Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(16):3232-3241. doi:10.7150/jca.20833 This issue Cite
Research Paper
Mismatch Repair Protein hMLH1, but not hMSH2, Enhances Estrogen-Induced Apoptosis of Colon Cancer Cells
Peng Jin1*, De-zhi Wang1, 2*, Chen-xi Lyu1, Ya-ting Wang1, Yu-qi He1, Jian-qiu Sheng1 ![]() , Xuhang Li2
, Xuhang Li2 ![]()
1. Department of Gastroenterology, PLA Army General Hospital, Beijing 100700, China.
2. Johns Hopkins University School of Medicine, Department of Medicine/GI Division, Baltimore, MD21205, USA.
* These authors contributed equally to this study.
Received 2017-5-2; Accepted 2017-8-25; Published 2017-9-15
Abstract
Background: Epidemiological studies suggest a protective role of estrogen against colon carcinogenesis; this effect appears to be dependent on mismatch repair (MMR) status. However, the underlying mechanism remains unclear. This study investigated the role of MMR proteins in apoptosis of colon cancer cells in the presence or absence of estrogen.
Methods: Two major MMR proteins, human mutL homolog 1 (hMLH1) and mutS homolog 2 (hMSH2), as well as estrogen receptor-β (ERβ), were transiently expressed in either hMLH1-deficient HCT116 cells or hMSH2-deficient LoVo cells. Effects of estradiol on cell viability and apoptosis were assessed. Furthermore, we examined the apoptotic status of epithelial cells in colonic mucosa taken from previous healthy female subjects with menopausal syndrome before and after 6-month hormone replacement therapy (HRT).
Results: In hMLH1-deficient HCT116 cells, re-expression of hMLH1 led to a significantly decreased cell viability and increased apoptosis, which were further enhanced by estradiol, including marked increase of activated caspase-3 and caspase-9, as well as Bax and P53. The effect of hMLH1 overexpression in LoVo cells resulted in a similar increase in apoptosis that was greatly stimulated by estradiol. The enhanced apoptosis by hMLH1 and estradiol was further validated by FACS analyses of Annexin V expression. Re-expression of hMSH2 or overexpression of ERβ in HCT116 cells also enhanced apoptosis; however, the effects were independent of estradiol. Furthermore, studies on healthy menopausal women before and after 6-month HRT demonstrated a significant HRT-mediated upregulation of the hMLH1 expression, with concomitant elevation of caspase-3 and caspase-9 activation in the colonic mucosa.
Conclusion: We present the first evidence that hMLH1 and hMSH2 have similar but distinct roles in the apoptosis of colon cancer cells: an increased expression of either one can promote apoptosis, while only the effect of hMLH1 but not hMSH2 is estradiol-dependent. Our data suggest that MMR status should be assessed before hormone replacement therapy or future application of estrogen-based chemoprevention.
Keywords: Colon cancer, hMLH1, hMSH2, Estradiol/Estrogen, Mismatch repair (MMR), Apoptosis.
Introduction
Colorectal cancer is one of the most common malignant tumors in China and the Western world. Large-scale population studies have shown that hormone replacement therapy (HRT) significantly reduces the risk of colorectal cancer1. Though benefits of estrogen or progesterone are difficult to separate, the protective effects are seen in some observational studies with estrogen treatment alone2. These findings strongly indicate that estrogen may lower the risk of colorectal cancer. It has been shown in rodents and cell lines that estrogen induces apoptosis and inhibits proliferation of the intestinal epithelial cells3, 4. However, the mechanism by which estrogen enhances apoptosis and prevents colorectal carcinogenesis remains unclear.
The DNA mismatch repair (MMR) system consists of proteins that detect and repair nucleotide base mismatches and slippage mistakes on newly synthesized DNA at microsatellite sequences. It plays a key role in maintaining genomic stability. MMR dysfunction causes microsatellite instability (MSI). It has been reported that postmenopausal hormone replacement therapy reduces risks of only MSI-low/MSI-stable colorectal cancer subtypes, but not of MSI-high tumors5, 6. Moreover, estrogen receptor β (ERβ), the predominant ER in the human colonic epithelium7, has been indicated to have a protective role in colorectal cancer8, 9. These findings suggest that MMR proteins may participate in an estrogen-mediated protective effect on colorectal cancer.
In this study, we investigated the roles of two major MMR proteins, human mutL homolog 1 (hMLH1) and mutS homolog 2 (hMSH2), in the apoptosis of colon cancer cells and the involvement of estrogen in the processes. Two MMR-deficient human colon carcinoma cell lines were used: the hMLH1 deficient HCT116 cells containing a homozygous nonsense mutation at codon 252 (stop codon) in exon 9, and the hMSH2 deficient LoVo cells containing a homozygous deletion in the hMSH2 gene from exon 3 to exon 8 10. The functions of the two MMR proteins in apoptosis were evaluated by overexpression of wild-type cDNA of hMLH1 and hMSH2, respectively, in HCT116 or LoVo in the presence and absence of estrogen. We demonstrated by both Western blot and FACS analysis that hMLH1 promote apoptosis that was further enhanced by estrogen. In addition, overexpression of hMSH2 or ERβ in HCT116 cells also promotes apoptosis, although the effects were estrogen-independent. We provide definite evidence that hMLH1 and hMSH2 exhibit similar yet distinctive role in estrogen-dependent and -independent apoptosis of colon cancer cells. Moreover, our studies of healthy post-menopausal female subjects demonstrated that 6-month HRT led to a significant elevation of hMLH1 expression and a concurrent increase in activation of caspase-3 and caspase-9 in the colonic mucosa.
