Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(13):2511-2522. doi:10.7150/jca.18161 This issue Cite
Research Paper
Rac3 Regulates Cell Invasion, Migration and EMT in Lung Adenocarcinoma through p38 MAPK Pathway
Chenlei Zhang1, Tieqin Liu1, Gebang Wang1, Huan Wang2, Xiaofang Che3, Xinghua Gao4 ![]() , Hongxu Liu1, 2
, Hongxu Liu1, 2 ![]()
1. Department of Thoracic Surgery, Cancer Hospital of China Medical University, Liaoning Cancer Hospital & Institute, No.44 Xiaoheyan Road, Dadong District, Shenyang 110042, Liaoning, P.R. China;
2. Department of Thoracic Surgery, The First Hospital of China Medical University, NO.155 North Nanjing Street, Heping District, Shenyang 110001, Liaoning, P.R. China;
3. Department of Medical Oncology, Key Laboratory of Anticancer Drugs and Biotherapy of Liaoning Province, the First Hospital of China Medical University, NO.155 North Nanjing Street, Heping District, Shenyang 110001, Liaoning, P.R. China;
4. Department of Dermatology, The First Hospital of China Medical University and Key Laboratory of Immunodermatology, Ministry of Education and Ministry of Health, NO.155 North Nanjing Street, Heping District, Shenyang 110001, Liaoning, P.R. China.
Received 2016-11-1; Accepted 2017-5-18; Published 2017-8-2
Citation:
Zhang C, Liu T, Wang G, Wang H, Che X, Gao X, Liu H. Rac3 Regulates Cell Invasion, Migration and EMT in Lung Adenocarcinoma through p38 MAPK Pathway. J Cancer 2017; 8(13):2511-2522. doi:10.7150/jca.18161. https://www.jcancer.org/v08p2511.htm
Other stylesAbstract
Background: The role of Rac3 in cell proliferation in lung adenocarcinoma has been tackled in our previous study. However, the role of Rac3 in cell invasion and migration of lung adenocarcinoma is still not clear.
Methods: The expression of Rac3 in lung adenocarcinoma specimens and paired noncancerous normal tissues were evaluated by immunohistochemistry. Lentivirus-mediated RNA interference (RNAi) was employed to silence Rac3 in lung adenocarcinoma cell lines A549 and H1299. A p38 MAPK inhibitor (LY2228820) was employed to inhibit activity of p38 MAPK pathway. Cell invasion and migration in vitro were examined by invasion and migration assays, respectively. PathScan® intracellular signaling array kit and western blot were employed in mechanism investigation.
Results: Rac3 expression was frequently higher in lung adenocarcinoma than paired noncancerous normal tissues. Rac3 expression was an independent risk factor for lymphonode metastasis, and was associated with worse survival outcome. Silencing of Rac3 inhibited cell invasion and cell migration in lung adenocarcinoma cell lines. Knockdown of Rac3 decreased activity of p38 MAPK pathway. LY2228820, which was an important p38 MAPK inhibitor, inhibited Rac3-induced cell invasion and migration of lung adenocarcinoma. E-cadherin expression was increased and vimentin expression was decreased after silencing of Rac3 or following the treatment of LY2228820.
Conclusions: Our findings suggest that Rac3 regulates cell invasion, migration and EMT via p38 MAPK pathway. Rac3 may be a potential biomarker of invasion and metastasis for lung adenocarcinoma, and knockdown of Rac3 may potentially serve as a promising therapeutic target for lung adenocarcinoma.
Keywords: Rac3, lung adenocarcinoma, invasion, migration, p38, EMT.
Introduction
Lung cancer is one of the most frequent causes of cancer-related deaths worldwide [1]. Non-small cell lung cancer (NSCLC) accounts for approximately 85% of lung cancer. The major histological subtype of NSCLC is lung adenocarcinoma which has poor 5-year survival rate because of tumor invasion and metastasis. There are many personalized treatments that have been developed for lung adenocarcinoma, such as gefitinib, erlotinib, crizotinib and ceritinib, but less than 15% of patients with lung adenocarcinoma are ideal candidates for these targeted therapies in the world [2]. Therefore, it is crucial to understand the molecular mechanism of invasion and migration of lung adenocarcinoma and find a novel therapy target.
The small G protein Rac3 (ras-related C3 botulinum toxin substrate 3) belongs to Rho subfamily of Ras proteins [3]. Rac3 proteins cycle between an active state bound to GTP and an inactive state bound to GDP. Rho and Rac-GTPases are known to be involved in human carcinogenesis, cancer cell proliferation, migration and aggression [4 -10]. Rac3 is highly expressed in a wide range of tissues [3], and correlates with poor prognosis of prostate cancer [11]. Rac3 gene has been mapped to chromosome 17q23-25, a region that is frequently deleted in breast and ovarian carcinomas [3,12]. Current studies indicate that Rac3 promotes cell proliferation and cell aggressiveness in breast cancer [9,10], and is also involved in esophagus and ovarian cancer development [13,14].
In our previous studies, we proved that Rac3 could promote cell proliferation and inhibit apoptosis in lung adenocarcinoma cell lines [15,16]. However, the role of Rac3 in lung adenocarcinoma cells invasion and migration is not clear. In this study, we analyzed clinical lung adenocarcinoma samples and determined the relationship between Rac3 expression and lymphonode metastasis. Furthermore, we investigated the role and mechanism of Rac3 in lung adenocarcinoma cells invasion and migration.
Materials and Methods
Patients and specimens
Eighty lung cancer specimens and paired noncancerous normal tissues were obtained from patients pathologically diagnosed as lung adenocarcinoma. All patients underwent complete surgical resections at the Department of Thoracic Surgery in the First Affiliated Hospital of China Medical University from 2008 to 2010. None of patients received chemotherapy or radiotherapy before surgery, and all patients received routine chemotherapy after surgery. All patients had completed survival follow-up data. The survival was defined as the time from the day of surgery to the day of death.
