Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(12):2369-2383. doi:10.7150/jca.19473 This issue Cite
Review
VSV based virotherapy in ovarian cancer: the past, the present and …future?
Beata Urszula Orzechowska1 ![]() , Marcin Jędryka2,3, Katarzyna Zwolińska1, Rafał Matkowski2,3
, Marcin Jędryka2,3, Katarzyna Zwolińska1, Rafał Matkowski2,3
1. Laboratory of Virology, Ludwik Hirszfeld Institute of Immunology and Experimental Therapy, Polish Academy of Sciences, Weigla 12, 53-114, Wroclaw, Poland
2. Division of Surgical Oncology, Gynaecological Oncology, Chemotherapy and Department of Oncology, Wroclaw Medical University, Plac Hirszfelda 12, 53-413 Wrocław, Poland
3. Lower Silesian Oncology Centre, Wroclaw, Plac Hirszfelda 12, 53-413 Wrocław, Poland
Received 2017-2-3; Accepted 2017-5-2; Published 2017-7-22
Abstract
The standard approach to treating patients with advanced epithelial ovarian cancer (EOC) after primary debulking surgery remains taxane and platinum-based chemotherapy. Despite treatment with this strategy, the vast majority of patients relapse and develop drug-resistant metastatic disease that may be driven by cancer stem cells (CSCs) or cancer initiating cells (CICs). Oncolytic viruses circumvent typical drug-resistance mechanisms, therefore they may provide a safe and effective alternative treatment for chemotherapy-resistant CSCs/CICs. Among oncolytic viruses vesicular stomatitis virus (VSV) has demonstrated oncolysis and preferential replication in cancer cells.
In this review, we summarize the recent findings regarding existing knowledge on biology of the ovarian cancer and the role of ovarian CSCs (OCSCs) in tumor dissemination and chemoresistance. In addition we also present an overview of recent advances in ovarian cancer therapies with oncolytic viruses (OV). We focus particularly on key genetic or immune response pathways involved in tumorigenesis in ovarian cancer which facilitate oncolytic activity of vesicular stomatitis virus (VSV). We highlight the prospects of targeting OCSCs with VSV. The importance of testing an emerging ovarian cancer animal models and ovarian cancer cell culture conditions influencing oncolytic efficacy of VSV is also addressed.
Keywords: epithelial ovarian cancer (EOC), high grade serous ovarian cancer (HGSOC), ovarian cancer stem cells (OCSCs), virotherapy, rhabdovirus, vesicular stomatitis virus (VSV)
Introduction
Despite significant improvements in therapy, cancer is still one of the leading causes of death worldwide. Epithelium ovarian carcinoma (EOC) ranks fifth in lethal tumors among women, accounting for more deaths than any other cancer of the female genital tract. The American Cancer Society estimates that in the United States, in 2016 approximately 22,000 women received a new diagnosis of ovarian cancer, and near 14,000 women succumbed to this disease [1]. Ovarian cancer can be classified into multiple types: serous, endometrioid, clear cell, mucinous, transitional cell, or any combination of these (mixed) with each type having widely different clinicopathologic properties. Among them, high grade serous ovarian cancer (HGSOC) is the most frequent histological type representing 70% of all EOC [2]. Metastasis of EOC involves shedding of malignant cells from the primary tumor into the peritoneal cavity in the form of multicellular 3D spheroids. The cells within spheroid exhibit up-regulated expression of CSC markers suggesting that ovarian CSCs are enriched in this population [3]. Emerging evidence supports the concept that during chemotherapy treatment the majority of differentiated ovarian cancer cells are initially chemosensitive and are eliminated. However platinum-based therapies are unable to eliminate OCSCs that have developed chemoresistance [4, 5]. Therefore, development of novel therapeutic strategies for ovarian cancer therapy is of increasing interest. In particular successful clinical eradication of ovarian CSCs or targeting signaling pathways that are unique to OCSCs would ideally provide effective therapeutics that can potentially eradicate tumors entirely and lead to the sustained remission for patients with ovarian cancer. More and more evidence is accumulating to support the idea that ovarian CSCs can be an excellent target for oncolytic viruses [6, 7]. Oncolytic viruses are natural or modified viruses that are highly selective to tumor cells.