Materials and Methods
Cell culture, transfection, and estradiol treatments
Both HCT116 and LoVo cells were purchased from ATCC (Manassas, VA, US). HCT116 cells were maintained in phenol red-free Dulbecco's modified Eagle's medium (DMEM, HyClone, UT, USA) containing 10% fetal bovine serum (FBS, HyClone), 100 U/ml Penicillin G and 100 μg/ml Streptomycin Sulphate (Sigma, St. Louis, MO, US); whereas LoVo cells were maintained in phenol red-free RPMI 1640 medium (HyClone) containing 10% FBS. Both cell lines were cultured in a humidity atmosphere at 37℃, with 5% CO2. In the experiments in which cells were transfected and subsequently treated with or without (controls) estrogen, the cells were cultured as following: Cells were passaged once from frozen stock, and grown in regular 10%FBS (HyClone, UT, USA) for 3 days, followed by another passage and grown for 6 days in 10% charcoal-stripped FBS (Sigma) DMEM (for HCT116) or RPMI 1640 medium (LoVo cells). HCT116 or LoVo cells were transiently transfected with hMLH1, MSH2, and hERβ, respectively, using plasmids pCMV2-hygro-MLH1 (containing full-length wild-type cDNA of hMLH1), pCMV2-hygro-MSH2 (containing full-length wild-type cDNA of hMSH2), and pCMV2-hygro-ERβ (containing full-length cDNA of hERβ). pCMV2-hygro (empty vector) was used as a vehicle control. All vectors and corresponding cDNAs were sequence-validated. Transfection was performed when cells reached 70-80% confluence, using VigoFect transient transfection kit (Vigorous Biotech, Beijing, China) according to the manufacturer's instructions. 24 hours after transfection, cells were incubated with or without (control) 17β-estradiol (E2; Sigma, 10-8 M) for 24 or 48 hours.
Western blot analysis
The proteins were extracted from the cells or colonic biopsies by RIPA buffer (Sigma, St. Louis, MO, US). Extract proteins (50 μg) were separated by 12% SDS-polyacrylamide gel electrophoresis, transferred to nitrocellulose membrane, probed with 1:1000 dilution of primary antibodies (below), and incubated with FITC-conjugated anti-mouse or anti-rabbit immunoglobulin (1:15000 dilution; Millipore, MA, USA). Primary antibodies used for Western blot analyses include: Anti-hMLH1, -hMSH2, and -ERβ, polyclonal antibodies were purchased from Epitomics (Burlingame, CA, USA); anti-caspase 3 and -caspase 9 were purchased from Cell Signaling Technology (Beverly, MA, USA); anti-Bax, -P53 and -GAPDH monoclonal antibodies were purchased from Santa Cruz Biotechnology (Dallas, TX, USA). Expression of proteins of interests was imaged using an ODYSSEY quantitative fluorescent imaging system (LI-COR, Lincoln, NE, USA), and quantified by Image StudioTM Lite Western Blot Analysis software (LI-COR, Lincoln, NE, USA).
Flow cytometric analysis
Apoptosis analysis by FACS was performed using Annexin V-FITC/Dead Cell Apoptosis Kit with Annexin V-FITC and PI (Invitrogen, Grand Island, NY, USA) according to manufacture's instructions. HCT116 cells plated in 24-well plates were transfected respectively with hMLH1, hMSH2, and ERβ. 24 hours after transfection, cells were incubated with E2 for 24 or 48 hours. Cells were then harvested and rinsed with cold PBS 3 times and resuspended in the Binding Buffer. To each 100 μl of cell suspension culture, 5 μl of Annexin V-FITC and 1 μl of PI working solution (100 μg/ml) were added. Cells were incubated at room temperature for 15 minutes. After incubation, 400 μl of 1X Annexin-binding buffer was added to the cells and the cells were immediately placed on ice. The cells were analyzed using a BD FACS Calibur flow cytometer (BD Biosciences, San Jose, CA, USA). The percentage of early apoptotic cells (Annexin V+, PI-, the lower right quadrant) was calculated.
Assessment of cell viability
Cell viability was assessed by a tetrazolium-based colorimetric assay using Cell Counting Kit-8 (CCK-8, Takara BIO, Dalian, China), according to the manufacturer's instructions. 24 hours after transfection, cells were seeded in 96-well plates and incubated with 10-8 M E2 for 48 hours. CCK-8 (10 μl) was then added to each well. After incubation in the dark for 2 hours at 37℃, the optical density (OD) at 450 nm was measured by GENios Pro microplate reader (Tecan, Männedorf, Switzerland). The percent viability was calculated as follows: relative viability (%) = [A450 (treated) - A450 (blank)] / [A450 (control) - A450 (blank)] × 100%.
Colonic mucosal samples
Colonic (sigmoid) mucosal biopsies were obtained from 3 healthy female subjects with menopausal syndrome before and after 6-month hormone replacement therapy (HRT) with conjugated equine estrogens (0.3 mg /day). Subjects underwent colonoscopy and MMR before and after HRT. All participating subjects signed informed consents/agreements approved by the hospital ethic committee.
