Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(16):3142-3153. doi:10.7150/jca.19273 This issue Cite
Research Paper
FH535 Inhibits Proliferation and Motility of Colon Cancer Cells by Targeting Wnt/β-catenin Signaling Pathway
Yanyan Chen1, 2, Xianping Rao2, Kangmao Huang3, Xiaoxia Jiang1, Haohao Wang1, Lisong Teng1 ![]()
1. Department of Surgical Oncology, First Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou 310003, China;
2. Department of Cell Biology and Program in Molecular Cell Biology, School of Medicine, Zhejiang University, Hangzhou, Zhejiang 310058, China;
3. Department of Orthopaedic Surgery, Sir Run Run Shaw Hospital, School of Medicine, Zhejiang University, Hangzhou, Zhejiang 310016, China;
Received 2017-1-20; Accepted 2017-8-28; Published 2017-9-12
Abstract
Aberrant Wnt/β-catenin pathway activation is frequently observed in human colorectal cancer (CRC) and has become a promising target for CRC treatment. Our study aimed to evaluate the effect of FH535, a small molecule inhibitor of Wnt/β-catenin pathway, on two colon cancer cell lines, HT29 and SW480. We found FH535 significantly inhibited colon cancer cell proliferation in vitro and induced cell cycle arrest. Moreover, FH535 inhibited colon cancer xenograft growth in vivo. Wound-healing assay and Transwell assay revealed that FH535 notably suppressed migration and invasion of SW480 cells. FH535 also repressed expression of cancer stem cell markers, CD24, CD44 and CD133 in HT29 cells. Real time-quantitative PCR and Western blotting revealed that targeting Wnt/β-catenin pathway using FH535 effectively downregulated target genes including cyclin D1 and survivin at mRNA and protein level, which contributed to the FH535-induced inhibitory effect on colon cancer cell proliferation. As mechanisms for suppressing cancer cell motility, FH535 downregulated expression of matrix metalloproteinase-7 and -9, Snail and vimentin. RNA sequencing revealed that FH535 prominently altered multiple biological pathways associated with DNA replication, cell cycle and metabolism. Our study highlights the anti-cancer effect of FH535 on colon cancer and presents its potential in colon cancer treatment.
Keywords: FH535, colon cancer, Wnt/β-catenin pathway
Introduction
Colorectal cancer (CRC) is the second most common type of cancer and the fourth cause of cancer‑related mortalities worldwide [1]. Aberrant Wnt/β-catenin pathway activation is frequently observed in human CRC. It is reported by The Cancer Genome Atlas that Wnt signalling pathway was altered in 93% of colorectal cancer, including biallelic inactivation of adenomatous polyposis coli (APC) (82%) and activating mutations of β-catenin (CTNNB1)(4.7%) genes [2, 3]. The high mutation rate of Wnt pathway components in human CRC suggests the promise of Wnt signaling inhibition as a therapeutic approach.
Wnt/β-catenin signaling pathway is a highly conserved pathway which plays key regulatory roles in cell proliferation, migration, apoptosis, stem cell growth and differentiation [4]. The pathway is activated by the binding of Wnt protein ligands to a frizzled family receptor (Fzd) and low-density lipoprotein-related protein (LRP) receptors, and the free cytosolic β-catenin will be translocated to the nucleus, where it binds to T cell factor (TCF)/lymphoid enhancer-binding factor (LEF) transcription factors, together with other coactivators, leading to the regulation of Wnt target genes transcription [5], including Cyclin D1, c-myc and surviving [6-8].
Numerous small molecules targeting Wnt/β-catenin pathway at the extracellular, cytoplasmic or nuclear level have been developed and applied to preclinical or clinical treatment of CRC [5, 9]. FH535 is a dual small molecule inhibitor of peroxisome proliferator-activated receptors (PPARs) and β-catenin/TCF/LEF [10]. Previous studies demonstrated that FH535 inhibits the growth of colon, lung, breast, pancreatic and hepatocellular cancer cells [10-13], and represses pancreatic cancer xenograft growth and angiogenesis [14].
The aim of this study is to evaluate the effect of FH535 on the growth and motility of two p53- and APC-mutated colon cancer cell lines, HT29 and SW480 [15-17]. In the present study, cell cycle, clonegenity, cancer cell stemness, migration and invasion were assayed in vitro, to evaluate the effect of FH535 on biological behaviors of colon cancer cell lines. Importantly, we tested its inhibitory effect of colon cancer growth in vivo through HT29 nude mice xenograft. Furthermore, expression of targeted genes and proteins were assayed and transcriptomic profile was analyzed based on RNA sequencing data, elucidating the molecular mechanism of the anti-cancer effect of FH535.
Materials and Methods
Chemicals and reagents
FH535 was purchased from Selleckchem (S7484) and soluted in DMSO (Sigma-Aldrich). In all in vitro assays, DMSO was used as control, at final concentrations no more than 0.1%. Primary antibodies against β-catenin, Cyclin D1, survivin, Snail, vimentin and β-actin for Western blotting were obtained from Cell Signaling Technology.
Cell lines and cell culture
The human colon carcinoma cells HT29 and SW480 were purchased from American Type Culture Collection (ATCC) and maintained in RPMI 1640 medium (HyClone) supplemented with 10% fetal bovine serum (FBS, Gibco) and 1% penicillin/streptomycin at 37°C in an atmosphere of 5% CO2 in a humidified incubator.
Cell viability assay
Cells (3×103/well) were seeded in 96-well plates, treated with different concentrations of FH535 or DMSO as control for 0, 24, 48, and 72 hours, respectively. Cell Counting Kit-8 (Dojindo) was used to detect the cell viability following the manufacturer's instructions. Results were measured according to the reference-subtracted absorbance at 450 nm using a microplate enzyme-linked immunosorbent assay reader (Bio-Rad). The concentration that causes 50% inhibition of cell proliferation (IC50) was calculated based on inhibition rate at 48 hours.
Plate colony formation assay
HT29 and SW480 cells were seeded in 6-well plates at 500 and 1000 cells/well, respectively, and treated by FH535 for 72 hours, then the media were renewed without adding FH535. After culturing for another 10 days, cells were fixed by 4% PFA and subsequently stained with 0.1% crystal violet. The number of visible colonies was counted. The colony formation ability was calculated as follows: (visible colonies/seeded cells) × 100%.