The anti-tumor activity of oncolytic viruses may rely on direct lysis of neoplastic cells or induction of anti-tumor immunity. New generation of oncolytic viruses has been engineered to target CSCs and the CSCs' environment [8]. Vesicular stomatitis virus (VSV) has demonstrated oncolysis or preferential replication in cancer cells and the topic has been addressed in several excellent reviews [9, 10]. Nonetheless, the selective ability of this virus to kill cancer cells while sparing the normal cells remains debatable among some researchers. Also the potential of VSV in targeting of CSCs, specifically ovarian CSCs has not yet been explored. Here we will briefly summarize the current state of our knowledge regarding ovarian cancer, some characteristics of ovarian cancer stem cells, mechanism of their chemoresistance, and current state of oncolytic virotherapy for ovarian cancer [Table 1]. We also review the key components of the innate immune system and signaling pathways that might be involved in VSV susceptibility. In recent years several ovarian cancer animal models and pathway-specific engineered mouse allograft models that functionally recapitulate human serous epithelial ovarian cancer have been developed [11]. Thus, future challenges and the potential of VSV in ovarian cancer treatment is discussed.
Summary of article content
| Section | Key points | References |
|---|---|---|
| 1 | Current diagnostic tools in early detection and treatment options for ovarian cancer patients are still very limited | [12-16] |
| High recurrence rate of chemoresistant ovarian cancer has been associated with self-renewing ovarian cancer stem cells (OCSCs) | [27-40] | |
| Multiple mechanisms have been identified for OCSCs associated chemoresistance | [41-66] | |
| 2 | Several clinical trials to treat ovarian cancer patients with oncolytic viruses have been initiated in recent years | [67-82] |
| 3 | Viruses that have been shown to have the potential of eradicating CSCs include adenovirus (Ads), herpes simplex virus-1 (HSV-1), vaccinia virus (VV), myxoma virus (MYXV), reovirus, measles virus (MV), Newcastle disease virus (NDV), and Maraba virus (MRBV) | [8, 83-98] |
| 4 | Vesicular stomatitis virus (VSV) is newly emerging and promising oncolytic agent for cancer treatment. Defects in innate immune responses involving the interferon (IFN) system and dysregulated translation machinery may contribute to VSV's intrinsic, oncolytic properties Several approaches have been undertaken to improve safety and selectivity of VSV | [99-137] |
| 5 | Efficacy of VSV in ovarian cancer mouse models varied considerably. Discrepancies in the outcome in these models may be related to the differences within tumors | [139-140, 142, 145-152] |
| 6 | The potential oncolytic effectiveness of VSV should be evaluated in newly emerged experimental models (in vivo or in vitro) that recapitulate different aspects of human ovarian cancer such as fallopian tube origins and ovarian cancer spheroids | [153-171] |
1. Ovarian Cancer
Detection and Current State of Ovarian Cancer Treatment
The best currently available protocol for early detection of ovarian cancer is transvaginal ultrasound (TVS) and the presence of elevated CA-125 [12, 13]. Among other biomarkers used for the detection of ovarian cancer is HE4 (human epididymis secretory protein 4), which together with CA125 is a part of the Risk of Ovarian Malignancy Algorithm (ROMA), a scoring model proven to be useful in excluding malignant diagnosis in premenopausal women [14]. New screening methods, including multiple serum markers are also being investigated [15, 16]. All newly diagnosed epithelial ovarian cancer (EOC) patients undergo surgery, which is required for diagnosis and staging, in addition to providing cytoreduction. Nevertheless conventional combinations of primary cytoreductive surgery and paclitaxel-platinum based chemotherapy have not had a significant impact on overall survival of ovarian cancer in last several decades [17]. The overall five-year survival rates of patients diagnosed at stage III and IV of this disease are 32% and 18%, respectively [18]. Patients with platinum-resistant/refractory ovarian cancer are good candidates for novel investigational approaches. Recently it has been shown that introducing an antiangiogenic agent, such as, bevacizumab may significantly prolong the disease free survival time and the response rate to both front-line chemotherapy and recurrent disease treatment of EOC patients [19, 20]. The strategy of antiangiogenic agents actions concentrates on the inhibition of the proangiogenic ligands with antibodies and receptors with tyrosine kinase inhibitors or receptors antibodies which should lead, in combination with chemotherapy, to antiangiogenic-induced tumor vasculature normalization and better chemotherapeutic agents elivery [21]. Recently hyperthermic intraperitoneal chemotherapy (HIPEC) has been investigated as a treatment option for EOC [22, 23]. The development of poly (adenosine diphosphate-ribose) polymerase (PARP) inhibitors also represents a novel therapeutic strategy, especially in patients with advanced, recurrent ovarian cancer who have mutations of the breast cancer 1 gene (BRCA1) or breast cancer 2 gene (BRCA2) [24].
