Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(14):2793-2801. doi:10.7150/jca.19787 This issue Cite
Research Paper
Villi-specific Gene Expression Reveals Novel Prognostic Biomarkers in Multiple Human Cancers
Botao Zhang1, Lin Feng1, Honglin Guo1, Hongxia Li2, Yuanjing Wang2, Kaitai Zhang1, Xuexin Yu3 ![]() , Shujun Cheng1
, Shujun Cheng1 ![]()
1. State Key Laboratory of Molecular Oncology, Department of Etiology and Carcinogenesis, National Cancer Center/Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, 100021, China;
2. Department of Obstetrics and Gynecology, Beijing Shijitan Hospital, Capital Medical University, Beijing, 100038, China;
3. College of Bioinformatics Science and Technology, Harbin Medical University, Harbin, 150081, China.
Received 2017-2-23; Accepted 2017-6-7; Published 2017-8-23
Abstract
Despite many striking connections, the biological similarities between embryonic development and tumorigenesis have not been well explored. Development of the placental villi is a crucial process involving many cellular activities, including immunity, proliferation, and cell adhesion. In this study, we designed a strategy to identify the gene expression pattern of villi development and to explore the corresponding features in tumors. We discovered villi-specific genes that are highly expressed in the villus as opposed to the mature placenta and then measured the expression levels of these genes in tumors. We found large changes in the expression of villi-specific genes in multiple types of cancer. These villi-specific genes showed distinct expression patterns and were primarily involved in three biological processes: immune-related (5), proliferation-related (6), and focal adhesion-related (8); these genes were extracted from the corresponding enriched Gene Ontology (GO) terms. We observed that these genes were also dysregulated at the transcriptional level across several tumor types. Moreover, the expression of these three gene groups was associated with poor prognosis in a subset of tumors. Based on villi-specific gene expression, this correlation study indicated the existence of common gene expression patterns between embryonic development and tumorigenesis. Therefore, a systematic analysis of villi-specific gene aberrations in various tumors could serve as an indicator for identifying novel prognostic biomarkers.
Keywords: Development, Human cancer, Prognosis, Villi.
Introduction
Early in 1858, Rudolf Virchow first proposed that neoplasms arise “in accordance with the same law, which regulates embryonic development.” Since then, a growing body of evidence supports the relationship between oncogenesis and embryonic development, which share many surprising similarities [1-3]. With respect to cellular mobility and invasiveness, both processes involve epithelial-to-mesenchymal transitions (EMTs) [4, 5]. To be able to grow in the host or mother, both tumors and embryos must escape monitoring by the immune system [6, 7] as well as promote the formation of blood vessels to meet nutritional requirements [8]. In addition, many of the pathways that are crucial for orchestrating cellular activities and morphogenesis during development, such as Wnt, FGF, Notch, BMP and Hedgehog signaling [9], are reactivated and contribute to cancer progression and metastasis during tumorigenesis [10]. Considering these many similarities, we decided to reexamine embryonic development to illuminate the trajectory of human cancer development.
Presently, studies on the similarities between embryogenesis and tumorigenesis are mainly performed in animal models—for example, development of the rhesus macaque and corresponding organic tumors [11, 12]—or using in vitro cell lines [13]. While investigating fetal lung and colon development, our group identified specific expression profiles during the transition from development to normal tissue homeostasis to cancer. Some of these expression profiles had a characteristic “V” shape, being up-regulated during both development and tumorigenesis, whereas others had an “A” shape, being down-regulated during these two stages. Furthermore, we identified two groups of genes that are associated with overall survival in lung cancer and colorectal cancer patients [14, 15]. In this study, we focused on the early embryonic process of placental villi development. The chorionic villus is the main component of the fetus derived from the morula, and it is crucial for embryonic development. These trophoblastic cells grow, proliferate, differentiate, implant into the uterine decidua and myometrium, and remodel the maternal spiral artery, all of which are necessary for fetal growth. For these reasons, trophoblasts have been defined as a “pseudo-malignant” tissue, and these processes have been described as a type of “physiological metastasis” [16, 17]. Indeed, most of these mechanisms are strikingly similar to others discovered in tumors [18-20]. Therefore, we expected that by studying villi development, we would be able to identify novel biomarkers and therapeutic targets for carcinogenesis.