Flow cytometry
For cell cycle analysis, HT29 and SW480 cells were serum starved for 24 hours for cell cycle synchronization, then cultured with media containing 10% FBS and different concentrations of FH535 for another 24 h. The cells were harvested, incubated with RNase A (Thermo Scientific) and stained with propidium iodide (Sigma-Aldrich), then analyzed using BD FACSCalibur flow cytometer.
For evaluation of CD24 and CD44 protein expression, cells were harvested after 24-hour FH535 treatment and incubated with anti-CD24 (phycoerythrin [PE]-conjugated, BioLegend), anti-CD44 (fluorescein isothiocyanate [FITC]-conjugated, BioLegend) or corresponding isotype control antibodies for 30 minutes at 4°C, then analyzed using flow cytometer.
Migration and invasion assays
Cell migration was evaluated by in vitro wound healing assay. Cells were grown to confluence in 6-well plates. Cell monolayers were scraped with a sterile micropipette tip and treated with different concentrations of FH535. The wound area was photographed by microscope (Olympus IX2-UCB) before and 24 hours after the treatment. The wound widths were measured using ImageJ.
Transwell invasion assay was carried out using 24-well Transwell chamber with an 8 µm pore size polycarbonate filter membrane (Corning). Before the assay, Matrigel (1:10 dilution, BD Biosciences) was coated in the upper chamber overnight. 1×105 cells in 200 µl RPMI 1640 with 1% FBS were incubated in the upper chamber, 900µl RPMI 1640 with 10% FBS were added in the lower chamber. After incubation for 36 hours, invaded cells were fixed by 4% PFA and stained with 0.1% crystal violet, then photographed by microscope. The results were presented as counted cells per field at 400× magnification.
Nude mice tumor xenograft model and treatment
Animal experiments were approved by the Institutional Animal Care and Use Committee of Zhejiang University (approval ID: SYXK(ZHE)-2005-0072). Colon cancer xenografts were established in 6- to 7-week-old male BALB/c nude mice. Single-cell suspensions (1×107 cells in 200 μl PBS) were injected subcutaneously into the nude mice. When tumors were grown to 100-200 mm3, the mice were randomly assigned to control and FH535 groups. For each treatment, FH535 group were injected intraperitoneally with 15 mg/kg FH535 dissolved in 100 μl DMSO / RPMI 1640 (1:1 mixture), and the control group were injected with the same volume of dissolvent. Treatment was conducted every 2 days for 14 days. Tumor volume was measured before each treatment and calculated using the formula: volume = length× width2 / 2. At the end of the experiment, mice were sacrificed using cervical dislocation and the xenograft tumor tissues were harvested, weighed, formalin-fixed and paraffin-embedded, ready for subsequent immunohistochemical stain.
Immunohistochemistry
Paraffin-embedded 4-μm tissue sections were stained for ki-67. In brief, tumor tissues were cut at 4-μm thickness, heated at 60 °C for 30 min, then deparaffinized by xylene, rehydrated in graded concentrations of alcohol. Antigen retrieval was performed using EDTA-Tris buffer (pH 9.0). The sections were rinsed in PBS and immersed in 3% H2O2 in methanol for 15 min. After blocking the sections with 3% bovine serum albumin for 1 hour, sections were incubated with anti-Ki-67 antibody (Servicebio) at 4 °C overnight. After washing with PBS, sections were incubated with horseradish peroxidase (HRP)-conjugated secondary antibody. Antibody binding was detected by using DAB chromogen solution (DAKO). Hematoxylin was used as the counterstain.
RNA isolation and real time-quantitative PCR (RT-qPCR)
Total RNA from cells was isolated by RNeasy Mini Kit (QIAGEN, Valencia, California, USA). After spectrophotometric quantification, reverse transcription was performed with 1 μg RNA using Reverse Transcription System (Promega). Real-time quantitative PCR was carried out with SYBR Premix Ex TaqTM (Tli RNaseH Plus) (Takara Bio Inc., Shiga, Japan) using StepOne Real-Time PCR System (Applied Biosystems) according to the manufacturer's instructions. Primers used are listed in Table 1. The reactions were performed in triplicate. Gene expression was normalized to GAPDH expression, and calculated based on the method of 2‑ΔΔCt.
List of the primer sequences for RT-qPCR [11, 14, 43, 56-58].
| Gene | Sense (5' to 3') | Antisense (5' to 3') |
|---|---|---|
| CTNNB1 | TGTGAATCCCAAGTACCAGTGT | CGTCAGACAAAGGAGAAACATT |
| LEF1 | AGCGAATGTCGTTGCTGAGTGTA | CTCTTGCAGACCAGCCTGGATAA |
| AXIN2 | TGTCTTAAAGGTCTTGAGGGTTGAC | CAACAGATCATCCCATCCAACA |
| Cyclin D1 | GGATGCTGGAGGTCTGCGA | TAGAGGCCACGAACATGCAAGT |
| Survivin | CAAGGAGCTGGAAGGCTGG | GTTCTTGGCTCTTTCTCTGTCC |
| MMP-9 | TGGGTGTACGACGGTGAAAA | CATGGGTCTCTAGCCTGATA |
| c-Myc | TCTGGATCACCTTCTGCTGG | AGGATAGTCCTTCCGAGTGG |
| CD24 | CAGGGCAATGATGAATGAGAAT | CCTGGGCGACAAAGTGAGA |
| CD44 | GTGATGGCACCCGCTATGTC | AACCTCCTGAAGTGCTGCTCC |
| CD133 | TCAGGATTTTGCTGCTTGTG | GCAGTATCTAGAGCGGTGGC |
| GAPDH | GGAGCGAGATCCCTCCAAAAT | GGCTGTTGTCATACTTCTCATGG |
RNA sequencing (RNA-seq)
RNA sequencing of control and FH535-treated HT29 cells (each with 3 biological replicates) was performed using BGISEQ-500 sequencing system [18]. Gene expression were quantified and normalized using RSEM tool [19]. Differentially expressed genes (DEGs) between control and FH535-treated HT29 were screened using NOISeq method [20]. KEGG pathway enrichment analysis [21] and Gene Ontology analysis [22] of DEGs were conducted.
Western Blotting
Cell lysates were extracted by RIPA lysis buffer and quantified by Pierce BCA Protein Assay Kit (Thermo Scientific). Samples containing equal amounts of protein were electrophoresed by sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE) on 8-12% gels and transferred to polyvinylidene fluoride membranes. Membranes were then blocked by 5% skim milk (BD BioSciences) and incubated overnight at 4˚C with primary antibodies. After washing with TBST, membranes were incubated with secondary antibody conjugated to HRP (Cell Signaling Technology) and were exposed with ECL Western Blotting Substrate (Bio-rad) Specific immune complexes were detected using G:BOX Chemiluminescence Imaging System (Syngene). Positive bands were semiquantified by densitometrical analyses using the ImageJ software.