Immunohistochemistry
Eighty surgically excised lung adenocarcinoma specimens and paired noncancerous normal tissues were fixed in 10% neutral buffered formalin and embedded in paraffin, and 4μm paraffin-embedded sections were prepared. The sections were firstly deparaffinized in xylene, and then dehydrated by graded alcohol. After retrieval using sodium citrate buffer (pH 6.0), the slides were washed with phosphate buffered saline (PBS), and incubated with rabbit anti-human Rac3 monoclonal primary antibody (Abcam, USA, 1:100 dilution) overnight at 4℃. Biotinylated anti-rabbit secondary antibody was used at 37℃ for 20min. Steptavidin peroxidase was conjugated at room temperature for 20min. The sections were visualized using 3, 3'-diaminobenzidine (DAB) solution, couterstained with hematoxylin, and dehydrated in ethanol in the end. Two independent pathologists examined the immune-stained slides randomly. Cytoplasmic or nuclear immunostaining was considered positive staining. The intensity of Rac3 staining was graded as follows: 0 for no staining, 1 for light brown color, 2 for medium brown color, and 3 for brown color. The percentage scores of positive cells were as follows: 0 (≤5%), 1 (>5%-25%), 3 (>50%).The final scores of each sample were multiplied to give a score ranging from 0 to 9. Rac3 expression levels were defined as follows: - (score 0), + (score 1-2), ++ (score 3-5), +++ (score 6-9). The Rac3 expression levels were categorized into negative expression (-) and positive expression (+~+++).
Cell culture and reagents
The human lung adenocarcinoma cell lines, A549 and H1299, were obtained from Shanghai Cell Bank (Chinese Academy of Sciences, Shanghai, China). The cells were routinely maintained in Dulvecco's modified Eagle's medium (DMEM Hyclon) supplemented with 10% fetal bovine serum (FBS), 100 units/ml penicillin and 100μg/ml streptomycin at 37℃ in a humidified atmosphere of 5% CO2.
LY2228820 were purchased from selleck company (Shanghai, China). The medium was supplemented with 5 μM LY2228820 for inhibition of p38 MAPK pathway, with DMSO as a control.
Recombinant lentivirus construction and transfection
The complementary DNA (cDNA) sequence of Rac3 was designed from the full-length Rac3 sequence by Shanghai GeneChem Company (Shanghai, China). After testing knockdown efficiencies, stem-loop oligonucleotides were synthesized and cloned into the lentivirus-based vector PscioR (Addgene, Boston, MA, USA). A nontargeting scrambled RNA PscioR vector was generated as a negative control. Lentivirus particles were prepared as described previously [17]. Lung adenocarcinoma cells were infected with the Rac3 siRNA-lentivirus or negative control virus at a multiplicity of infection (MOI) of 20 at Day 7 and examined at Day 10. Cells were incubated in serum-free DMEM at 37oC for 4 h, then 10% DMEM was added and incubated for the following experiments.
RNA extraction and real-time qPCR
Total RNA was extracted from cells with Trizol reagent (Invitrogen, Carlsbad, USA) according to manufacturer′s protocol and converted to cDNA with oligo dT using M-MLV reverse transciptase (Promega, USA). Quantitative Real-time PCR was performed using SYBR Premix Ex Taq (TaKaRa, Dalian, China) on the Real-time PCR system (TaKaRa, Dalian, China), as follows: 95℃ for 15 s, 45 cycles of 95℃ for 5 s and at 60℃ for 30s. A dissociation procedure was performed to generate a melting curve for confirmation of amplification specificity. The PCR primers pairs were as follows: for Rac3, sense 5′-CTCCAAAGTCATCGTCCGGTT and antisense 5′-TGAGTTGCACGTCAAATCTGG; for GAPDH, used as the endogenous control, sense 5′-TGACTTCAACAGCGACACCCA and antisense 5′-CACCCTGTTGCTGTAGCCAAA. The relative expression levels of Rac3 were normalized against GAPDH and analyzed using the 2-ΔΔCt method
[ΔΔCt=(CtRac3-CtGAPDH)sample-(CtRac3-CtGAPDH)control]
Transwell invasion assay and transwell migration assay
Transwell invasion assay was performed using 24-well invasion chambers (Corning, BioCoat, USA) with an 8 micron pore size PET membrane that has been treated with Matriel Matrx. Cells were trypsinized and resuspended, then inoculated into the upper Matrgel chamber in 500μl of serum-free DMEM medium at a concentration of 1×105 cells/well, and incubated for 24 hours in A549 cells or 36 hours in H1299 cells. DMEM medium containing 30% fetal bovine serum in the lower chamber served as the chemoattractant. At the end of incubation, the noninvading cells on the upper membrane surface were erased with cotton swabs. The invasive cells on the lower surface of the membrane were fixed and stained with Giemsa for 5 min. Nine visual fields of each chamber were randomly chosen under a X71 microscope (Olympus, Tokyo, Japan), and the number of invading cells was counted. The invasion activity was quantified by cell counter.
Transwell migration assay was performed according the protocol of cell invasion assay, except that the 24-well chambers (Corning, BioCoat, USA) were not treated with Matriel Matrx. After 72h transfection, A549 cells were incubated for an additional 24 hours for migration, the H1299 cells were cultured for 16 hours for migration. Nine random fields of vision for each chamber were chosen under a X71 microscope (Olympus, Tokyo, Japan), and the number of migratory cells was counted. The migration activity was quantified by cell counter.
Wound-healing assay
Cells migration was determined by wound-healing assay. Cells were seeded in 96-well plates at a concentration of 3×104 cells/well and incubated overnight. The center of cell monolayers was scraped with scratch instruments to create a scratch of constant width when the cultured cells reached a density of >90%. Then cellular debris was washed twice with serum-free DMEM medium, and DMEM medium with 0.5%FBS was added. Cells were cultured at 37℃ in a humidified atmosphere of 5% CO2. Wound area was observed and photographed at 0h, 8h and 24h with X71 microscope (Olympus, Tokyo, Japan). Quantitative analysis of cell migration was performed by using an average gap length from five random fields of vision and the percentage of change in the wound area was calculated using the following formula: % change = (average gap length at time 0h) - (average gap length at time 8h/24h) / average gap length at time 0h ×100%.
Intracellular signaling Array Kit
Cells were trypsinized and resuspended. Intracellular signaling molecules were detected using a PathScan® intracellular signaling antibody array kit (Cell Signaling Technology, CST) according to the manufacture's procedure.