In this study, we used microarrays to profile early placental villi development from 6 to 10 weeks of pregnancy. By comparing datasets from villi and mature placenta, we identified villi-specific genes that were highly expressed in the villus. A large fraction of these villi-specific genes was misregulated at the transcriptional level across tumor types, with human cancers composing the largest group with up-regulated expression. Based on the Gene Ontology (GO) enrichment results, we selected five immune-related genes, six proliferation-related genes, and eight focal adhesion-related genes that were reported to have genomic alterations in specific cancers. Expression of these three groups of genes could be used to predict prognosis in a subset of tumors. Therefore, this strategy provides a new method for discovering prognostic tumor biomarkers.
Materials and methods
Patients and samples
The study materials for villus development were obtained from Beijing Shijitan Hospital between March 2015 and August 2016. The samples included 36 cases of chorionic villus samples at 6 to 10 weeks of gestation (hereafter referred to as “6W”, “7W”, “8W”, “9W”, and “10W”) and eight cases of leaf chorionic samples from postpartum placental tissue representing mature placenta (detailed information regarding the number of samples is presented in Table 1). In the case of early gestation, women with a history of either spontaneous abortion or halted fetal development were excluded from this study. Regarding term pregnancies, women with gestational complications such as preeclampsia, fetal growth restriction and gestational diabetes and fetuses with known or suspected genetic disorders were excluded from the study. Gestational age was based on the first day of the last menstrual period. The villus and mature placenta samples were rinsed with normal saline and divided into two parts, one of which was placed in RNAlater RNA Stabilization Reagent (Ambion/Thermo Fisher, Waltham, MA, USA) at 4°C overnight and then stored at -80°C until use, and the other was placed in formaldehyde solution and stored at room temperature. All donors signed informed consent forms. The use of human tissue samples and experimental procedures for this study were reviewed and approved by the Ethics Committee of the Cancer Institute and Hospital, Chinese Academy of Medical Sciences.
Number of samples for each time point
| Time points | 6W | 7W | 8W | 9W | 10W | Mature Placenta |
|---|---|---|---|---|---|---|
| No. of samples | 8 | 9 | 6 | 8 | 5 | 8 |
RNA isolation
Total RNA was isolated from frozen tissues with TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. Samples allocated for microarray analysis were purified using the RNeasy kit (Cat No. 74106, Qiagen, Hilden, Germany). RNA concentrations were determined using an ND-1000 UV-VIS Spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA), and RNA integrity was evaluated using the RNA 6000 LabChip kit in combination with the Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). The RNA samples used in this study all exhibited RNA concentrations >40 ng/μl and RNA integrity numbers >6.0.
Microarray expression profiling and analysis
Following RNA concentration and integrity analysis, the 44 samples were analyzed using Agilent 4×44K Whole Human Genome Oligo Microarrays at the Cancer Institute and Hospital, Chinese Academy of Medical Sciences, according to the manufacturer's specifications. In brief, 500 ng purified total RNA was reversed transcribed in vitro using the Low RNA Input Linear Amplification Kit PLUS (Agilent) and then transcribed into cRNA labeled with Cy3. In total, 1.65 μg cRNA was hybridized to each microarray. After hybridization, the slides were washed and scanned with an Agilent G2505B Microarray Scanner System. The fluorescence intensities of the scanned images were extracted and preprocessed using Agilent Feature Extraction Software (v9.1). The raw data were normalized using the GeneSpring GX software program, version 11.5 (Silicon Genetics, Redwood City, CA, USA). The raw and processed data are publicly available on the Gene Expression Omnibus (GEO) website under the accession number GSE93520.
Downloading gene expression and clinical data from TCGA
We used the R package “TCGA2STAT” to download the RNA-Seq data and clinical data of patients representing 32 cancer types from The Cancer Genome Atlas (TCGA) database. For RNA-Seq data, we downloaded the RNAseqV2 data for each patient from TCGA. In the subsequent analysis, we used 20 cancer subtypes with at least five normal samples available to validate whether villi-specific genes were significantly dysregulated in tumors. The detailed information of all the cancer types tested (including the number of cancerous and normal samples) is shown in Table S2; furthermore, we used all 32 cancer types for the survival analysis.