Harvesting conditioned media and gelatin zymography
To harvest conditioned media of SW480 cells, cell culture supernatants were collected after centrifuged at 800 g for 5 minutes, then concentrated using Amicon Ultra Centrifugal Filter NMWL 3000 (Millipore). Gelatin zymography was carried out to detect MMP-9 using an MMP Gelatin Zymography assay kit (GenMed, Shanghai, China) following the manufacturer's instructions. MMP-9 activity was identified by clear bands at 92kDa appearing on the blue-stained background.
Statistical analysis
Data of experiments performed in triplicate were presented as means ± standard deviation (S.D.) and significant differences were determined using Student's t-test. Probability (p) values of < 0.05 were considered statistically significant.
Results
FH535 inhibits growth of colon cancer cells
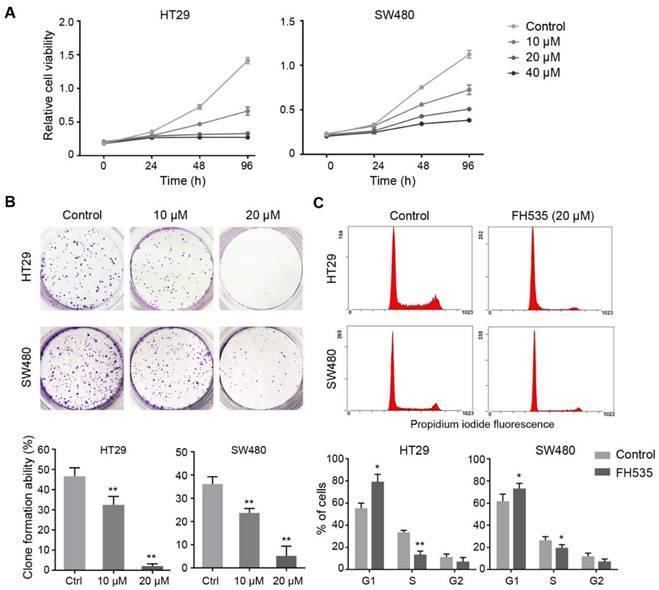
We evaluated the effect of FH535 on proliferation of colon cancer cell lines using CCK-8 method. Compared to control group, the proliferation of HT29 and SW480 treated with FH535 was significantly inhibited in a dose-dependent manner (Figure 1A). IC50 of FH535 was 18.6 μM and 33.2 μM in HT29 and SW480 cells, respectively. In the colony formation assay, cells treated with FH535 formed fewer and smaller colonies as compared to control group (Figure 1B), showing the inhibitory effect of FH535 on clonegenic ability of colon cancer cells. To explore the effect of FH535 on cell cycle regulation, cell cycle analysis was performed, which showed that FH535 significantly decreased the proportion of cells in S phase and increased that in G0/G1 (Figure 1C), The number of cells in G2 was decreased in FH535-treated group but did not reach statistical significance.
FH535 inhibited the growth of colon cancer cells in vitro. (A) Relative cell viability in FH535-treated and control colon cancer cell lines, assessed by CCK-8 assay. (B) In colony formation assay, HT29 and SW480 cells were seeded 500 and 1000 cells/well, respectively. Data were presented as colony formation rates. (C) Cell cycle analysis of FH535-treated and control colon cancer cells lines, performed by flow cytometry. Data were obtained from three independent experiments. *p < 0.05, **p < 0.01 verses control. Ctrl: Control.
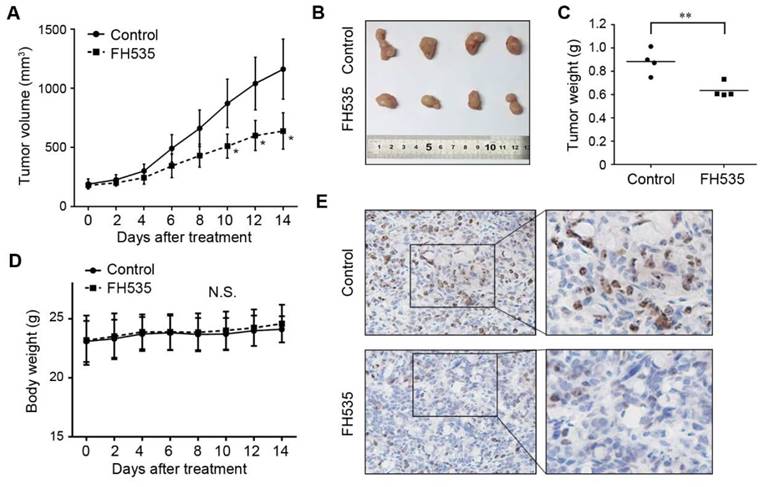
To investigate whether FH535 has inhibitory effect on colon cancer growth in vivo, we evaluated the effect of this chemical on HT29 nude mice xenografts. 12 days after the initiation of FH535 treatment, the calculated tumor volume of the FH535-treated group reached statistical significance as compared to the control group (Figure 2A). At the end of the experiment, the tumor size and weight of FH535-treated group was significantly smaller than the control group (Figure 2B-C). Importantly, no significant body loss was induced by FH535 (Figure 2D), suggesting the treatment was tolerable in mouse models. In immunohistochemistry of harvested tumor tissue, Ki-67 expression was tested as a cellular marker for proliferation. As a result, we observed lower expression of this protein in tumors from FH535-treated group than that of control group (Figure 2E), indicating that FH535 reduced proliferation rate of HT29 colon cancer xenograft.
Effects of FH535 on colon cancer growth in HT29 nude mice xenografts. (A) Tumor volume evaluation in FH535-treated and control group. (B) Photographs of tumor xenograft and (C) tumor weight in FH535-treated and control group. (D) Body weight of FH535-treated and control group. (E) Ki-67 staining in xenograft tissues from FH535-treated and control group. Magnifications: ×200 in left column, ×400 in right column. **p < 0.01. N.S., not significant.