Statistical analyses
Levels of proteins of interest evaluated by Western blot were quantified using Image StudioTM Lite Western Blot Analysis software (LI-COR, Lincoln, NE, USA) and normalized to GAPDH; and statistical analysis between groups was performed using ANOVA. The SPSS 13.0 software was used for statistical analyses of FACS data. P value <0.05 was considered statistically significant. At least 3 independent experiments were performed in each set of data presented.
Results
Effects of hMLH1, hMSH2 or ERβ on the changes of major apoptotic proteins in the presence or absence of estradiol/E2 in HCT116 cells
To evaluate the effects of hMLH1, hMSH2, or ERβ on the apoptotic status of colon cancer cells, we examined several major apoptosis-related proteins, including caspases-3, caspase-9, Bax, and P53 in colon cancer cells with over-expression of various genes of interest (Figure 1). Overall, overexpression of hMLH1, or hMSH2, or ERβ significantly increased the activated (cleaved) form caspase-3 and caspase-9, as well as the expression of Bax and P53, when compared with the controls (transfected with empty vector). However, only over-expression of hMLH1, but not hMSH2 or ERβ, resulted in additional E2-dependent activation of caspase-3 and caspase-9, and further enhanced expression of Bax and P53.
Apoptotic activities were analyzed by Western blot in HCT116 cells transfected respectively with or without hMLH1, hMSH2, and ERβ, in the presence or absence of estradiol (E2). HCT116 cells with corresponding transfections were treated or not with 10-8 M estradiol (E2) for 48 hours. Cells were harvested, and proteins were analyzed by SDS-PAGE and Western blot. (A). Western blot images showing the expression of proteins of interests as indicated. GAPDH was used as a loading control. E2 (-) = without estradiol; E2 (+) = with E2; hMLH1 (+) = transfected with hMLH1; hMSH2 (+) = transfected with hMSH2; ERβ (+) = transfected with ERβ. (B) Statistical analyses of P53, Bax, and activated caspase-3 and -9 from the results of at least three independent experiments as represented in (a). (* = P<0.05, ** = P<0.01, *** = P<0.001)
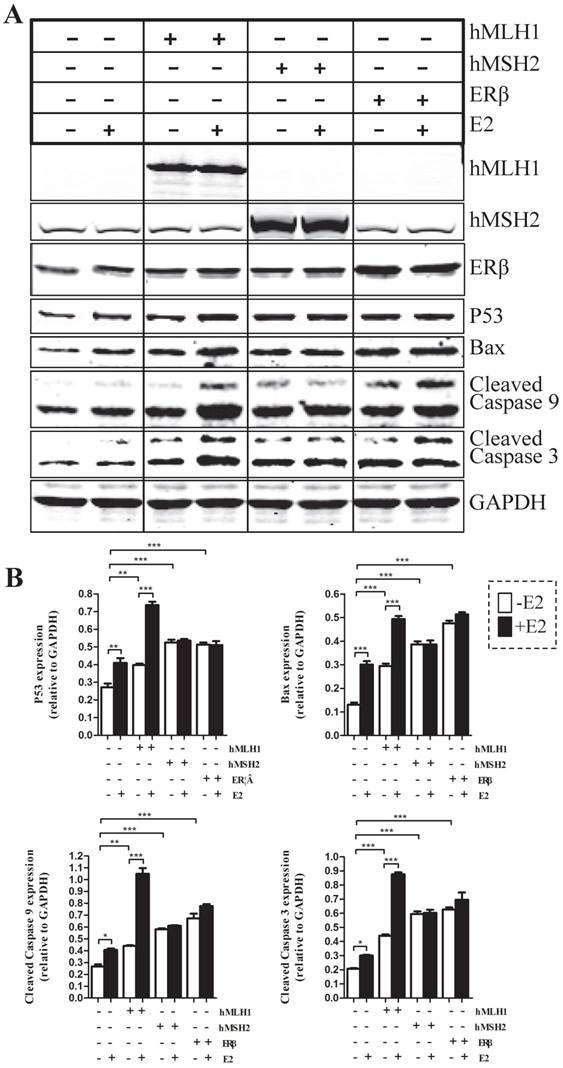
Expression of hMLH1, hMSH2 or ERβ decreased the cell viability in the presence or absence of estradiol/E2 in HCT116 cells
First, introducing either hMLH1, or hMSH2, or ERβ into HCT116 cells significantly decreased the cell viability, compared with the vehicle controls, regardless of presence or absence of E2 (Figure 2). However, E2-mediated further reduction in cell viability occurred only in cells with overexpression of hMLH1, but not hMSH2 or ERβ (Figure 2). These results are consistent with the apoptotic data described above.
The effects of hMLH1, hMSH2, or ERβ on the viability of HCT116 cells were evaluated by the WST-8-based Cell Counting Kits (CCK-8 assays) in the presence or absence of estradiol (E2). HCT116 cells transfected with hMLH1, hMSH2, or ERβ, were treated with 10-8 M estradiol for 48 hours described in Fig. 1. (A) Cell viability is illustrated as a bar graph (n= 3; * = P<0.05, ** = P<0.01). E2 (-) = without estradiol; E2 (+) = with E2; hMLH1 (+) = transfected hMLH1; hMSH2 (+) = transfected hMSH2; ERβ (+) = transfected ERβ.