Ovarian Cancer Stem Cells (OCSCs)
Relapse occurring after aggressive treatments such as surgical resection, chemotherapy, and radiotherapy has been attributed to a distinct tumor subpopulation of cancer stem cells (CSCs) or cancer initiating cells (CICs). The idea that cancer is driven by a smaller population of stem cells is not a new one. In the last century it was suggested that tumors contain a cellular subcomponent that retains key stem cell properties [25, 26]. In recent years the clonal evolution of cancer formation in which tumors are thought to be homogeneous masses of proliferating cells with identical genetic alterations was replacing by “cancer stem cell hypothesis”. According to the CSC theory, solid tumors, similarly to many adult tissues may also be hierarchically organized and contain cancer stem cells that sustain tumor growth and relapse after therapy [27, 28]. American Association for Cancer Research defined CSC as a malignant cancer cell with a stem cell phenotype [29]. CSCs are characterized by their ability to self-renew, asymmetric division and their capacity to initiate tumor formation [30]. Several excellent reviews provide comprehensive discussion of the presence of CSCs in many cancers [31, 32]. Ovarian cancer stem cells were first reported in the ascites of ovarian cancer patients [33]. Taking into a consideration the heterogeneity of ovarian tumor, it is possible that each of the types of ovarian cancer has its own unique ovarian CSC phenotype. The characteristic of tumor associated stem cells promises a challenge. Firstly, the expression of cell surface markers in the tissue of interest has to be known, secondly, the presence of normal adult stem cells in the tissue has to be taken into consideration as well. Currently there is no single, specific marker that definitively identify and enrich CSCs, rather a combination of multiple markers depending on the particular tumor type is used. However, with advancement of knowledge in this concept ovarian CSCs have been associated with the cell surface markers such as CD24, CD44, CD133, CD117, EpCAM and differential biochemical properties such as high expression of aldehyde dehydrogenase 1 (ALDH1) [34].
Among them expression rate of CD24, a membrane glycoprotein, positively correlated with higher histopathologic grade, clinical stage and omental metastasis in patients with epithelial ovarian cancer. Gao et al. revealed that CD24+ cells possess stem cell-like characteristics with elevated expression of related stem cell genes and proteins (including Nestin, β-catenin, Bmi-1, Oct4, Oct3/4, Notch 1, Notch 4, CD133, CD44 and CD117). Moreover, CD24+ cells had stronger resistance to chemotherapy drugs with higher self-renewal ability [35]. Another surface transmembrane glycoprotein highly expressed in ovarian CSCs is CD44, a receptor for hyaluronic acid (HA) involved in cell-cell and cell-matrix interactions. Meng et al. demonstrated that CD44+/CD24- cells in ovarian cancer cells exhibit cancer stem cell-like properties of enhanced differentiation, invasion, and resistance to chemotherapy [36]. Another widely described ovarian CSCs marker is CD133. It is a transmembrane glycoprotein associated with cell migration and invasion. It has been demonstrated that ovarian CD133+ CSCs express other stem cell markers, including Oct-4 and Nanog [37]. Chau et al. revealed that CD117, a type III receptor tyrosine kinase, is involved in cell signal transduction in ovarian carcinoma and its presence was associated with poor response to chemotherapy [38]. Nevertheless, it should be noted that ovarian cancer stem cell population may be composed of small overlapping cell fractions defined by mixed stem-like markers. There are several excellent reviews that address the more recent developments in ovarian CSCs research [39, 40].