FH535 inhibits the motility of SW480 cells
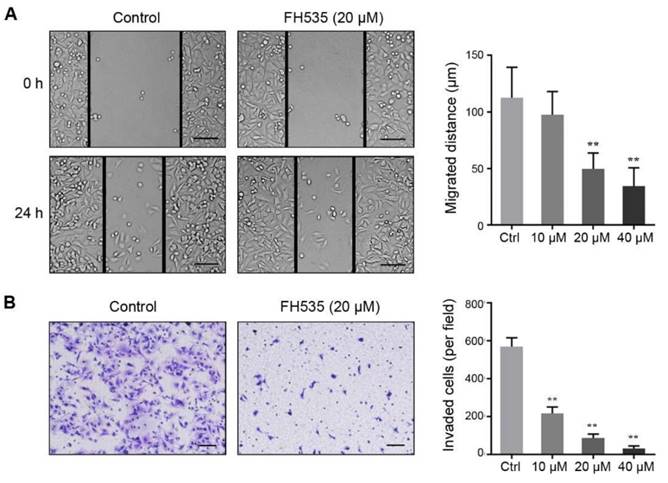
In our pilot experiments, SW480 showed much stronger motility than HT29 cells (data not shown), so we evaluated the effect of FH535 on cell motility of SW480. Wound-healing assay revealed that FH535 significantly decreased the migrating distance of SW480 (Figure 3A), suggesting its inhibitory effect on migration. Additionally, the result of Matrigel invasion assay indicated that FH535-treated cells had markedly weaker invading capacity as compared with the control cells (Figure 3B). Taken together, these data indicated that FH535 inhibits the motility of SW480.
FH535 suppressed cell migration and invasion in SW480 cells. (A) FH535-induced inhibition of cell migration assessed by wound healing assay. scale bar = 100 μm. (B) Transwell invasion assay showed that the FH535 suppressed the invasion ability of SW480 cells. scale bar = 100 μm. **p < 0.01, verses control. Ctrl: Control.
FH535 represses the stemness of HT29 cells
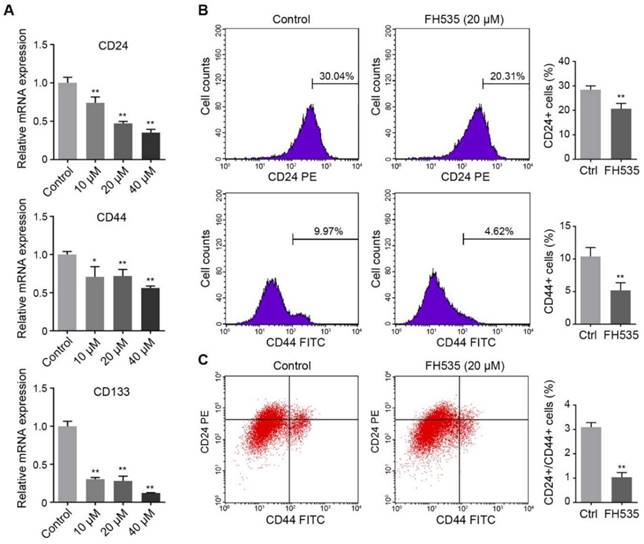
We further investigated the effect of FH535 on cancer cell stemness. The stemness of colon cancer cells was estimated by measuring expression of three cancer stem cell (CSC) markers, CD24, CD44 and CD133[23]. RT-qPCR showed that HT29 had significantly higher expression of these three markers than SW480 (Table S1), and FH535 dose-dependently downregulated expression of these markers in HT29 (Figure 4A), but not in SW480 (Figure S1). Next, protein expression of surface markers, CD24 and CD44 in HT29 were tested by flow cytometry, which revealed that FH535 treatment significantly reduced the CD24+, CD44+ and CD24+/CD44+ population (Figure 4B-C), suggesting its effect of stemness repression in HT29 cells.
FH535 reduced stemness of HT29 cells. (A) Relative mRNA levels of cancer stem cell markers, CD24, CD44 and CD133, in FH535-treated and control HT29 cells. (B) FH535 reduced CD24 and CD44 protein expression in HT29 cells. (C) FH535 reduced CD24+/CD44+ population of HT29 cells. *p < 0.05, **p < 0.01, verses control. Ctrl: Control.
FH535 inhibits the Wnt/β-catenin pathway and regulates downstream target genes
FH535 effectively suppressed TCF-dependent transcription in SW480 in dual luciferase reporter assay using TOPFlash, which confirmed FH535 as an inhibitor of Wnt/β-catenin pathway (Figure S2). Besides, consistent with RT-qPCR results, Western blotting showed FH535 downregulated total β-catenin in SW480 cells, but not in HT29 cells (Figure 5A). In both HT29 and SW480 cells, FH535 suppresses mRNA transcription of two vital target genes of Wnt/β-catenin pathway, LEF1 and AXIN2 [24-27] (Figure 5B).
FH535 inhibited Wnt/β-catenin pathway and its effect on target genes. (A) Relative mRNA and protein levels of β-catenin (CTNNB1) in FH535 treated vs control cells. (B) Relative mRNA levels of LEF1 and AXIN2 in FH535 treated vs control cells. (C) Relative mRNA and (D) protein levels of Cyclin D1and Survivin. (E) Relative mRNA levels of MMP-7 and MMP-9. (F) Secreted MMP-9 protein level determined by gelatin zymography. (G) Protein levels of vimentin and Snail in FH535-treated vs control cells. All mRNA levels were determined by RT-qPCR, normalized to GAPDH. *p < 0.05, **p < 0.01, verses control. Ctrl: Control. Semiquantification of positive bands were presented in Figure S3.