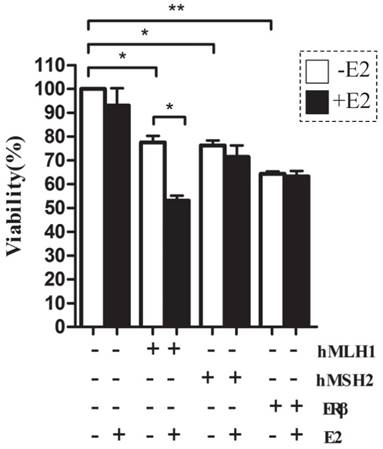
Apoptosis, measured by FACS with Annexin V-FITC, increased when cells transfected with either hMLH1, hMSH2 or ERβ; however, E2-dependent additional apoptosis occurs only in the in HCT116 cells overexpressing hMLH1
The apoptotic rates were potently enhanced in HCT116 cells transfected with either hMLH1, or hMSH2, or ERβ, compared with the vehicle controls (Figure 3). An additional increase in the apoptosis by E2 treatment at 48 hours was observed only in cells with expression of hMLH1 (Figure 3B & Figure 3C, right panel). There was no effect E2 treatment on apoptosis at 24 hours (Figure 3A & Figure 3C, left panel).
The effects of hMLH1, hMSH2, or ERβ on the viability of HCT116 cells were analyzed by flow cytometry with Annexin V-FITC and PI. Scatter-plots illustrate the percentages of early apoptotic cells (Annexin V+, PI-, the lower right quadrant) that were transfected with hMLH1, hMSH2, or ERβ, and incubated with E2 for 24 (A) or 48 (B) hours. Statistical analysis (n = 3) of FACS data is shown in (C). E2 (-) = without estradiol; E2 (+) = with E2; hMLH1 (+) = transfected with hMLH1; hMSH2 (+) = transfected with hMSH2; ERβ (+) = transfected with ERβ. (* = P<0.05).
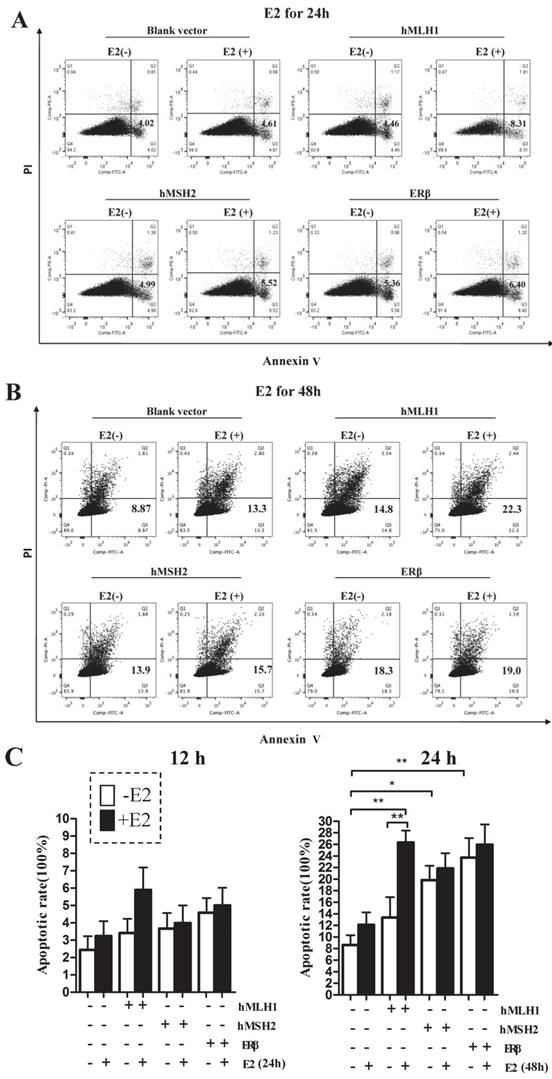
Overexpression of hMLH1 in hMSH2-deficient LoVo cells also enhances both basal level and estradiol-induced apoptosis
To provide additional evidence to support the observation in HCT116 cells that the estrogen-dependent pro-apoptotic effect is mediated by hMLH1, not MSH2, we overexpressed hMLH1 in hMSH2-deficient LoVo cells and evaluated the apoptotic status in the presence or absence of estradiol by examining the levels of activated (cleaved) forms of caspase-3 and caspase-9, as well as the expression of Bax and P53 (Figure 4). Clearly, deficiency of MSH2 in LoVo cells did not affect the E2-mediated enhancement of apoptosis, as E2 potently activated caspase -3 and -9 and increased the expression of P53 and Bax. This would suggest that MSH2 play little role in E2-mediated apoptosis. Importantly, however, overexpression of hMLH1 in LoVo cells promote apoptotic activity even in the absence of E2; and E2 could further enhance the apoptotic activity, as indicated by the increase of cleaved caspase 3 and caspase 9, as well as the expression of Bax and P53.
Enhanced apoptosis was also observed in colonic epithelial LoVo cells with overexpression of hMLH1. Cell treatment and Western blot (A) were done as described in Fig. 1. GAPDH was used as a loading control. E2 (-) = without estradiol; E2 (+) = with E2; hMLH1 (+) = transfected with hMLH1; hMSH2 (+) = transfected with hMSH2; ERβ (+) = transfected with ERβ. Statistical analysis (n = 3) was shown in (B). Representative of at least three independent experiments are shown.