Mechanisms of Ovarian CSCs Resistance to Chemotherapy
Standard treatment of epithelial ovarian cancer relies on the combination of cytoreductive surgery and combination chemotherapy with taxane and platinum. Recently, ovarian cancer stem cells (CSCs) population is accounted for temporary effects of such therapy and subsequent cancer recurrence in ovarian cancer patients [41, 42]. Multiple mechanisms have been identified for CSCs associated chemoresistance. Yang et al. hypothesized that chemotherapy not only enriches the cancer stem cells due to their intrinsic resistance but induces CSCs in hypoxic region of the tumor [43]. It is still unclear how CSCs survive DNA-damaging agent treatment, acquiring multidrug resistance (MDR) and enhanced the migration potential. Several pathways could be involved in these mechanisms. One of them is an increased rate of efflux of anti-cancer drugs which is mediated mainly by a group of transmembrane transporter glycoproteins known as ATP-binding cassette (ABC) proteins [44, 45]. Glutathione system (GSH), involved in cellular detoxification by conjugation with toxic chemicals and certain chemotherapeutic agents such as cisplatin promote their efflux from cells [46]. Moreover, the glutathione system is one of the intracellular scavenger systems for neutralizing free oxygen radicals (ROS) and has been found to maintain the lower levels of ROS in CSCs, which is essential to sustain CSC self-renewal and stemness [47]. Recently the aldehyde dehydrogenase (ALDH) has also been recognized for CSC associated chemoresistance. Landen et al. demonstrated that expression and activity ALDH which are responsible for oxidizing intracellular aldehydes was significantly higher in taxane and platinum-resistant ovarian cancer cell lines [48]. Deregulation of β-catenin, the key protein in the Wnt signalling pathway may also play a role in the pathogenesis of HGSOC. Furthermore, strong membranous β-catenin expression was associated with resistance to platinum-based chemotherapy [49]. Another marker that has been associated with chemoresistance of ovarian CSCs is a transcriptional receptor Bmi1 (B cell-specific Moloney murine leukemia virus integration site 1). Bmi1 belongs to the polycomb group (PcG) gene family involved in several cellular processes including cell cycle regulation, cell immortalization and senescence of adult, and neoplastic stem cells [50]. Zhang et al. revealed that ovarian tumor-derived spheroid cells overexpressed Bmi1 and furthermore, they were resistant to cisplatin [39]. Additionally it has been demonstrated that Bmi-1 is essential to promote epithelial to mesenchymal transition (EMT) and tumor-initiating capability [51, 52]. EMT is a crucial event for cell dissemination of epithelial tumors. Through this process epithelial cells lose their differentiated characteristics, acquire mesenchymal features, and finally become mesenchymal phenotype which facilitates migration and resistance to chemotherapy. Chiu et al. revealed that chemoresistant ovarian cancer cells exhibit an epithelial-mesenchymal transition (EMT) phenotype and high invasion ability [53]. Ip et al. pointed out the significance of hydrodynamics in ovarian cancer progression. With the use of a microfluidic model of the peritoneum and three-dimensional (3D) spheroids for mimicking tumor behavior, the researchers showed that shear stress enhanced stemness and chemoresistance of ovarian cancer spheroids through downregulation of miR-199a-3p expression and activation of PI3K/Akt signaling [54]. The NFκB pathway which is a major source of pro-inflammatory cytokines may also contribute to ovarian cancer chemoresistance [55]. Recently a higher expression of the survival protein Bcl-2, and a signaling kinase PKCδVIII in cisplatin-resistant ovarian cells TOV-112D has been reported [56]. Another key player which is influencing apoptosis is the p53 protein. Mutation of the p53 gene is a frequent event in tumorigenesis and the majority of high-grade serous carcinomas harbor mutations in this gene [57]. Yang-Hartwich et al. demonstrated that impaired p53 degradation in a population of ovarian cancer cells with cancer stem cell properties led to the imbalance of p53 turnover that promoted the formation of p53 aggregates and consequently to p53 inactivation and platinum resistance [58]. Another pathway, important in maintaining the cancer stem cell in a variety of cancers is Notch signaling. The Notch pathway regulates cell proliferation, cell fate, differentiation, and cell death. It has been found that particularly Notch3 contributes to the tumorigenesis in ovarian cancer. Notch3 is overexpressed in 66% of ovarian serous carcinomas [59]. The study of McAuliffe et al. demonstrated that Notch3 overexpression in ovarian CSCs results in their expansion and increased resistance to cisplatin [60]. Steg et al. has reported other signaling pathways that were overexpressed in recurrent ovarian tumors. Among them were members of the TGF-β superfamily, mainly ENG, and the primary mediators of Hedgehog (Hh) pathway (GLI1 and GLI2) [41]. Additionally, Zeng