Functional analysis and pathway enrichment of villi-specific genes
In an integrated and high-throughput data-mining environment, the Database of Annotation Visualization and Integrated Discovery (DAVID; http://david.abcc.ncifcrf.gov/) was used to analyze differentially expressed genes from established high-throughput experiments. Using the DAVID tool, GO functional analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis were performed for villi-specific genes. In addition, the GO functional enrichment analysis predominantly focused on genes involved in biological process (BP). P<0.05 was used as the cut-off criterion.
Survival analysis
To determine whether the differentially expressed genes were clinically relevant, we performed a survival analysis. For patients with the same cancer type, we used the k-means method to divide the patients into two groups based on established cut-off values for gene expression. Furthermore, we used the R package “survival” to perform a survival analysis based on the Kaplan-Meier method. The differences between the two survival curves were assessed using the log-rank test.
Results
Summary of microarray data
The microarray data were generated using Agilent 4×44K Whole Human Genome Oligo microarrays for the 44 samples, which included villus tissues from 6 to 10 weeks of gestation as well as mature placental tissues. Each time point had at least five samples (detailed information is shown in Table 1). The samples were evaluated by hematoxylin-eosin staining, and at least 90% of each sample was made up of placental chorion to exclude interference from other cell types. RNA was extracted from those samples, and its quality met all the requirements. We used GeneSpring software to normalize and filter the raw microarray data. First, the normalization process resulted in 41,093 probes, among which 15,427 were detected in all the tested samples; these probes were used in subsequent analyses. The raw and processed data are publicly available on the GEO website under the accession number GSE93520.
Villi-specific genes dominate distinct functions with different expression patterns
To determine the primary features of villus development, it was first necessary to identify villi-specific genes that are predominantly expressed in the villus instead as opposed to the mature placenta. We defined villi-specific genes as those with mean expression levels in the villus samples greater than three standard deviations (SDs) above the mean of the mature placenta samples. Ultimately, we identified 237 villi-specific genes (Table S1).
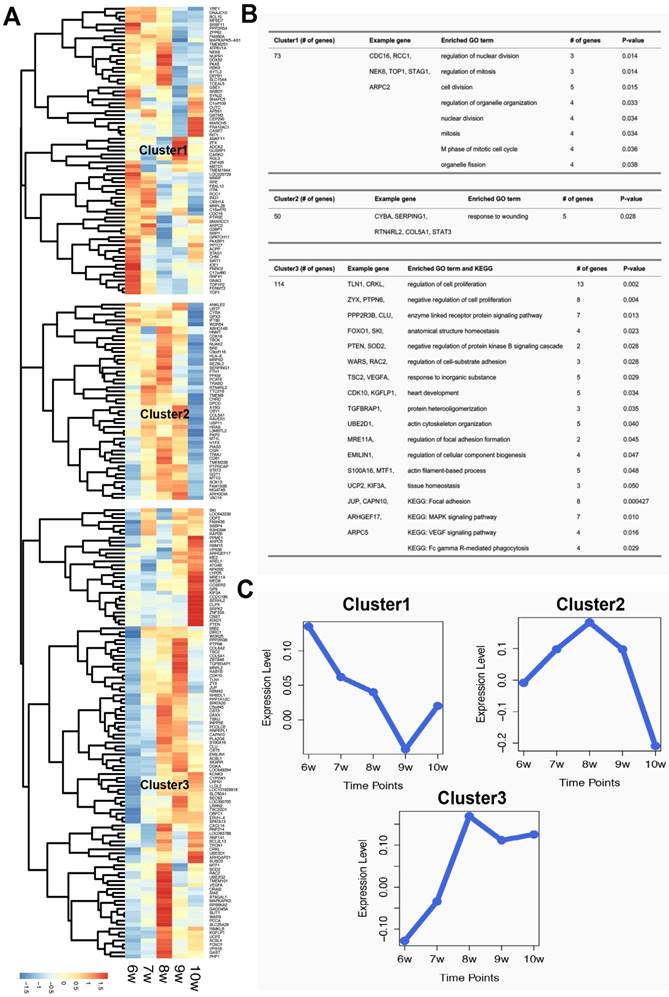
After hierarchical clustering of the identified villi-specific genes, we observed three clusters with distinct expression profiles (Figure 1). The expression levels of genes in Cluster1 decreased from 6 to 9 weeks of gestation. Based on the GO analysis, the significant biological processes of genes in Cluster1 were primarily associated with proliferation, including cell division, nuclear division, and mitosis. In contrast, the expression levels of Cluster2 genes increased from 6 to 8 weeks of gestation and then sharply decreased. Cluster2 genes were significantly enriched for a specific immune-associated biological process: response to wounds. Finally, Cluster3 contained 114 genes whose expression levels increased from 6 to 8 weeks of gestation and then remained steady. These 114 genes were involved in many biological processes, including the regulation of cell proliferation, regulation of cell adhesion formation, and actin filament-based processes. Moreover, eight of these 114 genes were involved in the 'focal adhesion' KEGG pathway. These results suggest that villi-specific genes are linked to proliferation, immunity, and focal adhesions, and these functions may not only be important for embryonic development but also for tumorigenesis and metastasis.