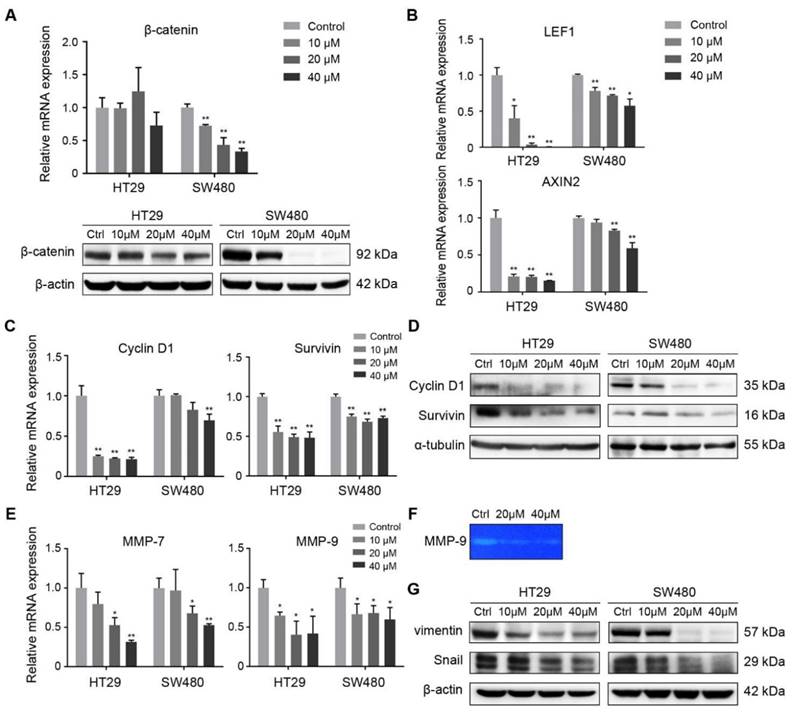
Wnt/β-catenin pathway regulates cell proliferation and survival through various target genes. Genes encoding Cyclin D1(CCND1) and survivin (BIRC5) were both downstream targets of β-catenin, regulated in their promoter regions through TCF/LEF components [6, 8]. Cyclin D1 is a critical target of proliferative signals that is required in the transition from G1 to S-phase in the cell cycle [28]. Survivin is an anti-apoptotic protein which promotes tumor cell growth by inhibiting apoptosis (IAP) protein family [29, 30]. RT-qPCR was performed to reveal the effect of FH535 on these target genes in HT29 and SW480 cells. The result showed that FH535 induces dose-dependent downregulation of Cyclin D1 and survivin in both cells (Figure 5C). In correspondence with the RT-qPCR results, expression of Cyclin D1 and survivin proteins were downregulated by FH535 (Figure 5D).
FH535 suppresses colon cancer cell motility by downregulating MMPs, Snail and vimentin
To further investigate the effect of FH535-induced cell motility suppression, the expression of MMPs were evaluated. Expression of MMP-7 and MMP-9 were dose-dependently downregulated at mRNA level by FH535 in HT29 and SW480 (Figure 5E). To test the gelatinase activity of secreted MMP-9, gelatin zymography was performed on SW480 cells, and the result indicated that FH535 significantly suppressed the gelatinase activity of MMP-9 (Figure 5F).
The transcription factor Snail is an important mediator of tumor invasion. Wnt signaling inhibits Snail phosphorylation and consequently increases Snail protein levels and activity [31]. Vimentin is an intermediate filament protein and a widely used marker of epithelial-mesenchymal transition (EMT) [32]. In our study, a notable decrease at protein level of Snail and vimentin was induced by FH535 (Figure 5G), which may be contributing factor for the suppression of migrating and invading capacity.
FH535 alters multiple cancer-associated biological processes
To acquire a clearer insight into the effecting mechanism of FH535, RNA sequencing (RNA-seq) was conducted to detect its impact on transcriptomic profile. All samples passed quality control and showed high consistency within replicates (Figure S4).1826 differentially expressed genes (DEGs) were screened out between FH535-treated and control HT29 by NOISeq method using the filtering criteria: fold change ≥ 2 and diverge probability ≥ 0.8 (Figure 6A-B). KEGG pathway enrichment analysis of DEGs suggested that FH535 altered a wide range of biological pathways involved in cancer, including DNA replication, cell cycle, mismatch repair, miRNAs and multiple pathways of metabolism (Figure 6C, Table S2). Similarly, Gene Ontology (GO) analyses revealed that the filtered DEGs were involved in various functions, especially in DNA replication and metabolic processes (Table S3).
Analysis of differentially expressed genes (DEGs) induced by FH535 treatment. (A) Scatter plot of global gene expression pattern in control and FH535-treated HT29 cells. Down- (blue dots) and up-regulated genes (orange dots) are presented as DEGs. (B) Hierarchical clustering of DEGs. Gradient color barcode at the right top indicates fold change of expression (log2). (C) Pathway enrichment analysis of DEGs based on KEGG database. 20 most enriched pathways (with lowest Q value) are displayed. Rich factor is the ratio of DEGs to all genes in this pathway.
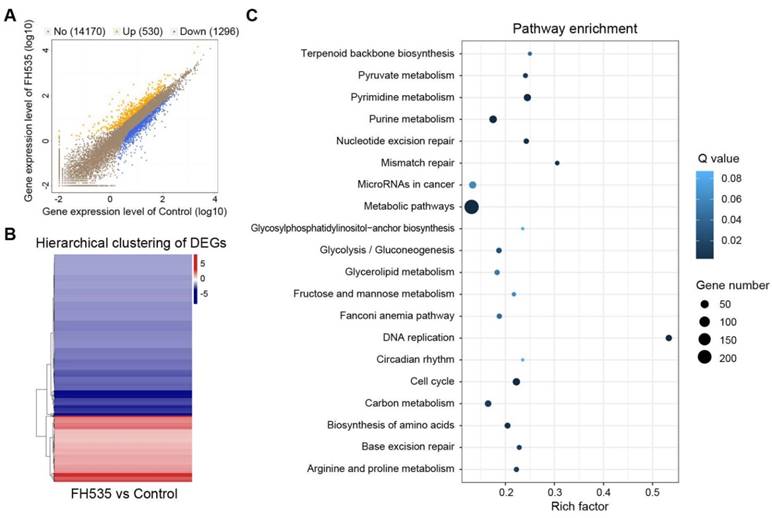
Moreover, we validated our previous results determined by RT-qPCR in RNA-seq data. CD24, CD133 and MMP7 were among the downregulated DEGs filtered by our criteria. CD44, AXIN2, CCDN1 (Cyclin D1), BIRC5 (survivin) were also significantly downregulated, with a diverge probability ≥ 0.8 but fold change lower than 2. Downregulation of LEF1 had a fold change of -3.46 but its diverge probability did not reach threshold due to its low expression level. CTNNB1 (β-catenin) expression did not show significant change (Table S4). MMP9 was not identified in the RNA-seq profile. In summary, differential analysis of RNA-seq data showed consistency with our RT-qPCR results.
Discussion
Activation of Wnt signaling through beta-catenin/TCF complexes is a critical event in CRC progression and has become a promising target for treatment. A variety of negative modulators of this pathway, such as LGK974, OMP-18R5, OMP‑54F28, PRI-724, have been developed and entered clinical trials for cancer treatment [5, 33]. Unlike the inhibitors mentioned above, FH535 inhibits recruitment of the coactivators GRIP1 and β-catenin to PPARγ and PPARδ along with antagonizing β-catenin/TCF-mediated transcription, indicating a unique effecting mechanism. The study of Handeli, S et al. showed that FH535 was selectively toxic to colon carcinoma cells RKO, LoVo and COLO205, but not to HCT116 and SW48 (HT29 and SW480 not tested) [10]. However, the effects of FH535 on cell cycle and cell motility of colon cancer cells were not evaluated, and its effecting mechanism was unclear.