Western blot and simon automated western blot analysis
Western blot analyses were performed 4 days after lentivirus infection. Cells were harvested and lysed in 2×Lysis Buffer (100mM Tris-HCl, 2% mercaptoethanol, 20% glycerol, 4% SDS). Proteins were quantified using a BCA Protein Assay Kit (HyClone-Pierce, Cat no.). Equal amount of proteins (20μg) were separated by 10% SDS-PAGE and transferred onto PVDF membranes (Millipore, Billerica, MA, USA) at 80 V for 150 min. The membranes were blocked with TBST containing 5% nonfat milk for 1h at room temperature and incubated with primary antibodies overnight at 4℃. Secondary antibodies were then incubated with membranes for 2h at room temperature. Signals were detected using the ECL-PLUS/Kit (Amersham). GAPDH was served as a loading control. The Primary antibodies used were as follows: Rac3 (Rabbit, Abcam, 1:500 dilution), E-cadherin (Mouse, CST, 1:500 dilution), vimentin (Rabbit, CST, 1:500 dilution), GAPDH (Mouse, Santa Cruz, 1:2000 dilution). The secondary antibodies were anti-mouse IgG (Santa Cruz, 1:5000 dilution) and anti-rabbit IgG (Santa Cruz, 1:5000 dilution).
Simon automated western blot was performed by using the SimonTM machine (ProteinSimple, San Jose, CA, www.proteinsimple.com/simon.html) according the manufacture's procedure. Briefly, Samples were diluted to final concentration to 1-3μg/μl with sample buffer and master mix (containing fluorescent standards and DTT) and then heated at 95℃ for 5min. The samples, blocking reagent, wash buffer, primary antibodies, secondary antibodies and chemiluminescent substrate were loaded into proper wells in microplates according the manufacturer's instructions. The procedures of plate loading, the separation and immunodetection were fully automated. The Primary antibodies used were as follows: p38 (Mouse, Abcam, 1:50 dilution), p-p38 (Rabbit, CST, 1:50 dilution), β-actin (Mouse, Santa Cruz, 1:20 dilution). The secondary antibodies were anti-mouse IgG (ProteinSimple) and anti-rabbit IgG (ProteinSimple). Data analysis was performed by the Compass Software (ProteinSmiple).
Statistical analysis
All cell culture experiments were performed in triplicate. The immunohistochemistry results and univariate analysis of lymphonode metastasis were performed by Chi-square test. Multivariate analysis of lymphonode metastasis was performed by logistic regression. The overall survival rate was estimated by Kaplan-Meier method and was analyzed by log-rank test. Other data were expressed as the mean±S.D. by Student's t test. Statistical analysis was performed using SPSS 16.0 software (SPSS, IL, USA) and the level of statistical significance was set at P<0.05.
Results
High-expression of Rac3 in lung adenocarcinoma is correlated with lymphonode metastasis and poor survival
Rac3 was positively expressed mainly in the cytoplasm, and significantly expressed in lung adenocarcinoma tissues (Fig. 1A). Rac3 expression was significantly higher expressed in lung adenocarcinoma specimens compared with paired noncancerous normal tissues (P<0.0001, Table 1). Rac3 expression in different tissues was shown in Fig.1 and Table 1. The relationships between lymphonode metastasis and clinical pathological features including Rac3 expression were investigated, we found that lymphonode metastasis was significantly associated with Rac3 expression, gender, TNM stage, differentiation and smoking history (P<0.05, Table 2) in univariate analysis. Furthermore, Rac3 expression and TNM stage are independent risk factors for lymphonode metastasis in multivariate analysis (P<0.05, Table 3). Rac3 positive expression group in lung adenocarcinoma patients had lower overall survival than Rac3 negative expression group (P<0.05, Fig.2).
Figure 1
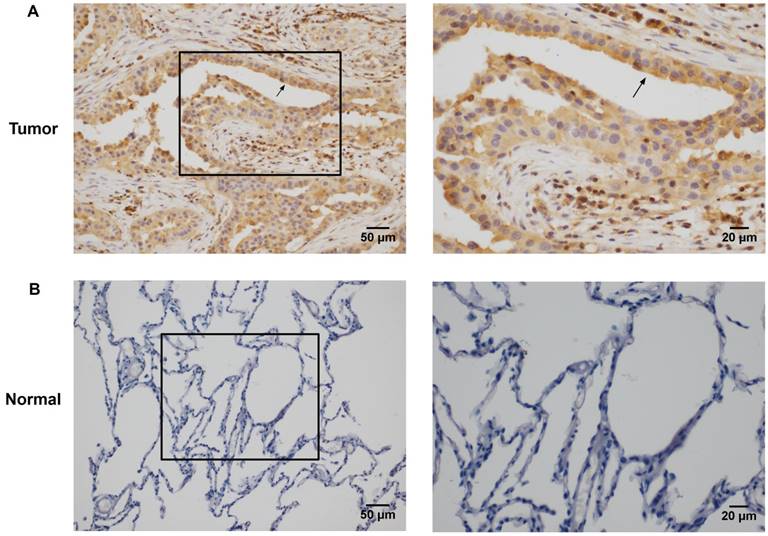
Immunohistochemistry staining of Rac3 expression in lung adenocarcinoma tissues (A magnification ×200, ×400) and paired noncancerous normal tissues (B magnifcation ×200, ×400). Bar=50μm, 20μm
Table 1
Rac3 expression in different tissues (n=80).
| N | Rac3 Expression | Chi-Square Value | P Value | ||
|---|---|---|---|---|---|
| Positive (%) | Negative (%) | ||||
| Lung adenocarcinoma | 80 | 41 (51.3) | 39 (48.7) | 29.789 | <0.0001 |
| Paired noncancerous normal tissues | 80 | 9 (11.3) | 71 (88.7) | ||
Table 2
Univariate analysis of influence of variables on lymphonode metastasis
| Total (n=80) | Lymphonode metastasis | Chi-Square | P Value | ||
|---|---|---|---|---|---|
| Positive | Negative | ||||
| Rac3 expression | 4.086 | 0.043 | |||
| Positive | 41 | 30 | 11 | ||
| Negative | 39 | 20 | 19 | ||
| Age | 0.004 | 0.951 | |||
| ≤60 | 53 | 33 | 20 | ||
| >60 | 27 | 17 | 10 | ||
| Gender | 4.364 | 0.037 | |||
| Male | 36 | 18 | 18 | ||
| Female | 44 | 32 | 12 | ||
| TNM Stage | 30.131 | <0.001 | |||
| I-II | 46 | 17 | 29 | ||
| III-IV | 34 | 33 | 1 | ||
| T Stage | 0.010 | 0.919 | |||
| T1-2 | 73 | 45 | 28 | ||
| T3-4 | 7 | 5 | 2 | ||
| Differentiation | 6.015 | 0.014 | |||
| Well | 34 | 16 | 18 | ||
| Moderate/Poor | 46 | 34 | 12 | ||
| Smoking history | 6.492 | 0.011 | |||
| YES | 31 | 14 | 17 | ||
| NO | 49 | 36 | 13 | ||
Rac3 is effectively knocked down by lentivirus-mediated RNAi
Lentivirus-mediated RNAi technology was employed for Rac3 knock-down. High infection efficiency was confirmed by GFP fluorescence 3 days after lentivirus infection (Fig. 3A). The effect of lentivirus-mediated RNAi on the expression of Rac3 was examined by qRT-PCR and Western blot analysis in A549 and H1299 cell lines. Compared with control group, the mRNA level of Rac3 in Rac3-siRNA groups was greatly decreased (P<0.001 Fig. 3B). A similar decrease was found in protein expression in Rac3-siRNA groups (Fig. 3C). The results indicated that the down-regulation of Rac3 via lentivirus-mediated RNAi was specific and efficient.