Characteristics of villi-specific genes. (A) Hierarchical clustering of villi-specific gene expression values. (B) Significant GO terms and KEGG pathways for each gene cluster. (C) Expression profiles of the three clusters.
Villi-specific genes are significantly dysregulated in tumors
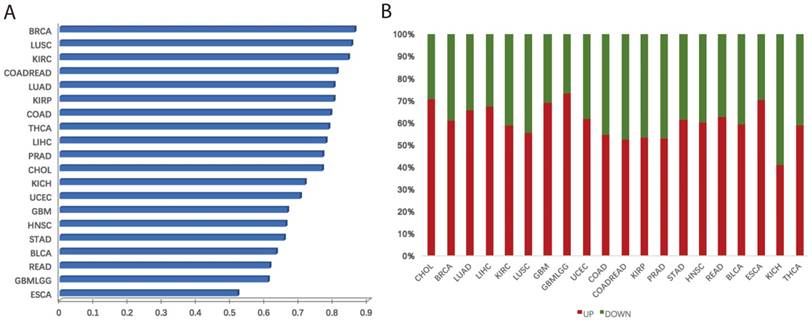
We systematically investigated the expression levels of villi-specific genes in human cancers. This analysis was based on a large set of transcriptomic data derived from 20 cancer types from TCGA (Table S2). Each cancer type had at least five normal samples, and we used t-tests to determine whether the genes were differentially expressed between the cancer and normal samples. If the t-test p value of a gene was less than 0.05, we considered this gene to be differentially expressed [8]. Results revealed that a large fraction of villi-specific genes are aberrantly expressed across cancer types (Figure 2A). For example, 86%, 85%, and 84% of villi-specific genes were differential expressed in breast cancer, lung adenocarcinoma, and renal clear cell carcinoma, respectively. Furthermore, we found that many important villi-specific genes were up-regulated in the cancer samples (Figure 2B). Specifically, 61%, 59%, and 65% of the differentially expressed villi-specific genes were up-regulated in breast cancer, lung adenocarcinoma, and kidney renal clear cell carcinoma, respectively. Large-scale changes in the expression of villi-specific genes across cancer types indicate that embryonic development and cancer share many important mechanisms, including proliferation, immune response, and focal adhesion. We hypothesized that these villi-specific genes might represent an invaluable source of diagnostic and prognostic cancer biomarkers.
Aberrations in villi-specific genes across cancer types. (A) Frequencies of misregulated villi-specific genes in each cancer type. (B) Fraction of up- and down-regulated villi-specific genes in each cancer type.
Immune-, proliferation-, and focal adhesion-related villi-specific genes associated with prognosis
Because many villi-specific genes are systematically dysregulated in a variety of cancers and the main functions of these genes involve the immune response, proliferation, and focal adhesion, we next investigated whether these genes were useful biomarkers.