et al. demonstrated that Hedgehog signaling is also involved in the regulation of CD24 expression, which is one of the most widely described ovarian CSCs markers [61]. Liao et al. pointed out that resistance of ovarian cancer stem cells to conventional chemotherapies may be due to their metabolic properties, mainly their resistance to oxygen deprivation. His group observed that spheroid cells have defects in complete glucose oxidation in the tricarboxylic acid (TCA) cycle and utilize glucose via anaerobic glycolysis. According to the authors, such cells are more likely to survive in severely hypoxic microenvironments and have an increased chemoresistance [62]. Another mechanism involved in acquiring drug resistance is regional gene activation as was demonstrated in taxane-selected human ovarian cancer cell lines [63]. Srivastava et al. showed recently that ovarian CSCs have intrinsically enhanced Pol η-mediated translesion DNA synthesis (TLS) which permitting CSCs to survive cisplatin treatment and leading to tumor relapse [64]. Despite these findings however, successful cytoreduction and chemotherapy is currently a first, and sometimes the only line of defense. It may destroy the CSCs environment preventing tumor recurrence. Paclitaxel for example has been shown to induce cell cycle arrest in the G2-M phase, and activating proapoptotic signaling in ovarian cells [65]. Jia et al. showed that ovarian cell line- SKOV3 treated with paclitaxel, acquired a benign fibroblast-like phenotype and have decreased proliferative potential and impaired capacity to form colonies [66]. It is crucial to fully understand the basic biology of CSCs and mechanisms causing chemoresistance. Furthermore, identification of specific therapeutic targets in key signaling pathways of CSCs may improve the development of novel treatment strategies involving oncolytic viruses.
2. Oncolytic Virotherapy for Ovarian Cancer - Current Clinical Testing
Oncolytic viruses (OVs) are defined as genetically engineered or naturally occurring viruses that selectively replicate and kill cancer cells, spreading within the tumor while sparing surrounding normal, healthy tissue. OVs can be injected intratumorally or intravenously. Systemic distribution of the oncolytic virus allows for the opportunity to treat even distant, metastatic lesions. Another hallmark of oncolytic viruses, besides oncolysis is initiating or augmenting existing anti-tumor immunity. Several virus families have served as backbones for the development of OVs for treatment ovarian cancer [67, 68]. Currently 9 active phase I or II oncolytic clinical trials (as of end of 2016) have been conducted for cancers affecting ovary, fallopian tube and peritoneal cavity. Three study employed measles virus encoding thyroidal sodium iodide symporter IP (NCT02364713, NCT00408590, NCT02068794), no results are yet available [69-71]. The trial NCT02017678 with vaccinia virus (JX-594) with inactivated viral thymidine kinase gene (TK) and encoding human granulocyte-macrophage colony-stimulating factor (GM-CSF) has been withdrawn prior to enrollment, however promising results of yet another study using JX-594 in unresectable primary hepatocellular carcinoma (NCT00554372) gives hope for the treatment of ovarian carcinoma [72]. GL-ONC1- vaccinia virus with the insertion of beta-galactosidase and beta-glucuronidase in place of TK and hemagglutinin, and vaccinia virus Ankara expressing p53 (p53MVA) are also currently evaluated in oncolytic immunotherapy in patients with recurrent ovarian cancer (NCT02759588 and NCT02275039) [73, 74].
Adenovirus (Enadenotucirev, ColoAd1), a chimeric unarmed Ad11p/Ad3 group B - is currently under evaluation in patients with recurrent, platinum-resistant ovarian cancer (NCT02028117), no results are yet available [75]. The safety, potential efficacy, and possible gene transfer imaging capacity of adenovirus expressing a therapeutic thymidine kinase suicide gene -Ad5.SSTR/TK.RGD was confirmed in patients with recurrent gynecologic cancer (NCT00964756) [76]. Another genetically modified adenovirus Ad5Delta24RGD binds to integrin αvβ3 and αvβ5, and it has been shown to be well tolerated by ovarian cancer patients (NCT00562003) [77]. Several replication defective adenoviral vectors (Adp53) to restore of wild-type p53 function have been conducted in various clinical trials. However, clinical studies fail to provide an adequate safety and antitumor efficacy for adenoviral vectors [78]. There are new, promising studies underway utilizing adenoviral vectors in targeting ovarian cancer (NCT02435186 and NCT02140996) [79, 80]. An additional prominent example of OV is reovirus. Used already in many clinical trials as monotherapy currently is also evaluated in combination with chemotherapeutic drug in patients with ovarian epithelial cancer (NCT01199263) [81]. Although reovirus oncotherapy has been shown to have a potential in treatment of ovarian cancer, researchers have to combat the problem of an antiviral host immune responses induced by reovirus and refine delivery of the virus to cancer cells [82].