Importantly, our study first evaluated the effect of FH535 in nude mice colon cancer xenografts. Although no significant acute or chronic toxicity was observed in our mouse model, further evaluation for the safety of FH535 is still required.
Though there are still no well-established stem cell markers in CRC, CD24, CD44 and CD133 have been previously implicated as putative CSC markers in CRC [23]. Previous study showed that CD24+ CRC cells were susceptible to EMT induction, which also indicated a stem-cell-like phenotype [34]. CD24 expression was found to be correlated with differentiation of CRC [35]. CD44 was implicated to be a robust CSC marker and is of importance for cancer initiation and growth [36]. CD133 promotes β-catenin signaling and therefore inhibits cancer cell differentiation [37], and indicates infinitive growth [38]. Besides, it is reported that FH535 represses cancer cell stemness in pancreatic cancer, reducing the CD24+/CD44+ population [14]. Based on the knowledge above, we estimated cancer cell stemness by testing expression of CD24, CD44 and CD133. Therefore, FH535-induced downregulation of these markers could suggest an inhibitory effect of CSC-like phenotype.
It was previously revealed that FH535 inhibited growth and motility in breast [12] and pancreatic [39] cancer cell lines regardless of the difference in β-catenin regulation, which were similar cases with our results, suggesting the effecting mechanisms of this inhibitor were possibly cell-type dependent. Moreover, cell-type dependent c-Myc expression induced by FH535 was observed, namely, downregulation in HT29 and upregulation in SW480 (Figure S5). C-Myc is a critical target gene of Wnt/β-catenin pathway controling multiple aspects associated with cell proliferation and cellular metabolism regulation [7, 40]. Therefore, different effects of c-Myc expression may be one mechanism for the disparity between HT29 and SW480 in sensitivity to FH535.
LEF1 and AXIN2 were both well-established downstream genes of Wnt/β-catenin pathway [26, 27, 41, 42]. Activation of Wnt/β-catenin pathway leads to activation of LEF1 transcription factors, and facilitates cancer cell invasion [24, 25], and its expression could be a predictor for poor prognosis in CRC patients [43]. AXIN2 was reported to be induced by Wnt/β-catenin signaling and acts as a negative regulator of this pathway [26], and its elevation could indicate Wnt/β-catenin pathway activation in colon cancers [27]. Therefore, downregulation of LEF and AXIN2 could indicate Wnt/β-catenin pathway inhibition in colon cancer cells.
Cyclin D1 and survivin are both critical regulators of cell cycle. Cyclin D1 binds with cyclin-dependent kinases CDK4 and CDK6 to form a complex targeting transcriptional factor E2F1, which is responsible for inducing G1/S transition [44]. Accordingly, downregulation of Cyclin D1 induces G1/S phase arrest [28, 45]. Survivin orchestrates multiple networks promoting tumor cell proliferation and inhibiting apoptosis [29]. Our data suggested that the change of distribution in G1 and S phase was possibly mediated by the FH535-induced inhibition of cyclin D1 and survivin.
MMPs are a prominent family of proteases which degrade extracellular matrix (ECM) proteins, modulate tumor microenvironment and promote cancer cell invasion and metastasis [46]. MMP-7, a target of the Wnt/β-catenin pathway, has substrate specificity for a wide range of ECM components and is implicated to mediate proliferation, invasion and metastasis of CRC [47, 48]. Importantly, MMP-7 expression has been shown to be a negative predictor of prognosis in CRC [49]. MMP-9 was shown to play critical roles in regulating migration and invasion, metastasis and angiogenesis [50, 51]. Downregulation of MMP-9 leads to reduced capacity of degrading gelatin, and represses invasion consequently. Vimentin regulates filament formation, induces mesenchymal shape and promotes cell motility as well as focal adhesion dynamics [32]. Previous study showed that silencing Snail efficiently suppresses MMP-9 secretion and vimentin expression in cancer cells, strongly reducing their invasive capacity [52]. Our data suggest that FH535-induced downregulation of Wnt pathway target genes, including Snail, vimentin and two important MMPs, may contribute to the motility suppression of colon cancer cells.
It is well-established that Wnt/β-catenin pathway plays role in inducing EMT. Wnt signalling promotes EMT by inhibiting GSK3β to stabilize β‑catenin, which translocates to the nucleus to engage LEF and TCF, thus promote a gene expression pattern that favours EMT [53]. Su H. et al reported that FH535 reversed EMT in a radioresistant esophageal cancer cell line [54]. Additionally, the study of Lee SC et al. revealed that suppressing survivin expression could enhance the EMT reversal effects by a Wnt/β-catenin pathway inhibitor, IWR-1 [55]. In our results, FH535 induced downregulation of two widely-used mesenchymal-like markers, Snail and vimentin, which were also downstream effects of inhibiting Wnt/β-catenin pathway. However, we did not observe significant upregulation of E-cadherin expression or downregulation of N-cadherin in FH535-treated HT29 and SW480 (data not shown), suggesting FH535 did not induce EMT reversal in these cell lines. Crosstalk of other signaling pathways possibly played roles in regulating EMT phenotype.
In KEGG pathway and GO analysis of RNA-seq data, the most prominent biological processes impacted by FH535 were associated with DNA replication, cell cycle and metabolism, which could account for growth inhibition and stemness repression induced by FH535. The impact on pathways relating cell motility was not significant, which could be due to the low-motility phenotype of HT29 cells. To note, genes including MMP-9 were not identified in the mapping of sequencing data for several possible reasons: 1) gene database used in pipeline was not complete; 2) new transcripts in sequencing data; 3) sequencing reads from non-coding regions. Generally, RNA-seq validated our previous data and provided clearer insight into the anti-cancer effect of FH535.
In summary, our study demonstrated the anti-proliferation effect of FH535 on colon cancer cells in vitro, and for the first time in vivo. Moreover, the inhibitory effect of FH535 on colon cancer cell motility and stemness was observed. Expression measurement at mRNA and protein levels of Wnt/β-catenin pathway target genes suggested that FH535 inhibited cell proliferation through downregulating cyclin D1 and survivin. FH535 also downregulated MMP-7, MMP-9, Snail and vimentin, contributing to the inhibition of motility. Analysis of RNA-seq data revealed that FH535 prominently affected pathways relating to DNA replication, cell cycle and metabolism. Our study provides new evidence for the anti-cancer effect of FH535 on colon cancer. Further evaluation of the efficacy and safety of this therapeutic approach is still required.