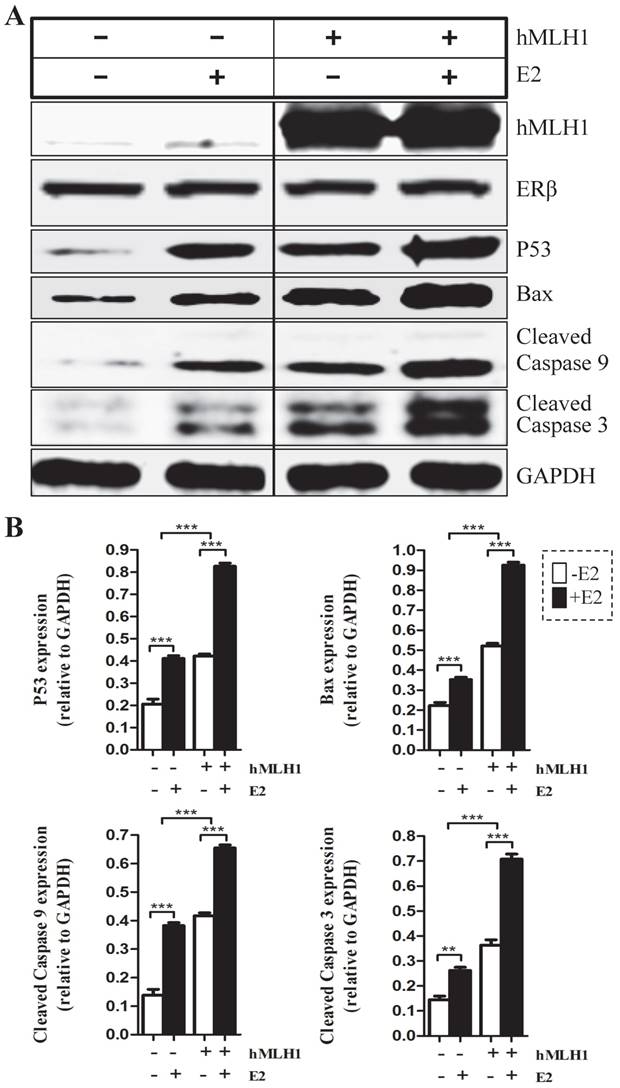
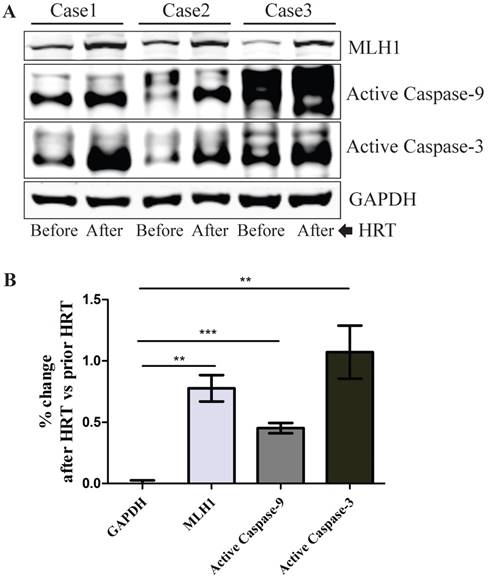
In vivo hormone replacement therapy (HRT) in healthy post-menopausal women led to increased expression of hMLH1, with concurrent elevation of cleaved caspase 3 and caspase 9 in the colonic mucosa
To determine the relevance of our in vitro cell culture data to human subjects in vivo, we investigated if the hMLH1-mediated and estrogen-dependent pro-apoptotic effects observed with colonic epithelial cells could be recapitulated in the colonic biopsies from postmenopausal women after 6-month HRT. As shown in Figure 5, HRT consistently enhanced the expression of hMLH1, with a significant concurrent increase in the activation of caspase 3 and caspase 9.
Healthy postmenopausal female subjects (n=3) underwent 6-month HRT exhibited a significantly enhanced the apoptotic activity in the colonic mucosal biopsy specimens. (A) The expression of hMLH1 and the activation of apoptotic proteins, caspase 3 and caspase 9, were analyzed by Western blot analysis in colonic biopsies before and after 6-month HRT [conjugated equine estrogens (0.3 mg /day]. (B) Statistical analysis (n = 3) of the data represented in (A) was shown as the percent changes (HRT vs prior HRT). GAPDH was used as a control.
Discussion
We present the first evidence, both in vitro and in vivo to demonstrate that the protective role of estrogen in colorectal cancer may be exerted specifically by MMR protein hMLH1-mediated programmed cell death of colon cancer cells. This estrogen-induced apoptosis occurs only through hMLH1, but not hMSH2, although overexpression of either can promote apoptosis.
Accumulating indirect evidence indicates that MMR proteins may involve in estrogen-mediated prevention or delaying of colorectal carcinogenesis. Important in maintaining genomic stability, MMR dysfunction can lead to microsatellite instability (MSI). MSI-high has been shown to be significantly associated with very low levels of estrogen receptor in colorectal carcinoma tissues, suggesting that MMR-deficient tumors might lose an estrogenic modulation mechanism 11. The fact that postmenopausal HRT reduces risks of only MSI-low/MSI-stable colorectal cancer subtypes further indicates a role of estrogen in colorectal cancer prevention 5. Moreover, a cohort study has shown that hormone replacement therapy only reduces the risk of MMR-proficient colorectal cancer, but not of MMR-deficient tumors 6. We have previously demonstrated that estradiol increases the expression of hMLH1 in colonic epithelial cells via ERβ 12, 13. We now demonstrated here that HRT significantly enhances hMLH1 expression in the colonic biopsies, suggesting that the HRT [conjugated equine estrogens (0.3 mg /day) may work as ERβ-agonist to increase MLH1 expression.