3. Targeting Cancer Stem Cells with Oncolytic Viruses
A growing number of researchers worldwide question whether CSCs can be eradicated by oncolytic virus therapy. Viruses that have been shown to have the potential of eliminating CSCs include adenovirus (Ads), herpes simplex virus-1 (HSV-1), vaccinia virus (VV), myxoma virus (MYXV), reovirus, measles virus (MV), Newcastle disease virus (NDV) and Maraba virus (MRBV) Tong [8]. Recent study utilizing GP73-regulated oncolytic adenoviruses demonstrated their effectiveness in destroying human liver cancer stem-like cells [83]. Yano et al. have shown that genetically engineered, oncolytic adenovirus, OBP-301 efficiently killed CD133+ GCSCs resistant to chemoradiotherapy [84]. Lv et al. revealed that adenovirus expressing 11R-P53 and GM-CSF targets hepatocellular carcinoma and teratoma stem cells [85]. Among herpesvirus family, HSV G47Δ was found to be highly effective to the CD44+CD24-/low breast cancer stem-like cells population in vitro [86]. An oncolytic HSV-1 mutant rQNestin34.5 has been shown kill neuroblastoma CSCs [87]. Belonging to the poxvirus family-oncolytic vaccinia virus (CVV) effectively suppressed stem cell-like colon cancer cells (SCC) [88]. Recent study of Wang et al. demonstrated that oncolytic VV, GLV-1h68, replicated more efficiently in breast cancer stem-like cells that were characterized by higher ALDH1 activity [89]. Myxoma virus (MYXV), another virus from the Poxviridae family has been shown to effectively infect neuroblastoma CIC cultures and human brain tumor-initiating cells (BTICs) [90, 91]. Moreover, MYXV was potent ex vivo to remove CICs from samples obtained from AML patients [92]. CSCs of breast cancers expressing CD24- CD44+ and ALDH1+ have been susceptible to oncolytic reovirus in vitro and in vivo [93]. Reovirus has been also shown to be effective in human glioblastoma cell line grown in spheroid cultures and in glioblastoma stem-like cells (GSC) [94, 95]. The attenuated strains of measles virus (MV), MV-141.7 and MV-AC133 have been shown to infect and selectively lyse CD133+ tumor cells of primary glioma and colon cancer [96]. Another promising oncolytic virus is Maraba virus (MRBV) belonging to Rhabdoviridae family. Different point mutations in the strain of MRBV were introduced to increase its oncolytic efficiency. The specific tropism of MRBV for cancer cells remains unclear, however MRBV has been shown to be a promising oncolytic agent for cancer virotherapy in a broad range of cancer cells, including EOC [97]. Moreover, Maraba virus effectively replicates in epithelial ovarian cancer (EOC) tumor spheroids cells suggesting that VSV might also be a potential therapeutic agent for eradication of ovarian CSCs [98].
4. Oncolytic properties of vesicular stomatitis virus (VSV)
Vesicular stomatitis virus (VSV) of the Rhabdoviridae family as an oncolytic agent has been studied recently by numerous investigators [99]. VSV's single-stranded RNA genome is relatively simple, fully understood, and easy to manipulate. The 11 kb VSV genome encodes five genes; these include the nucleocapsid (N) protein, phosphoprotein (P), matrix (M) protein, G protein and large polymerase (L). Bullet-shaped virion is enveloped and encodes all five virus-encoded proteins [100]. VSV causes a relatively harmless nature of oral diseases in livestock and symptomatic human infection is usually rare and is characterized by mild, flu-like symptoms [101]. VSV replicates within the cytoplasm of infected cells, and does not undergo genetic recombination; moreover, it does not integrate any part of its genome into the host, and it is not known to have transformation potential. Another advantage of VSV is fast growth to very high titers in vitro, which eases a purification of large amounts of virus and viral proteins in infected cell lysates [102]. Although VSV rarely causes symptomatic human infection, seroconversion with no obvious signs of illness was reported among laboratory workers, in human populations in endemic regions, as well as animal handlers [103]. VSV as an oncolytic agent is currently being considered by a number of insightful research groups in the USA and Canada. Recent findings revealed that several obstacles for successful oncolytic virotherapy with VSV must be overcome. Although preferential tropism of VSV to the tumor cells has been examined in the case of melanoma, sarcoma, glioblastoma, cervical cancer, breast cancer and leukemia [104 and Ref. within], some scientists are not fully convinced in regards to VSV oncoselectivity [105]. We have been exploited VSV in the study of innate immunity in humans for many years. In our