Rac3 silencing inhibits cell invasion and migration in A549 and H1299 cells
We performed transwell migration assay and wound-healing assay for cell migration assays in vitro. Compared with control group, the number of migratory cells were significantly decreased after 16 or 24 hours incubation in transwell migration assay (P<0.01 Fig. 4A-B). In wound-healing assay, cells migrated into the wound area more slowly, and the migration rate was significantly decreased than those in control group at 24h (P<0.01 Fig. 4C-D). These results indicated that Rac3 silencing inhibited the migratory abilities of lung adenocarcinoma cells in vitro.
To investigate the effect of Rac3 silencing on the invasion of lung adenocarcinoma cells in vitro, we performed invasion assay using 24-well invasion chambers. The number of invading cells were significantly decreased compared with control group after 24 or 36 hours incubation (P<0.01 Fig 4E-F), which indicated that Rac3 silencing inhibited the invasive abilities of lung adenocarcinoma cells in vitro.
Figure 2
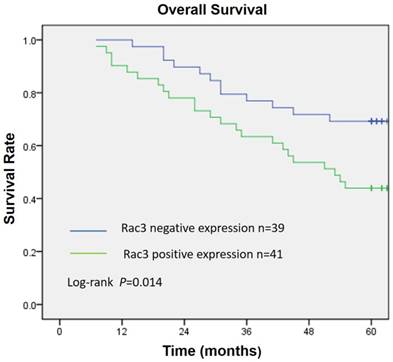
Overall survival curve according to Rac3 expression in 80 lung adenocarcinoma patients. Kaplan-Meier method estimated overall survival. P value was obtained by log-rank test.
Table 3
Multivariate analysis of influence of variables on lymphonode metastasis
| B | S.E. | Wald | P Value | OR | 95% CI | |
|---|---|---|---|---|---|---|
| Rac3 expression | 1.865 | 0.741 | 6.343 | 0.012 | 6.457 | 1.512-27.571 |
| TNM Stage | 4.380 | 1.164 | 14.155 | <0.001 | 79.822 | 8.151-781.682 |
| Differentiation | 1.163 | 0.692 | 2.825 | 0.093 | 3.199 | 0.824-12.415 |
| Smoking history | -0.596 | 0.871 | 0.467 | 0.494 | 0.551 | 0.100-3.041 |
| Gender | 0.306 | 0.941 | 0.106 | 0.745 | 1.358 | 0.215-8.586 |
| Constant | -9.173 | 3.715 | 6.096 | 0.014 | 0.000 |
Figure 3
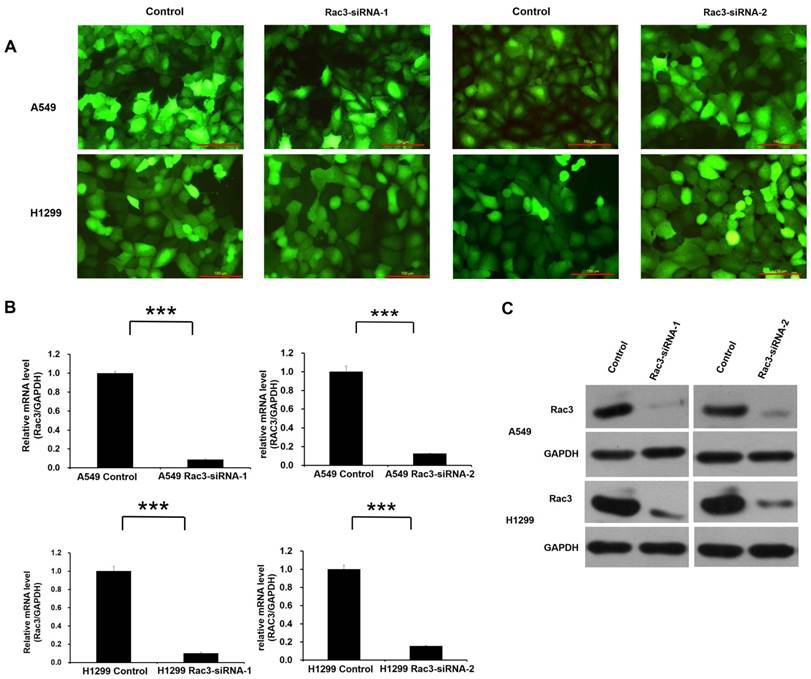
RNAi-mediated knockdown of Rac3 in A549 and H1299 cell lines. A: Transfection efficiency was evaluated by GFP fluorescence in both cell lines. B: The relative mRNA levels of Rac3 were detected by qRT-PCR in both cell lines. C: The protein expression of Rac3 was detected by western blot analysis in both cell lines. GAPDH was used as internal control for western blot analysis. Bar=150μm. *** P<0.001
Rac3 knockdown inhibits activity of p38 MAPK pathway
To further explore the molecular mechanisms by which Rac3 affects lung adenocarcinoma cells invasion and migration, a PathScan® intracellular signaling antibody array kit was used to detect the changes of signaling molecules in A549 cells before and after Rac3 knockdown. The data indicated that the phosphorylation of p38(Thr180/Thr182) was down-regulated in Rac3-RNAi group (P<0.05 Fig. 5A-B). Furthermore, western blot analysis showed that knock down of Rac3 significantly down-regulated p-p38 expression (Fig. 5C). By contrast, no Rac3 silencing-induced changes were observed in p38 expression (Fig. 5C). The results indicated that Rac3 knock down could significantly inhibit invasion and migration of lung adenocarcinoma cells, possibly via p38 MAPK pathway.