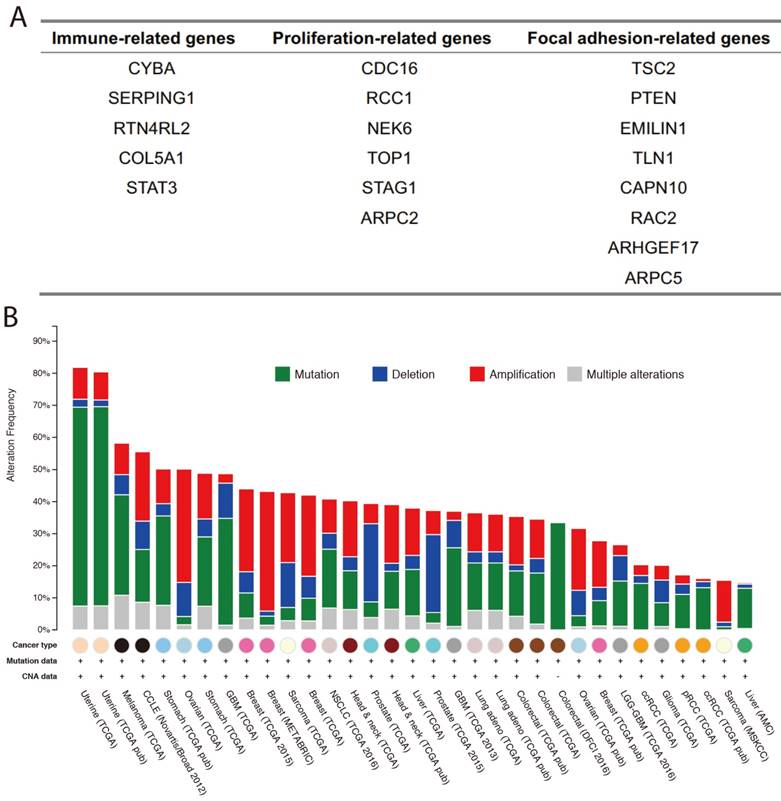
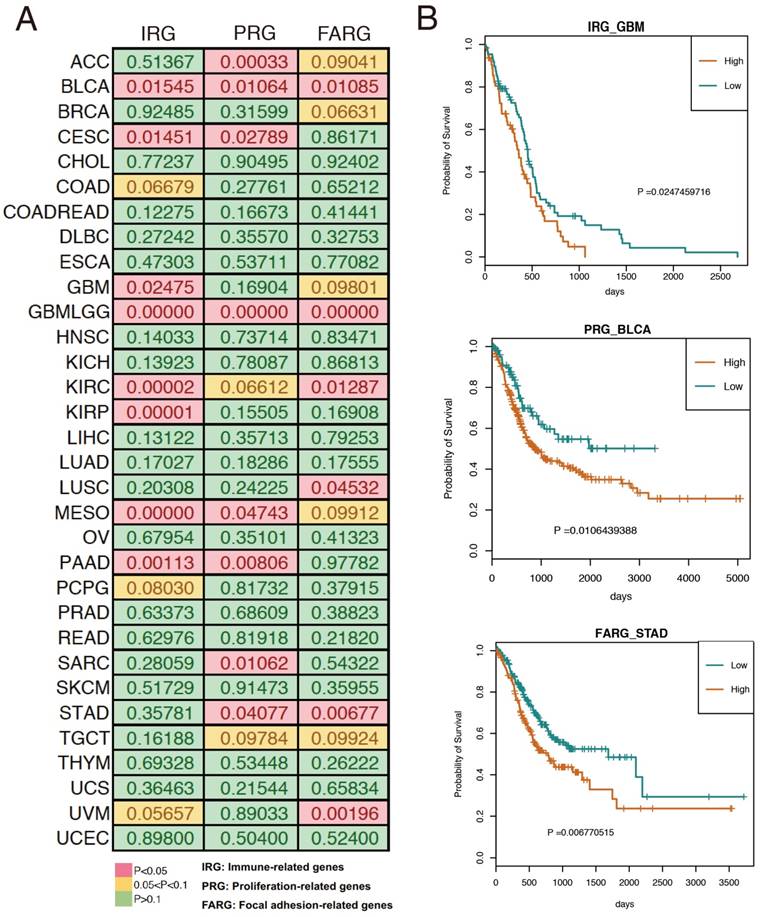
Based on the results of our functional enrichment analysis, we selected five, six, and eight genes from the immune, proliferation, and focal adhesion categories (Figure 3A), respectively. To evaluate the genomic variations of these three groups of genes in cancer, we summarized the mutations and copy number variants found in 33 cancer datasets using cBioPortal [21]. Results indicated that these genes tend to have genomic alterations across many cancer types, including somatic mutations, amplification, and deletion. For example, TCGA GBM (glioblastoma multiforme) data indicated that the COL5A1 gene was mutated, amplified and deleted, and previous studies demonstrated that COL5A1 expression is altered in GBM patients [22]. Moreover, the mutation and amplification of the TLN1 gene was identified in TCGA STAD (stomach adenocarcinoma), and Philip R. Taylor et al. observed that TLN1 was significantly associated with the risk of stomach cancer [23]. We also used the expression levels of each group of genes to split the cancer samples into two groups by hierarchical clustering, and we found that expression of these three groups of genes was associated with patient survival in various cancer types (Figure 4A). For example, high expression of immune-related genes was significantly associated with poor survival in GBM patients (Figure 4B). We also found a similar situation for proliferation- and focal adhesion-related genes in BLCA (bladder carcinoma) and STAD, respectively.