MMR protein-dependent pro-death signaling in response to various DNA-damaging agents have been well characterized, including 5-fluorouracil 14, 15, cisplatin 16, 17, and methylation agents 18, 19. However, MMR protein-dependent pro-apoptotic response to estrogen is largely unknown. There are two predominant hypotheses on the MMR protein-dependent initiation of cell death in response to DNA damage 15. The first hypothesis assumes that the repairing capacity of MMR proteins is required for MMR protein-dependent cell death, while the second suggests that it may not necessarily be required in damage-induced signaling. Since both hMLH1 and hMSH2 are indispensable for DNA mismatch repair function 20, the fact that overexpression of hMLH1 in hMSH2-deficient LoVo cells could also enhance estradiol-induced apoptosis would strongly suggest that the DNA repair capacity of MMR is not essential for estradiol-induced apoptosis. Moreover, in methylation agent- or cisplatin-induced cells apoptosis, it has been shown that hMSH2 is required for the formation of either hMSH2/hMSH6 or hMSH2/hMSH3 heterodimer that recognizes the DNA damage and subsequently triggers pro-death signaling 16, 21. However, our study demonstrated that estrogen-induced apoptosis of colonic cancer cells was mediated by hMLH1, but not hMSH2. Our data support a distinct role of MMR proteins in estrogen-induced apoptosis vs DNA-damaging agent-mediated cell death.
How estrogen/estradiol, which is not a DNA-damaging agent, can induce programmed cell death? Although the mechanism is not well understood, certain metabolites of estrogen have been shown to enhance the rate of oxidative DNA damage in breast and endometrial cells 22, 23, suggesting a possible role of estrogen in potentiating oxidative cytotoxicity. However, we do not believe this is the mechanism how estrogen enhances hMLH1-dependent apoptosis in colon cancer cells because if it is, overexpression of MMR protein hMLH1 would have promoted DNA repair, thereby decreasing apoptosis. Therefore, the exact molecular mechanism by which estrogen induces apoptosis through hMLH1 remains to be elucidated.
ERβ is the predominant ER in the human colonic epithelium and its expression decreases during colon carcinogenesis 24, 25. Lack of ERβ expression is associated with loss of differentiation and decreased apoptosis 26. We demonstrate that overexpression of ERβ in HCT116 cells spontaneously induces apoptosis that is independent of estrogen. Together with our hMLH1 and hMSH2 data, our study strongly supports previous reports that ERβ induces an extensive anti-tumorigenic effect in colon cancer cells through perhaps interaction with the DNA repair and p53 genes 27, 28.
Our in vivo study with healthy postmenopausal female subjects demonstrates that HRT could significantly enhance the apoptosis in the colonic biopsies, as indicated by the robust activation of caspase 3 and caspase 9. Previous reports have demonstrated that estrogen induces apoptosis in malignant and non-malignant colonocytes of human or rodents 28-33. Various mechanisms of action have been proposed, including p53-mediated signaling 28, 30, 32, increased Bax expression 33, and decreased Bcl-2 31. Moreover, overexpression of hMLH1 or hMSH2 could induce apoptosis in either MMR-proficient or -deficient cells 34, indicating that hMLH1 and hMSH2 may play a similar or redundant role in enhancing apoptosis through increased expression. This observation has also been validated in our studies using both HCT116 and LoVo cells. Interestingly, however, our study revealed that hMLH1 also mediated estrogen-induced apoptosis that is distinctively different from hMSH2, as lack of or overexpression of hMSH2 has no significant influence on estradiol-induced apoptosis. Mechanism dictating these distinctive functions is currently unknown. It is possible that hMSH2 may have other unrecognized functions unrelated to estrogen regulation, as it has been shown to interact with ERβ in a ligand-independent manner 35.
In summary, we present compelling evidence that, while increased expression of MMR protein, either hMLH1 or hMSH2, promotes apoptosis in colonic cancer cells, estrogen-induced apoptosis of these cells is mediated by hMLH1, but not hMSH2. Our human colonic mucosal biopsies data demonstrate that a 6-month HRT can potently enhance hMLH1 expression and concurrently activate apoptosis in vivo. Considering the previous reports that HRT-mediated reduction on the risk of colorectal cancer is dependent on MSI status 5, 6, these findings have important implications for clinical practice: MMR status should be evaluated in patients being considered for hormone replacement therapy or in the future application of estrogen-based chemoprevention against colorectal cancer to maximize the benefit/risk ratio.
Acknowledgements
This study was supported by the National Natural Science Foundation of China, Grant No. 2014BAI09B05; and NIH/ NIH-NIDDK R21 DK077064 and NIH/NIDDK P30DK089502, USA
Competing Interests
The authors have declared that no competing interest exists. The manufacturers were not involved in either the design of the study or the analysis of the data.