LY2228820 inhibited Rac3-induced cell invasion and migration in lung adenocarcinoma cells
LY2228820, which was an important p38 MAPK inhibitor, was used to inhibit activity of p38 MAPK pathway. Transwell migration assay and invasion assay were performed to test cell migration and invasion activities by the treatment with LY2228820, with DMSO as a control. Our results showed that compared with control group, the number of migratory and invasion cells were markedly decreased after 18 or 20 hours incubation following the treatment of LY2228820 at 5μM in A549 cells (P<0.01 Fig. 6A-B).
Figure 4
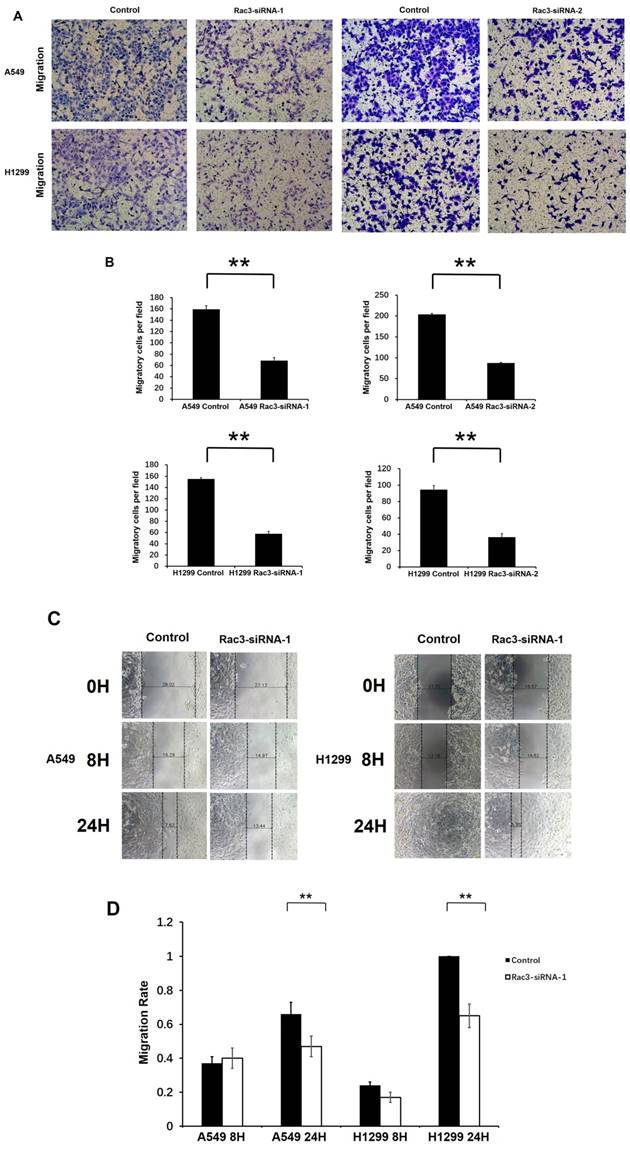
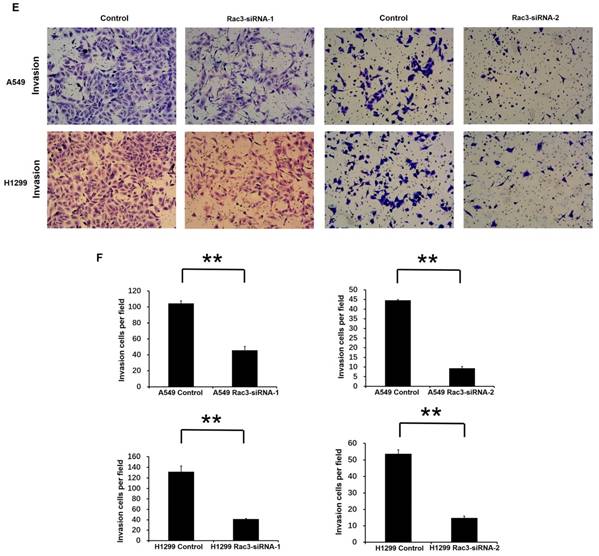
Effect of Rac3 on cell migation and invasion in A549 and H1299 cell lines. A-B: Transwell migration assay was employed to detect migtation ability of control cells and trasnfected Rac3-siRNA cells in both cell lines. C-D: Wound-healing assay was employed to detect migration ability of control cells and transfected Rac3-siRNA cells in both cell lines. E-F: Transwell invasion assay was employed to detect invasion ability of control cells and trasnfected Rac3-siRNA cells in both cell lines. **P<0.01
Figure 5
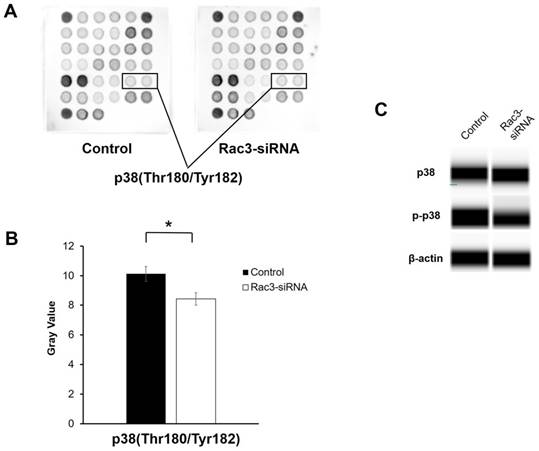
Effects of silencing of Rac3 on cell signaling molecules in A549 cells. A-B: PathScan® intracellular signaling antibody array kit was used to detect the changes of signaling molecules in A549 cells before and after Rac3 knockdown. C: Simon automated western blot analysis was emplyed to detect the protein expression of p38 and p-p38 in A549 cells before and after Rac3 knockdown. β-actin was used as internal control for western blot analysis. *P<0.05
Figure 6
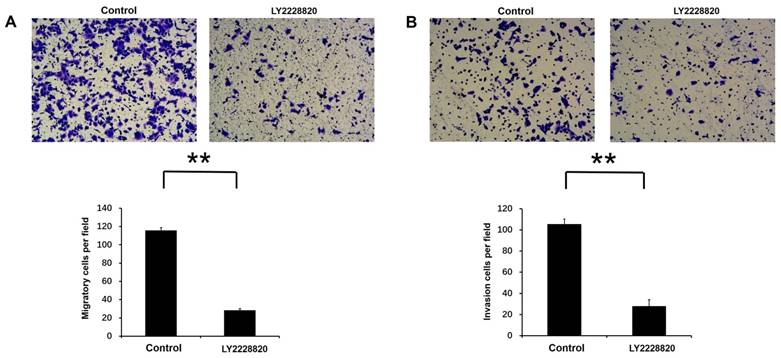
Effect of p38 MAPK pathway on Rac3-induced cell migation and invasion in A549 cells. A: Transwell migration and invasion assays were employed to detect the ability of cell migration and invasion following the treatment of LY2228820 at 5μM. B: The numbers of migrated and invaded cells were counted. **P<0.01
Rac3 regulates EMT through p38 MAPK pathway
Western blot analysis was performed to explore the relationships of Rac3, EMT and p38 MAPK pathway. Our results showed that E-cadherin expression was markedly increased and vimentin expression was decreased after silencing of Rac3 in A549 cells (Fig.7A). Following the treatment of LY2228820 at 5μM for 24h, the expressions of E-cadherin and vimentin were similar to Rac3 knockdown (Fig.7B). These data strongly suggested that Rac3 regulated EMT through p38 MAPK pathway.