Supplementary Material
Additional File 1Supplementary figures and tables.
Supplementary table s2.
Supplementary table s3.
Acknowledgements
The authors thank lab members of Department of Cell Biology and Program in Molecular Cell Biology, School of Medicine, Zhejiang University, who provided academic and technical assistance for this study. This study was supported by National Natural Science Foundation of China (No. 81272676), National Science and Technology Major Project of the Ministry of Science and Technology of China (No. 2013ZX09506015) and Ministry of Health Ministry of construction project (No. WKJ-ZJ-1717).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA: a cancer journal for clinicians. 2015;65:87-108
2. Cancer Genome Atlas N. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330-7
3. Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C. et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333-9
4. Polakis P. Wnt signaling and cancer. Genes Dev. 2000;14:1837-51
5. Novellasdemunt L, Antas P, Li VS. Targeting Wnt signaling in colorectal cancer. A Review in the Theme: Cell Signaling: Proteins, Pathways and Mechanisms. American journal of physiology Cell physiology. 2015;309:C511-21
6. Shtutman M, Zhurinsky J, Simcha I, Albanese C, D'Amico M, Pestell R. et al. The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:5522-7
7. He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT. et al. Identification of c-MYC as a target of the APC pathway. Science. 1998;281:1509-12
8. Ma H, Nguyen C, Lee KS, Kahn M. Differential roles for the coactivators CBP and p300 on TCF/beta-catenin-mediated survivin gene expression. Oncogene. 2005;24:3619-31
9. Song L, Li Y, He B, Gong Y. Development of Small Molecules Targeting the Wnt Signaling Pathway in Cancer Stem Cells for the Treatment of Colorectal Cancer. Clinical colorectal cancer. 2015;14:133-45
10. Handeli S, Simon JA. A small-molecule inhibitor of Tcf/beta-catenin signaling down-regulates PPARgamma and PPARdelta activities. Molecular cancer therapeutics. 2008;7:521-9
11. Gedaly R, Galuppo R, Daily MF, Shah M, Maynard E, Chen C. et al. Targeting the Wnt/beta-catenin signaling pathway in liver cancer stem cells and hepatocellular carcinoma cell lines with FH535. PloS one. 2014;9:e99272
12. Iida J, Dorchak J, Lehman JR, Clancy R, Luo C, Chen Y. et al. FH535 inhibited migration and growth of breast cancer cells. PloS one. 2012;7:e44418
13. Tomizawa M, Shinozaki F, Motoyoshi Y, Sugiyama T, Yamamoto S, Ishige N. FH535 suppresses the proliferation and motility of hepatocellular carcinoma cells. International journal of oncology. 2016;48:110-4
14. Liu L, Zhi Q, Shen M, Gong FR, Zhou BP, Lian L. et al. FH535, a beta-catenin pathway inhibitor, represses pancreatic cancer xenograft growth and angiogenesis. Oncotarget. 2016;7:47145-62
15. Brabletz T, Jung A, Reu S, Porzner M, Hlubek F, Kunz-Schughart LA. et al. Variable beta-catenin expression in colorectal cancers indicates tumor progression driven by the tumor environment. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:10356-61
16. Morin PJ, Vogelstein B, Kinzler KW. Apoptosis and APC in colorectal tumorigenesis. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:7950-4
17. Rodrigues NR, Rowan A, Smith ME, Kerr IB, Bodmer WF, Gannon JV. et al. p53 mutations in colorectal cancer. Proceedings of the National Academy of Sciences of the United States of America. 1990;87:7555-9
18. Fehlmann T, Reinheimer S, Geng C, Su X, Drmanac S, Alexeev A. et al. cPAS-based sequencing on the BGISEQ-500 to explore small non-coding RNAs. Clin Epigenetics. 2016;8:123
19. Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323
20. Tarazona S, Furio-Tari P, Turra D, Pietro AD, Nueda MJ, Ferrer A. et al. Data quality aware analysis of differential expression in RNA-seq with NOISeq R/Bioc package. Nucleic Acids Res. 2015;43:e140
21. Kanehisa M, Sato Y, Kawashima M, Furumichi M, Tanabe M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016;44:D457-D62
22. Ye J, Fang L, Zheng H, Zhang Y, Chen J, Zhang Z. et al. WEGO: a web tool for plotting GO annotations. Nucleic Acids Res. 2006;34:W293-7
23. Abetov D, Mustapova Z, Saliev T, Bulanin D. Biomarkers and signaling pathways of colorectal cancer stem cells. Tumour biology: the journal of the International Society for Oncodevelopmental Biology and Medicine. 2015;36:1339-53
24. Huang FI, Chen YL, Chang CN, Yuan RH, Jeng YM. Hepatocyte growth factor activates Wnt pathway by transcriptional activation of LEF1 to facilitate tumor invasion. Carcinogenesis. 2012;33:1142-8
25. Nguyen A, Rosner A, Milovanovic T, Hope C, Planutis K, Saha B. et al. Wnt pathway component LEF1 mediates tumor cell invasion and is expressed in human and murine breast cancers lacking ErbB2 (her-2/neu) overexpression. International journal of oncology. 2005;27:949-56
26. Jho EH, Zhang T, Domon C, Joo CK, Freund JN, Costantini F. Wnt/beta-catenin/Tcf signaling induces the transcription of Axin2, a negative regulator of the signaling pathway. Mol Cell Biol. 2002;22:1172-83
27. Yan D, Wiesmann M, Rohan M, Chan V, Jefferson AB, Guo L. et al. Elevated expression of axin2 and hnkd mRNA provides evidence that Wnt/beta -catenin signaling is activated in human colon tumors. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:14973-8
28. Baldin V, Lukas J, Marcote MJ, Pagano M, Draetta G. Cyclin D1 is a nuclear protein required for cell cycle progression in G1. Genes Dev. 1993;7:812-21
29. Altieri DC. Survivin, cancer networks and pathway-directed drug discovery. Nat Rev Cancer. 2008;8:61-70
30. Kim PJ, Plescia J, Clevers H, Fearon ER, Altieri DC. Survivin and molecular pathogenesis of colorectal cancer. Lancet. 2003;362:205-9