References
1. Chlebowski RT, Wactawski-Wende J, Ritenbaugh C, Hubbell FA, Ascensao J, Rodabough RJ, Rosenberg CA, Taylor VM, Harris R, Chen C, Adams-Campbell LL, White E; Women's Health Initiative Investigators. Estrogen plus Progestin and colorectal cancer in postmenopausal women. N Engl J Med. 2004;350(10):991-1004
2. Hildebrand JS, Jacobs EJ, Campbell PT, McCullough ML, Teras LR, Thun MJ, Gapstur SM. Colorectal cancer incidence and postmenopausal hormone use by type, recency, and duration in cancer prevention study II. Cancer Epidemiol Biomarkers Prev. 2009;18(11):2835-41
3. Schleipen B, Hertrampf T, Fritzemeier KH, Kluxen FM, Lorenz A, Molzberger A, Velders M, Diel P. ERβ-specific agonists and genistein inhibit proliferation and induce apoptosis in the large and small intestine. Carcinogenesis. 2011;32(11):1675-83
4. Qiu Y, Waters CE, Lewis AE, Langman MJ, Eggo MC. Estrogen-induced apoptosis in colonocytes expressing oestrogen receptor beta. J Endocrinol. 2002;174(3):369-77
5. Limsui D, Vierkant RA, Tillmans LS, Wang AH, Weisenberger DJ, Laird PW, Lynch CF, Anderson KE, French AJ, Haile RW, Harnack LJ, Potter JD, Slager SL, Smyrk TC, Thibodeau SN, Cerhan JR, Limburg PJ. Postmenopausal hormone therapy and colorectal cancer risk by molecularly defined subtypes among older women. Gut. 2012;61(9):1299-305
6. Newcomb PA, Zheng Y, Chia VM, Morimoto LM, Doria-Rose VP, Templeton A, Thibodeau SN, Potter JD. Estrogen plus progestin use, microsatellite instability, and the risk of colorectal cancer in women. Cancer Res. 2007;67(15):7534-9
7. Konstantinopoulos PA, Kominea A, Vandoros G, Sykiotis GP, Andricopoulos P, Varakis I, Sotiropoulou-Bonikou G, Papavassiliou AG. Estrogen receptor beta (ERbeta) is abundantly expressed in normal colonic mucosa, but declines in colon adenocarcinoma paralleling the tumour's dedifferentiation. Eur J Cancer. 2003;39(9):1251-8
8. Giroux V, Lemay F, Bernatchez G, Robitaille Y, Carrier JC. Estrogen receptor beta deficiency enhances small intestinal tumorigenesis in ApcMin/+ mice. Int J Cancer. 2008;123(2):303-11
9. Giroux V, Bernatchez G, Carrier JC. Chemopreventive effect of ERβ-Selective agonist on intestinal tumorigenesis in Apc(Min/+) mice. Mol Carcinog. 2011;50(5):359-69
10. Umar A, Boyer JC, Thomas DC, Nguyen DC, Risinger JI, Boyd J, Ionov Y, Perucho M, Kunkel TA. Defective mismatch repair in extracts of colorectal and endometrial cancer cell lines exhibiting microsatellite instability. J Biol Chem. 1994;269(20):14367-70
11. Notarnicola M, Gristina R, Messa C, Cariola F, Fiorente P, Caruso ML, Gentile M, Di Leo A. Oestrogen receptors and microsatellite instability in colorectal carcinoma patients. Cancer Lett. 2001;168(1):65-70
12. He YQ, Sheng JQ, Ling XL, Fu L, Jin P, Yen L, Rao J. Estradiol regulates miR-135b and mismatch repair gene expressions via estrogen receptor-β in colorectal cells. Exp Mol Med. 2012;44(12):723-32
13. Jin P, Lu XJ, Sheng JQ, Fu L, Meng XM, Wang X, Shi TP, Li SR, Rao J. Estrogen stimulates the expression of mismatch repair gene hMLH1 in colonic epithelial cells. Cancer Prev Res (Phila). 2010;3(8):910-6
14. Iwaizumi M, Tseng-Rogenski S, Carethers JM. DNA mismatch repair proficiency executing 5-fluorouracil cytotoxicity in colorectal cancer cells. Cancer Biol Ther. 2011;12(8):756-64
15. Valeri N, Gasparini P, Braconi C, Paone A, Lovat F, Fabbri M, Sumani KM, Alder H, Amadori D, Patel T, Nuovo GJ, Fishel R, Croce CM. MicroRNA-21 induces resistance to 5-fluorouracil by down-regulating human DNA MutS homolog 2 (hMSH2). Proc Natl Acad Sci U S A. 2010;107(49):21098-103
16. Topping RP, Wilkinson JC, Scarpinato KD. Mismatch repair protein deficiency compromises cisplatin-induced apoptotic signaling. J Biol Chem. 2009;284(21):14029-39
17. Papouli E, Cejka P, Jiricny J. Dependence of the cytotoxicity of DNA-damaging agents on the mismatch repair status of human cells. Cancer Res. 2004;64(10):3391-4
18. Li LS, Morales JC, Hwang A, Wagner MW, Boothman DA. DNA mismatch repair-dependent activation of c-Abl/p73alpha/GADD45alpha-mediated apoptosis. J Biol Chem. 2008;283(31):21394-403
19. Hirose Y, Katayama M, Stokoe D, Haas-Kogan DA, Berger MS, Pieper RO. The p38 mitogen-activated protein kinase pathway links the DNA mismatch repair system to the G2 checkpoint and to resistance to chemotherapeutic DNA-methylating agents. Mol Cell Biol. 2003;23(22):8306-15