Discussion
The rapid progression, recurrence and metastasis of lung adenocarcinoma which is the main subtype of lung cancer are the main reasons for poor prognosis and treatment rate. Therefore, it is of great important to find the precise molecular mechanism involved in lung adenocarcinoma invasion and metastasis. Rac3 was highly expressed in several malignant tumors, such as brain, breast and prostate tumors, which played an important role in cancer development [9-11,18,19]. Accumulating evidence has suggested that Rac3 might act as an oncogene in various human malignant tumors. Most studies were mainly focused on the role of Rac3 in cell proliferation, except for the report that Rac3 promoting cell aggressiveness in breast cancer cells through Rac3/ERK-2/NF-κB signaling pathway [9]. The roles of Rac3 in tumor invasion and migration were seldom investigated, especially in lung cancer. To the best of our knowledge, our study reported for the first time that silencing of Rac3 inhibited lung adenocarcinoma cells invasion and migration.
We examined Rac3 expression in lung adenocarcinoma patients, and found that Rac3 was significantly highly expressed in lung adenocarcinoma tissues than paired noncancerous normal tissues. Rac3 expression was significantly correlated with worse overall survival. These results were similar to our previous study [16]. Moreover, we identified that Rac3 expression was an independent risk factor for lymphonode metastasis of lung adenocarcinoma. These data suggested that Rac3 could be a potential biomarker of lung adenocarcinoma for metastasis. The number of patient specimens was small and there were no data on recurrence and distant metastasis of lung adenocarcinoma, which brought limitations to this study. More patient specimens and clinical data on metastasis of lung adenocarcinoma should be involved in the future.
In this study, we detected modifications of some signaling molecules in cancer cell growth, invasion and migration by Intracellular signaling array kit. Selected signaling molecules were further detected by simon western blot analysis. We found that knock down of Rac3 inhibited activity of p38 MAPK pathway. In terms of the clinical importance of our findings, we found that a selective p38 MAPK inhibitor, LY2228820, reduced the Rac3-induced cell invasion and migration in lung adenocarcinoma cells. LY2228820 demonstrated antitumor activity of various cancers in vitro and vivo, including NSCLC[20], and the safety, tolerability and pharmacokinetics of LY2228820 were acceptable for patients with advanced cancer in phase I study[21]. LY2228820 enhanced the efficacy of standard chemotherapeutic agents used in clinical treatment of ovarian cancer, and was currently being investigated in combination with gemcitabine and carboplatin for women with platinum-sensitive ovarian cancer in phase II study [22].
P38 MAPKs play important roles in tumor cells invasion and metastasis [23]. Numerous studies have suggested that P38 MAPK signaling regulates epithelial-mesenchymal transition (EMT) [24-27], which is associated with invasion and metastasis of tumor [28-30]. Evidence in literatures implicates that p38 regulates EMT protein marker E-cadherin and vimentin expression [31,32]. So we further detected E-cadherin and vimentin expression, and found that vimentin expression was decreased and E-cadherin expression was increased by silencing of Rac3 or the treatment of LY2228820. Previous studies have proven that p38 downregulates E-cadherin induced promotion of EMT in head and neck squamous cell cancer [31], and plays an important role in downregulation E-cadherin during gastrulation [33]. Evidence in the literatures indicates that E-cadherin acts as a suppressor of invasion and metastasis in various cancers, including lung cancer [34-37]. Silencing the expression of P38 and JNK inhibited TGFβ1-mediated EMT of A549 cells, accompanied by the down-regulation of vimentin [32]. Recent studies have demonstrated that vimentin expression was closely related with invasion and metastasis of gallbladder carcinoma [38], prostate carcinoma [39], breast cancer [40-42], cervical carcinoma [43] and NSCLC [44]. Dauphin M et al. [45] reported that vimentin expression was correlated with the distant metastasis of NSCLC after surgery, although there was no significant association between vimentin expression and the lymphonode status in NSCLC. We speculated that Rac3 was not only associated with lymphonode metastasis, but also the distant metastasis of lung adenocarcinoma since Rac3 regulated vimentin expression in lung adenocarcinoma cells. Additionally, in vivo experiments should be performed to confirm the role of Rac3 on tumorigenesis and metastasis of lung adenocarcinoma in the future. The Walker MP et al. [46] found that Rac3, as an estrogen receptor (ER)α co-activator, promoted cell migration of ERα positive breast cancer. Recent research showed that estrogen promoted lung adenocarcinoma cell metastasis through ER-EMT signaling pathway and reduced intercellular adhesion force by PI3K/AKT signaling pathway [47]. Therefore, it is worthy for us to explore the relationships of Rac3, ER, ER-EMT and PI3K/AKT signaling pathways in future studies.