31. Yook JI, Li XY, Ota I, Fearon ER, Weiss SJ. Wnt-dependent regulation of the E-cadherin repressor snail. J Biol Chem. 2005;280:11740-8
32. Mendez MG, Kojima S, Goldman RD. Vimentin induces changes in cell shape, motility, and adhesion during the epithelial to mesenchymal transition. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2010;24:1838-51
33. Sawa M, Masuda M, Yamada T. Targeting the Wnt signaling pathway in colorectal cancer. Expert Opin Ther Targets. 2016;20:419-29
34. Okano M, Konno M, Kano Y, Kim H, Kawamoto K, Ohkuma M. et al. Human colorectal CD24+ cancer stem cells are susceptible to epithelial-mesenchymal transition. International journal of oncology. 2014;45:575-80
35. Choi D, Lee HW, Hur KY, Kim JJ, Park GS, Jang SH. et al. Cancer stem cell markers CD133 and CD24 correlate with invasiveness and differentiation in colorectal adenocarcinoma. World J Gastroenterol. 2009;15:2258-64
36. Du L, Wang H, He L, Zhang J, Ni B, Wang X. et al. CD44 is of functional importance for colorectal cancer stem cells. Clin Cancer Res. 2008;14:6751-60
37. Mak AB, Nixon AM, Kittanakom S, Stewart JM, Chen GI, Curak J. et al. Regulation of CD133 by HDAC6 promotes beta-catenin signaling to suppress cancer cell differentiation. Cell reports. 2012;2:951-63
38. Grunt TW, Hebar A, Laffer S, Wagner R, Peter B, Herrmann H. et al. Prominin-1 (CD133, AC133) and dipeptidyl-peptidase IV (CD26) are indicators of infinitive growth in colon cancer cells. Am J Cancer Res. 2015;5:560-74
39. Wu MY, Liang RR, Chen K, Shen M, Tian YL, Li DM. et al. FH535 inhibited metastasis and growth of pancreatic cancer cells. OncoTargets and therapy. 2015;8:1651-70
40. Miller DM, Thomas SD, Islam A, Muench D, Sedoris K. c-Myc and cancer metabolism. Clin Cancer Res. 2012;18:5546-53
41. Gehrke I, Gandhirajan RK, Kreuzer KA. Targeting the WNT/beta-catenin/TCF/LEF1 axis in solid and haematological cancers: Multiplicity of therapeutic options. Eur J Cancer. 2009;45:2759-67
42. Boras-Granic K, Chang H, Grosschedl R, Hamel PA. Lef1 is required for the transition of Wnt signaling from mesenchymal to epithelial cells in the mouse embryonic mammary gland. Dev Biol. 2006;295:219-31
43. Wang WJ, Yao Y, Jiang LL, Hu TH, Ma JQ, Ruan ZP. et al. Increased LEF1 expression and decreased Notch2 expression are strong predictors of poor outcomes in colorectal cancer patients. Dis Markers. 2013;35:395-405
44. Chiron D, Martin P, Di Liberto M, Huang X, Ely S, Lannutti BJ. et al. Induction of prolonged early G1 arrest by CDK4/CDK6 inhibition reprograms lymphoma cells for durable PI3Kdelta inhibition through PIK3IP1. Cell cycle. 2013;12:1892-900
45. Liu L, Zhang H, Shi L, Zhang W, Yuan J, Chen X. et al. Inhibition of Rac1 activity induces G1/S phase arrest through the GSK3/cyclin D1 pathway in human cancer cells. Oncology reports. 2014;32:1395-400
46. Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141:52-67
47. Jia ZC, Wan YL, Tang JQ, Dai Y, Liu YC, Wang X. et al. Tissue factor/activated factor VIIa induces matrix metalloproteinase-7 expression through activation of c-Fos via ERK1/2 and p38 MAPK signaling pathways in human colon cancer cell. Int J Colorectal Dis. 2012;27:437-45
48. Sui H, Xu H, Ji Q, Liu X, Zhou L, Song H. et al. 5-hydroxytryptamine receptor (5-HT1DR) promotes colorectal cancer metastasis by regulating Axin1/beta-catenin/MMP-7 signaling pathway. Oncotarget. 2015;6:25975-87
49. Xing XJ, Gu XH, Ma TF. Relationship of serum MMP-7 levels for colorectal cancer: a meta-analysis. Tumour biology: the journal of the International Society for Oncodevelopmental Biology and Medicine. 2014;35:10515-22
50. Bergers G, Brekken R, McMahon G, Vu TH, Itoh T, Tamaki K. et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol. 2000;2:737-44
51. Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C. et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature. 2005;438:820-7
52. Olmeda D, Jorda M, Peinado H, Fabra A, Cano A. Snail silencing effectively suppresses tumour growth and invasiveness. Oncogene. 2007;26:1862-74
53. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nature reviews Molecular cell biology. 2014;15:178-96
54. Su H, Jin X, Zhang X, Zhao L, Lin B, Li L. et al. FH535 increases the radiosensitivity and reverses epithelial-to-mesenchymal transition of radioresistant esophageal cancer cell line KYSE-150R. Journal of translational medicine. 2015;13:104
55. Lee SC, Kim OH, Lee SK, Kim SJ. IWR-1 inhibits epithelial-mesenchymal transition of colorectal cancer cells through suppressing Wnt/beta-catenin signaling as well as survivin expression. Oncotarget. 2015;6:27146-59
56. Piotrowski WJ, Gorski P, Pietras T, Fendler W, Szemraj J. The selected genetic polymorphisms of metalloproteinases MMP2, 7, 9 and MMP inhibitor TIMP2 in sarcoidosis. Med Sci Monit. 2011;17:CR598-607
57. Hamada S, Urakawa H, Kozawa E, Arai E, Ikuta K, Sakai T. et al. Characteristics of cultured desmoid cells with different CTNNB1 mutation status. Cancer medicine. 2016;5:352-60
58. Tao K, Xiao D, Weng J, Xiong A, Kang B, Zeng H. Berberine promotes bone marrow-derived mesenchymal stem cells osteogenic differentiation via canonical Wnt/beta-catenin signaling pathway. Toxicology letters. 2016;240:68-80
Author contact
![]() Corresponding author: Lisong Teng, Professor, Department of Surgical Oncology, First Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou 310003, China, E-mail: lstengedu.cn.
Corresponding author: Lisong Teng, Professor, Department of Surgical Oncology, First Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou 310003, China, E-mail: lstengedu.cn.