Immune-, proliferation-, and focal adhesion-related villi-specific genes. (A) Detailed gene lists for the three groups. (B) Genomic alterations in the three groups of genes from various cancer datasets.
Associations between the three gene groups and patient survival. (A) Log-rank p value for each cancer type. (B) Representative examples of survival curves for the three groups of genes in cancer.
Discussion
The close relationship between carcinogenesis and embryonic development has led to the emergence of a new research approach for studying cancer. Embryonic development is a reliable and strictly regulated model that can provide critical clues for studying tumor development [2]. In this study, transcriptomic data from multiple time points were used to investigate differential gene expression profiles during placental villi development. By analyzing villi microarray data, we identified 237 villi-specific genes that were primarily classified into immune-, proliferation-, and focal adhesion-related genes using hierarchical clustering. Overall, we found that tumor cells often mimic the biological behaviors of trophoblastic cells.
We discovered that these three groups of genes were associated with patient prognosis in a variety of human cancers. All three groups were associated with BLCA, glioblastoma multiforme lower grade glioma (GBMLGG), kidney renal clear cell carcinoma (KIRC), and mesothelioma (MESO) survival. Furthermore, these four cancers showed increased expression of these genes and were all related to poor prognosis.
Based on placental villi development data analysis, we identified three groups of genes that predominantly exert immune-, proliferation-, and focal adhesion-related activities. Because the expression of these genes could predict the prognosis of cancer patients, placental villi development might be a useful tool for cancer research. Previous studies demonstrated that the above mentioned three gene groups all played essential roles in the development and progression of various cancer types. For example, STAT3, an immune-related gene, has been reported to be aberrantly expressed in human intrahepatic cholangiocarcinoma [24] and colorectal cancer [25]; moreover, elevated STAT3 expression in both these cancers is associated with poor prognosis. NEK6 and TOP1 are genes involved in proliferation. NEK6 is highly expressed in gastric cancer [26] and colorectal cancer [27], and some researchers believe that this gene may be an important diagnostic and prognostic marker. Similarly, TOP1 is a novel tumor-associated antigen, and autoantibodies against TOP1 were detected at a relatively high frequency in the sera of individuals with early-stage non-small-cell lung cancer, gastric cancer, colorectal cancer and esophageal squamous cell carcinoma [28]. TSC2 and RAC2 are focal adhesion-related genes that have been reported in several cancers. Clinically relevant genomic alterations of TSC2 has been observed in pure mucinous breast carcinoma [29]; RAC2 regulates the actin cytoskeleton during breast cancer metastasis [30] and is associated with gastrointestinal carcinogenesis and progression in gastric cancer [31]. However, to the best of our knowledge, RTN4RL2 has not been reported to be associated with human cancers and requires further investigation. Taken together, these findings indicated that the three gene groups not only are associated with some human cancers at the genomic level (to some extent) but also can predict the prognosis of cancer patients. Accordingly, our findings suggest that the signature of these three gene groups could be associated with a fundamental oncogenic mechanism and that they may cooperate to generate aggressive tumors.
Of course, the transcriptome represents only one level of biology, and these studies should be expanded to other “-omics,” such as proteomics and epigenomics. We have begun investigating protein expression in villus tissues, and preliminary data indicate the existence of differential protein expression between chorionic villi and the mature placenta, which are also associated with protein expression in tumors (to be published). Additionally, DNA methylation also plays a critical role in embryonic development and tumor development. Sophile et al. found that up-regulated genes in embryonic stem cells/germ cells were associated with poor prognosis and metastasis in lung cancer [32], and Schroeder, D.I., et al. suggested that large partially methylated domains are a common developmentally dynamic feature in both normal embryonic development and cancer and that these domains could serve as epigenetic biomarkers [33].