Figure 7
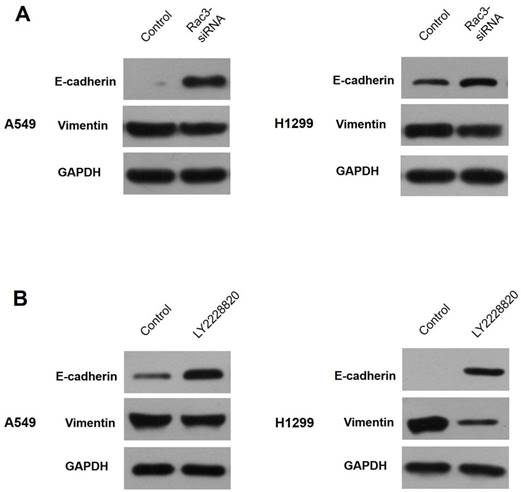
Rac3 regulates EMT through p38 MAPK pathway. A: Western blot analysis was employed to detecte the protein expression of E-cadherin and vimentin after sliencing of Rac3 in A549 cells. B: Western blot analysis was employed to detecte the protein expression of E-cadherin and vimentin following the treatment of LY2228820 at 5μM for 24h in A549 cells. GAPDH was used as internal control for western blot analysis.
In summary, we explore the role of Rac3 in invasion and metastasis of lung adenocarcinoma. Our results suggest that Rac3 was overexpressed in lung adenocarcinoma tissue, and was closely associated with the lymphonode metastasis and worse overall survival rate. Furthermore, we demonstrate that Rac3 promotes cells invasion, migration and EMT of lung adenocarcinoma through p38 MAPK pathway. Our results indicate the crucial role of Rac3 in invasion and metastasis of lung adenocarcinoma, and Rac3 is a potential therapeutic target for lung adenocarcinoma.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 30700821), the Liaoning Province Natural Science Foundation (Grant No. 2015020464), the Liaoning Bai Qian Wan Talents Program (Grant No. 2011921038), the Liaoning Province Science and Technology (S&T) Project (Grant No. 2013225585) and Project for Construction of Major Discipline Platform in Universities of Liaoning province (Grant No. 2016009).
Ethical Standard
This study protocol was approved by the ethics committee of the First Hospital of China Medical University (IRB Approval No. AF-SOP-07-1.0-01). All samples were obtained from the BioBank of the First Hospital of China Medical University and had been previously collected according to official ethical guidelines. Informed consent was obtained from all individual participants included in the study.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Torre LA, Bray F, Siegel RL. et al. Global Cancer Statistics, 2012. CA Cancer J Clin. 2015;65(2):33-64
2. Roviello G. The distinctive nature of adenocarcinoma of the lung. Onco Targets. 2015;8:2399-406
3. Haataja L, Groffen J, Heisterkramp N. Characterization of RAC3, a Novel Member of the Rho Family. J Biol Chem. 1997;272(33):20384-8
4. Fritz G, Just I, Kaina B. Rho GTPases are over-expressed in human tumors. Int J Cancer. 1999;81(5):682-7
5. Li H, Perollier K, Kilic G. et al. Rho GTPases and cancer. Biofactors. 2014;40(2):226-35
6. Ridley AJ. Rho GTPase signalling in cell migration. Curr Opin Cell Biol. 2015;36:103-12
7. Baugher PJ, Krishnamoorthy L, Price JE. et al. Rac1 and Rac3 isoform activation is involved in the invasive and metastatic phenotype of human breast cancer cells. Breast Cancer Res. 2005;7(6):R965-74
8. Ridley AJ. RhoA, RhoB and RhoC have different roles in cancer cell migration. J Microsc. 2013;251(3):242-9
9. Gest C, Joimel U, Huang L. et al. Rac3 induces a molecular pathway triggering breast cancer cell aggressiveness: differences in MDA-MB-231 and MCF-7 breast cancer cell lines. BMC Cancer. 2013;13:63
10. Mira Jp, Benard V, Groffen J. et al. Endogenous, hyperactive Rac3 controls proliferation of breast cancer cells by a p21-activated kinase-dependent pathway. Pro Natl Acad Sci USA. 2000;97(1):185-9
11. Engers R, Ziegler S, Mueller M. et al. Prognostic relevance of increased Rac GTPase expression in prostate carcinomas. Endocrine-Related Cancer. 2007;14(2):245-256
12. Morris CM, Haataja L, McDonald M. et al. The small GTPase RAC3 gene is located within chromosome band 17q25.3 outside and telomeric of a region commonly deleted in breast and ovarian tumours. Cytogenet Cell Genet. 2000;89(1-2):18-23
13. Dong S, Zhao J, Wei J. et al. F-box protein complex FBXL19 regulates TGF β 1-induced E-cadherin down-regulation by mediating Rac3 ubiquitination and degradation. Molecular Cancer. 2014;13:76
14. Junping Li, Yang Liu, Yihua Yin. Inhibitory effects of Arhgap6 on cervical carcinoma cells. Tumor Biol. 2016;37(2):1411-25
15. Liu TQ, Wang GB, Li ZJ. et al. Silencing of Rac3 Inhibits Proliferation and Induces Apoptosis of Human Lung Cancer Cells. Asian Pac J Cancer Prev. 2015;16(7):3061-5
16. Wang G, Wang H, Zhang C. et al. Rac3 regulates cell proliferation through cell cycle pathway and predicts prognosis in lung adenocarcinoma. Tumour Biol. 2016;37(9):12597-12607
17. Lois C, Hong EJ, Pease S. et al. Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science. 2002;295(5556):868-72
18. Chatterjee M, Sequeira L, Jenkins-Kabaila M. et al. Individual rac GTPases mediate aspects of prostate cancer cell and bone marrow endothelial cell interactions. J Signal Transduct. 2011;2011:541851
19. Hwang SL, Chang JH, Cheng TS. et al. Expression of Rac3 in human brain tumors. J Clin Neurosci. 2005;12(5):571-4
20. Campbell RM, Anderson BD, Brooks NA. et al. Characterization of LY2228820 dimesylate, a potent and selective inhibitor of p38 MAPK with antitumor activity. Mol Cancer Ther. 2014;13:364-74
21. Patnaik A, Haluska P, Tolcher AW. et al. A First-in-Human Phase I Study of the Oral p38 MAPK Inhibitor, Ralimetinib (LY2228820 Dimesylate), in Patients with Advanced Cancer. Clin Cancer Res. 2016;22(5):1095-102
22. S Pratt, R Gilmour, N Brooks. et al. LY2228820 Dimesylate, a P38 MAPK Inhibitor, Demonstrates Anti-Neoplastic Activity in Mouse Models of Human Ovarian Cancer. Eur J Cancer. 2012;48(Suppl 6):123