20. Gruber SB. New developments in Lynch Syndrome (hereditary nonpolyposis colorectal cancer) and mismatch repair gene testing. Gastroenterology. 2006Feb;130(2):577-587
21. O'Brien V, Brown R. Signalling cell cycle arrest and cell death through the MMR system. Carcinogenesis. 2006Apr;27(4):682-692
22. Wang Z, Wijewickrama GT, Peng KW, Dietz BM, Yuan L, van Breemen RB, Bolton JL, Thatcher GR. Estrogen receptor alpha enhances the rate of oxidative DNA damage by targeting an equine estrogen catechol metabolite to the nucleus. J Biol Chem. 2009;284(13):8633-42
23. Singh B, Chatterjee A, Ronghe AM, Bhat NK, Bhat HK. Antioxidant-mediated up-regulation of OGG1 via NRF2 induction is associated with inhibition of oxidative DNA damage in estrogen-induced breast cancer. BMC Cancer. 2013;13:253
24. Barone M, Scavo MP, Papagni S, Piscitelli D, Guido R, Di Lena M, Comelli MC, Di Leo A. ERβ expression in normal, adenomatous and carcinomatous tissues of patients with familial adenomatous polyposis. Scand J Gastroenterol. 2010;45(11):1320-8
25. Di Leo A, Barone M, Maiorano E, Tanzi S, Piscitelli D, Marangi S, Lofano K, Ierardi E, Principi M, Francavilla A. ER-beta expression in large bowel adenomas: implications in colon carcinogenesis. Dig Liver Dis. 2008;40(4):260-6
26. Elbanna HG, Ebrahim MA, Abbas AM, Zalata K, Hashim MA. Potential value of estrogen receptor beta expression in colorectal carcinoma: interaction with apoptotic index. J Gastrointest Cancer. 2012;43(1):56-62
27. Edvardsson K, Ström A, Jonsson P, Gustafsson JÅ, Williams C. Estrogen receptor β induces antiinflammatory and antitumorigenic networks in colon cancer cells. Mol Endocrinol. 2011;25(6):969-79
28. Weige CC, Allred KF, Armstrong CM, Allred CD. P53 mediates estradiol induced activation of apoptosis and DNA repair in non-malignant colonocytes. J Steroid Biochem Mol Biol. 2012;128(3-5):113-20
29. Principi M, Di Leo A, Pricci M, Scavo MP, Guido R, Tanzi S, Piscitelli D, Pisani A, Ierardi E, Comelli MC, Barone M. Phytoestrogens/insoluble fibers and colonic estrogen receptor β: randomized, double-blind, placebo-controlled study. World J Gastroenterol. 2013;19(27):4325-33
30. Zhang Z, Wang CZ, Du GJ, Qi LW, Calway T, He TC, Du W, Yuan CS. Genistein induces G2/M cell cycle arrest and apoptosis via ATM/p53-dependent pathway in human colon cancer cells. Int J Oncol. 2013;43(1):289-96
31. Wilkins HR, Doucet K, Duke V, Morra A, Johnson N. Estrogen prevents sustained COLO-205 human colon cancer cell growth by inducing apoptosis, decreasing c-myb protein, and decreasing transcription of the anti-apoptotic protein bcl-2. Tumour Biol. 2010;31(1):16-22
32. Hsu HH, Cheng SF, Wu CC, Chu CH, Weng YJ, Lin CS, Lee SD, Wu HC, Huang CY, Kuo WW. Apoptotic effects of over-expressed estrogen receptor-beta on LoVo colon cancer cell is mediated by p53 signalings in a ligand-dependent manner. Chin J Physiol. 2006;49(2):110-6
33. Linsalata M, Russo F, Notarnicola M, Guerra V, Cavallini A, Clemente C, Messa C. Effects of genistein on the polyamine metabolism and cell growth in DLD-1 human colon cancer cells. Nutr Cancer. 2005;52(1):84-93
34. Zhang H, Richards B, Wilson T, Lloyd M, Cranston A, Thorburn A, Fishel R, Meuth M. Apoptosis induced by overexpression of hMSH2 or hMLH1. Cancer Res. 1999;59(13):3021-3027
35. Wada-Hiraike O, Yano T, Nei T, Matsumoto Y, Nagasaka K, Takizawa S, Oishi H, Arimoto T, Nakagawa S, Yasugi T, Kato S, Taketani Y. The DNA mismatch repair gene hMSH2 is a potent coactivator of oestrogen receptor alpha. Br J Cancer. 2005;92(12):2286-91
Author contact
![]() Corresponding authors: Xuhang Li, PhD, Johns Hopkins University School of Medicine, Department of Medicine/GI Division, 720 Rutland Avenue, 918 Ross Research Bldg, Baltimore, MD21205, USA Email: xuhangedu Tel: 410-868-7156 Fax: 410-955-9677 Or Jian-qiu Sheng, MD, Department of Gastroenterology, PLA Army General Hospital, #5 Nanmencang, Dongcheng District, Beijing 100700, China E-mail: jianqiunet Tel: +86-10-66721014 Fax: +86-10-66721299
Corresponding authors: Xuhang Li, PhD, Johns Hopkins University School of Medicine, Department of Medicine/GI Division, 720 Rutland Avenue, 918 Ross Research Bldg, Baltimore, MD21205, USA Email: xuhangedu Tel: 410-868-7156 Fax: 410-955-9677 Or Jian-qiu Sheng, MD, Department of Gastroenterology, PLA Army General Hospital, #5 Nanmencang, Dongcheng District, Beijing 100700, China E-mail: jianqiunet Tel: +86-10-66721014 Fax: +86-10-66721299