Our findings point the way toward many new avenues of investigation and speculation. Specifically, we also plan to investigate the involvement of processes such as angiogenesis, energetic metabolism, and oxidative stress during chorionic villi development. By using developmental models, we believe that we can uncover novel mechanisms in tumorigenesis and discover novel biomarkers to assist with the diagnosis and treatment of human cancers.
Supplementary Material
Supplementary tables.
Acknowledgements
This work was supported by the Peking Union Medical College Graduate Student Innovation Fund in 2016 [0710-17] and the CAMS Innovation Fund for Medical Sciences (CIFMS2016-I2M-3-005).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Naxerova K, Bult CJ, Peaston A, Fancher K, Knowles BB, Kasif S. et al. Analysis of gene expression in a developmental context emphasizes distinct biological leitmotifs in human cancers. Genome biology. 2008;9:R108
2. Beaman KD, Jaiswal MK, Katara GK, Kulshreshta A, Pamarthy S, Ibrahim S. et al. Pregnancy is a model for tumors, not transplantation. American journal of reproductive immunology (New York, NY: 1989). 2016;76:3-7
3. Iles RK, Delves PJ, Butler SA. Does hCG or hCGbeta play a role in cancer cell biology? Molecular and cellular endocrinology. 2010;329:62-70
4. Nieto MA. Epithelial plasticity: a common theme in embryonic and cancer cells. Science. 2013;342:1234850
5. DaSilva-Arnold S, James JL, Al-Khan A, Zamudio S, Illsley NP. Differentiation of first trimester cytotrophoblast to extravillous trophoblast involves an epithelial-mesenchymal transition. Placenta. 2015;36:1412-8
6. Wilczynski JR. Cancer and pregnancy share similar mechanisms of immunological escape. Chemotherapy. 2006;52:107-10
7. Ridolfi L, Petrini M, Fiammenghi L, Riccobon A, Ridolfi R. Human embryo immune escape mechanisms rediscovered by the tumor. Immunobiology. 2009;214:61-76
8. Pavlov N, Frendo JL, Guibourdenche J, Degrelle SA, Evain-Brion D, Badet J. Angiogenin expression during early human placental development; association with blood vessel formation. BioMed research international. 2014;2014:781632
9. Wang Y, Steinbeisser H. Molecular basis of morphogenesis during vertebrate gastrulation. Cellular and molecular life sciences: CMLS. 2009;66:2263-73
10. Katoh M. Networking of WNT, FGF, Notch, BMP, and Hedgehog signaling pathways during carcinogenesis. Stem cell reviews. 2007;3:30-8
11. Yu X, Feng L, Liu D, Zhang L, Wu B, Jiang W. et al. Quantitative proteomics reveals the novel co-expression signatures in early brain development for prognosis of glioblastoma multiforme. Oncotarget. 2016;7:14161-71
12. Yu X, Feng L, Han Z, Wu B, Wang S, Xiao Y. et al. Crosstalk of dynamic functional modules in lung development of rhesus macaques. Molecular bioSystems. 2016;12:1342-9
13. Eastham AM, Spencer H, Soncin F, Ritson S, Merry CL, Stern PL. et al. Epithelial-mesenchymal transition events during human embryonic stem cell differentiation. Cancer research. 2007;67:11254-62
14. An N, Yang X, Zhang Y, Shi X, Yu X, Cheng S. et al. Cell cycle related genes up-regulated in human colorectal development predict the overall survival of late-stage colorectal cancer patients. Molecular bioSystems. 2016;12:541-52
15. Feng L, Wang J, Cao B, Zhang Y, Wu B, Di X. et al. Gene expression profiling in human lung development: an abundant resource for lung adenocarcinoma prognosis. PloS one. 2014;9:e105639
16. Ferretti C, Bruni L, Dangles-Marie V, Pecking AP, Bellet D. Molecular circuits shared by placental and cancer cells, and their implications in the proliferative, invasive and migratory capacities of trophoblasts. Human reproduction update. 2007;13:121-41