23. del Barco Barrantes I, Nebreda AR. Roles of p38 MAPKs in invasion and metastasis. Biochem Soc Trans. 2012;40(1):79-84
24. Hong J, Zhou J, Fu J. et al. Phosphorylation of serine 68 of Twist1 by MAPKs stabilizes Twist1 protein and promotes breast cancer cell invasiveness. Cancer Res. 2011;71(11):3980-90
25. Hipp S, Berg D, Ergin B. et al. Interaction of Snail and p38 mitogen-activated protein kinase results in shorter overall survival of ovarian cancer patients. Virchows Arch. 2010;457(6):705-13
26. Leng R, Liao G, Wang H. et al. Rac1 expression in epithelial ovarian cancer: effect on cell EMT and clinical outcome. Med Oncol. 2015;32(2):329
27. Wei J, Li Z, Chen W. et al. AEG-1 participates in TGF-beta1-induced EMT through p38 MAPK activation. Cell Bio Int. 2013;37(9):1016-21
28. Micalizzi DS, Farabaugh SM, Ford HL. Epithelial-mesenchymal transition in cancer: parallels between normal development and tumor progression. J Mammary Gland Biol Neoplasia. 2010;15(2):117-34
29. Schopferer M, Bär H, Hochstein B. et al. Desmin and vimentin intermediate filament networks: their viscoelastic properties investigated by mechanical rheometry. J Mol Biol. 2009;388(1):133-43
30. Shirahata A, Sakata M, Sakuraba K. et al. Vimentin methylation as a marker for advanced colorectal carcinoma. Anti-cancer Res. 2009;29(1):279-81
31. Maie St John. p38 Downregulates E-Cadherin in HNSCC. Otolaryngology Head & Neck Surgery. 2010;143(2):P68
32. Chen HH, Zhou XL, Shi YL. et al. Roles of p38 MAPK and JNK in TGF- b 1-induced Human Alveolar Epithelial to Mesenchymal Transition. Arch Med Res. 2013;44(2):93-8
33. Zohn IE, Li Y, Skolnik EY. et al. p38 and a p38-Interacting Protein Are Critical for Downregulation of E-Cadherin during Mouse Gastrulation. Cell. 2006;125(5):957-69
34. Christofori G, Semb H. The role of the cell-adhesion molecule E-cadherin as a tumour-suppressor gene. Trends Biochem Sci. 1999;24(2):73-6
35. Cheng JC, Klausen C, Leung PC. Hydrogen Peroxide Mediates EGF-Induced Down-Regulation of E-Cadherin Expression via p38 MAPK and Snail in Human Ovarian Cancer Cells. Mol Endocrinol. 2010;24(8):1569-80
36. Liu ZL, Wang Q, Huang LN. E-cadherin gene methylation in lung cancer. Tumour Biol. 2014;35(9):9027-33
37. Zhang B, Zhang H, Shen G. Metastasis-uassociated protein 2 (MTA2) promotes the metastasis of non-small-cell lung cancer through the inhibition of the cell adhesion molecule Ep-CAM and E-cadherin. Jpn J Clin Oncol. 2016;46(4):393
38. Dong P, He XW, Gu J. et al. Vimentin significantly promoted gallbladder carcinoma metastasis. Chin Med J (Engl). 2011;124(24):4236-44
39. Wei J, Xu G, Wu M. et al. Overexpression of Vimentin Contributes to Prostate Cancer Invasion and Metastasis via Src Regulation. Anticancer Res. 2008;28(1A):327-34
40. Tanaka K, Tokunaga E, Inoue Y. et al. Impact of Expression of Vimentin and Axl in Breast Cancer. Clin Breast Cancer. 2016;16(6):520-526
41. Calaf GM, Balajee AS, Montalvo-Villagra MT. et al. Vimentin and Notch as biomarkers for breast cancer progression. Oncol Lett. 2014;7(3):721-727
42. Hendrix MJ, Seftor EA, Seftor RE. et al. Experimental co-expression of vimentin and keratin intermediate filaments in human breast cancer cells results in phenotypic interconversion and increased invasive behavior. Am J Pathol. 1997;150(2):483-95
43. Gilles C, Polette M, Piette J. et al. Vimentin expression in cervical carcinomas: association with invasive and migratory potential. J Pathol. 1996;180(2):175-80
44. Tadokoro A, Kanaji N, Liu D. et al. Vimentin Regulates Invasiveness and Is a Poor Prognostic Marker in Non-small Cell Lung Cancer. Anticancer Res. 2016;36(4):1545-51
45. Dauphin M, Barbe C, Lemaire S. et al. Vimentin expression predicts the occurrence of metastases in non small cell lung carcinomas. Lung Cancer. 2013;81(1):117-22
46. Walker MP, Zhang M, Le TP. et al. RAC3 is a pro-migratory co-activator of ERα. Oncogene. 2011;30(17):1984-94
47. ZhaoXZ LiuY, ZhouLJ WangZQ. et al. Role of estrogen in lung cancer based on the estrogen receptor-epithelial mesenchymal transduction signaling pathways. Onco Targets Ther. 2015;8:2849-63
Author contact
![]() Corresponding authors: Hongxu Liu, E-mail: hxliuedu.cn; Xinghua Gao, E-mail: gaobarrycom
Corresponding authors: Hongxu Liu, E-mail: hxliuedu.cn; Xinghua Gao, E-mail: gaobarrycom
Citation styles
APA
Zhang, C., Liu, T., Wang, G., Wang, H., Che, X., Gao, X., Liu, H. (2017). Rac3 Regulates Cell Invasion, Migration and EMT in Lung Adenocarcinoma through p38 MAPK Pathway. Journal of Cancer, 8(13), 2511-2522. https://doi.org/10.7150/jca.18161.
ACS
Zhang, C.; Liu, T.; Wang, G.; Wang, H.; Che, X.; Gao, X.; Liu, H. Rac3 Regulates Cell Invasion, Migration and EMT in Lung Adenocarcinoma through p38 MAPK Pathway. J. Cancer 2017, 8 (13), 2511-2522. DOI: 10.7150/jca.18161.
NLM
Zhang C, Liu T, Wang G, Wang H, Che X, Gao X, Liu H. Rac3 Regulates Cell Invasion, Migration and EMT in Lung Adenocarcinoma through p38 MAPK Pathway. J Cancer 2017; 8(13):2511-2522. doi:10.7150/jca.18161. https://www.jcancer.org/v08p2511.htm
CSE
Zhang C, Liu T, Wang G, Wang H, Che X, Gao X, Liu H. 2017. Rac3 Regulates Cell Invasion, Migration and EMT in Lung Adenocarcinoma through p38 MAPK Pathway. J Cancer. 8(13):2511-2522.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.