (MMR) genes in endometrial cancer patients with HNPCC-associated familial predisposition to cancer. Cancer science. 2008;99:1715-9
56. Gylling A, Abdel-Rahman WM, Juhola M, Nuorva K, Hautala E, Jarvinen HJ. et al. Is gastric cancer part of the tumour spectrum of hereditary non-polyposis colorectal cancer? A molecular genetic study. Gut. 2007;56:926-33
57. <breast tumor.pdf>.
58. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD. et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. New England Journal of Medicine. 2015;372:2509-20
59. Llosa NJ, Cruise M, Tam A, Wicks EC, Hechenbleikner EM, Taube JM. et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer discovery. 2015;5:43-51
60. Liang JT, Huang KC, Lai HS, Lee PH, Cheng YM, Hsu HC. et al. High-frequency microsatellite instability predicts better chemosensitivity to high-dose 5-fluorouracil plus leucovorin chemotherapy for stage IV sporadic colorectal cancer after palliative bowel resection. International journal of cancer. 2002;101:519-25
61. Vaughn CP, Baker CL, Samowitz WS, Swensen JJ. The frequency of previously undetectable deletions involving 3' Exons of the PMS2 gene. Genes, chromosomes & cancer. 2013;52:107-12
62. Senter L, Clendenning M, Sotamaa K, Hampel H, Green J, Potter JD. et al. The clinical phenotype of Lynch syndrome due to germ-line PMS2 mutations. Gastroenterology. 2008;135:419-28
63. Wimmer K, Etzler J. Constitutional mismatch repair-deficiency syndrome: have we so far seen only the tip of an iceberg? Human genetics. 2008;124:105-22
64. Goodenberger ML, Thomas BC, Riegert-Johnson D, Boland CR, Plon SE, Clendenning M. et al. PMS2 monoallelic mutation carriers: the known unknown. Genetics in medicine: official journal of the American College of Medical Genetics. 2016;18:13-9
65. Bacher JW, Flanagan LA, Smalley RL, Nassif NA, Burgart LJ, Halberg RB. et al. Development of a fluorescent multiplex assay for detection of MSI-High tumors. Disease markers. 2004;20:237-50
Author contact
![]() Corresponding author: Liangping Xia, MD, PhD, Department of the VIP region, Sun Yat-sen University Cancer Centre, Guangzhou, China, 651# East Dongfeng Road, Guangzhou 510000, Guangdong Province, PR China. Fax: (+86) 020-87343107. E-mail: xialporg.cn.
Corresponding author: Liangping Xia, MD, PhD, Department of the VIP region, Sun Yat-sen University Cancer Centre, Guangzhou, China, 651# East Dongfeng Road, Guangzhou 510000, Guangdong Province, PR China. Fax: (+86) 020-87343107. E-mail: xialporg.cn.