17. Strickland S, Richards WG. Invasion of the trophoblasts. Cell. 1992;71:355-7
18. Holtan SG, Creedon DJ, Haluska P, Markovic SN. Cancer and pregnancy: parallels in growth, invasion, and immune modulation and implications for cancer therapeutic agents. Mayo Clinic proceedings. 2009;84:985-1000
19. Perry JK, Lins RJ, Lobie PE, Mitchell MD. Regulation of invasive growth: similar epigenetic mechanisms underpin tumour progression and implantation in human pregnancy. Clinical science (London, England: 1979). 2010;118:451-7
20. Ma Y, Zhang P, Wang F, Yang J, Yang Z, Qin H. The relationship between early embryo development and tumourigenesis. Journal of cellular and molecular medicine. 2010;14:2697-701
21. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science signaling. 2013;6:pl1
22. Tso CL, Freije WA, Day A, Chen Z, Merriman B, Perlina A. et al. Distinct Transcription Profiles of Primary and Secondary Glioblastoma Subgroups. Cancer Research. 2006;66:159-67
23. Li WQ, Hu N, Wang Z, Yu K, Su H, Wang L. et al. Genetic variants in epidermal growth factor receptor pathway genes and risk of esophageal squamous cell carcinoma and gastric cancer in a Chinese population. Plos One. 2013;8:e68999
24. Xin-Wei Y, Liang L, Guo-Jun H, Xin-Zhou Y, Qin-Guo X, Lei C. et al. STAT3 overexpression promotes metastasis in intrahepatic cholangiocarcinoma and correlates negatively with surgical outcome. Oncotarget. 2016
25. Park JH, van Wyk H, McMillan DC, Quinn J, Clark J, Roxburgh CS. et al. Signal Transduction and Activator of Transcription-3 (STAT3) in Patients with Colorectal Cancer: Associations with the Phenotypic Features of the Tumor and Host. Clin Cancer Res. 2017;23:1698-709
26. Lin YL, Chen HL, Cheng SB, Yeh DC, Huang CC, P'Eng F K. et al. Methylation-silencing RCC1 expression is associated with tumorigenesis and depth of invasion in gastric cancer. International journal of clinical and experimental pathology. 2015;8:14257-69
27. Gerceker E, Boyacioglu SO, Kasap E, Baykan A, Yuceyar H, Yildirim H. et al. Never in mitosis gene A-related kinase 6 and aurora kinase A: New gene biomarkers in the conversion from ulcerative colitis to colorectal cancer. Oncology reports. 2015;34:1905-14
28. Yie SM, Ye SR, Ma XL, Xie K, Zhang JB, Cao M. et al. A protein fragment derived from DNA-topoisomerase I as a novel tumour-associated antigen for the detection of early stage carcinoma. British Journal of Cancer. 2016
29. Ross JS, Gay LM, Nozad S, Wang K, Ali SM, Boguniewicz A. et al. Clinically advanced and metastatic pure mucinous carcinoma of the breast: a comprehensive genomic profiling study. Breast Cancer Res Treat. 2016;155:405-13
30. Li H, Yang L, Fu H, Yan J, Wang Y, Guo H. et al. Association between Gαi2 and ELMO1/Dock180 connects chemokine signalling with Rac activation and metastasis. Nature Communications. 2013;4:1706
31. Pan YL, Bi F, Liu N, Du JP, Jiang HP, Xue Y. et al. Rac subfamily expression and activity in gastrointestinal cancer cell lines. Chinese Journal of Oncology. 2003:25
32. Rousseaux S, Debernardi A, Jacquiau B, Vitte AL, Vesin A, Nagy-Mignotte H. et al. Ectopic activation of germline and placental genes identifies aggressive metastasis-prone lung cancers. Science translational medicine. 2013;5:186ra66
33. Schroeder DI, Blair JD, Lott P, Yu HO, Hong D, Crary F. et al. The human placenta methylome. Proc Natl Acad Sci U S A. 2013;110:6037-42
Author contact
![]() Corresponding author: Shujun Cheng, Tel.: +86 10 6776 6703, Fax: +86 10 6776 6703, E-mail: chengshjcn Xuexin Yu, E-mail:yuxuexin2011com
Corresponding author: Shujun Cheng, Tel.: +86 10 6776 6703, Fax: +86 10 6776 6703, E-mail: chengshjcn Xuexin Yu, E-mail:yuxuexin2011com