extensive research on the population of 300 people we determined the ability of VSV to replicate within human peripheral blood leukocyte populations obtained from healthy individuals as well as from people with immunocompromised immune system. We have shown that sensitivity among people to VSV is differentiated and depends on the condition of the immune system [106-109]. Thus health status of blood donors as well as their age contributes to the sensitivity of PBL to VSV infection [110]. In addition, we have also shown that VSV replication capacity reflects the progress of HIV infection in peripheral blood leukocytes [111]. Oncolytic virotherapy exploits live viruses and the fact that VSV has been shown to cause viral encephalitis in animal models make the safety the highest priority, especially in individuals with immune system compromised by the disease or chemotherapy [112]. Therefore the current key challenge for successful virotherapy is to generate VSV which is resistant to the host's antiviral immune responses, with better tumor specificity, enhanced oncolytic efficacy, and the most important- has an excellent safety profile. Furthermore, a therapeutic synergy with currently available cancer therapeutics would be also desirable. An additional problem associated with the use of VSV in oncolytic therapy is the fact that individual cancers may vary in their sensitivity to VSV even when these cancers arise from the same tissue type. Carey et al. discovered that the resistance to VSV is related primarily to the constitutive expression of antiviral genes, mainly IFN-inducible genes, prior to infection [113]. To improve safety and selectivity of VSV an attenuated matrix protein mutant (M51RVSV) was generated. Tumor cells often harbor defects in the IFN system, therefore VSV replicates to high levels in many transformed cells. Moreover, continuously proliferating cancer cells often have dysregulated translation machinery conducive to VSV replication [9, 114]. Lyles at al. discovered that wild type VSV inhibits hosts antiviral responses through interfering of the VSV matrix (M) protein with the functions of all the three RNA polymerases (RNAP) [115]. Moreover, M protein interacts with the mRNA export factor Rae1 and Nup98 protein in nucleus inhibiting the export of a subset of mRNAs and triggering a shutoff of host gene expression [116]. However, this M- protein mediated inhibition of mRNA export can be antagonized by IFN, which upregulates the expression of Nup98, Nup96 and Rae1 [117]. In contrast to wild type VSV, M51RVSV is defective for inhibition of the host antiviral response, however it can efficiently induce type I IFN production by normal cells. M51RVSV can produce progeny virions and is capable of inducing apoptosis facilitating selective killing of IFN- sensitive cancer cells, therefore M protein mutants have been proposed as strong candidates for oncolytic viral therapy [104]. To further improve selectivity of VSV different approaches have been undertaken. Recent studies have shown that VSV can be efficiently retargeted to different cellular receptors using measles virus envelope glycoproteins, non-neurotropic envelope glycoprotein of lymphocytic choriomeningitis virus (LCMV), Lassa virus, or human immunodeficiency virus type 1 (HIV-1) gp160 [118-121]. Another engineering strategy focused on controlling post-entry replication of VSV. Kelly et al. demonstrated ablated neurotoxicity of recombinant VSV which had incorporated neuron-specific microRNA target sequences without negatively affecting viral oncolysis [122]. Neuroattenuation of VSV, without perturbing its oncolytic potency has been achieved also through engineering of IRES elements from human rhinovirus type 2 (HRV2) and foot-and-mouth disease virus (FMDV) into VSV to control the translation of the matrix gene (M) [123]. Several studies were also conducted on attenuation of VSV through deleting viral genes or disruption of their order [124-126]. The safety and oncoselectivity of VSV was significantly enhanced in VSVs expressing murine interferon beta (VSV-mIFNβ) or human IFN-β (VSV-hIFNβ) and furthermore, such viruses retained their oncolytic activity against tumor cells in vitro and against tumor xenografts in mice [127, 128]. Safety studies on intravenous administration of oncolytic recombinant VSV confirmed that intravenous administration of VSV-mIFNβ-NIS is well tolerated in C57BL/6 mice as well as in purpose-bred dogs [129, 130]. Moreover, a phase I clinical study is currently underway in patients with liver cancer (NCT01628640), mainly to evaluate the safety of intratumoral administration of VSV expressing human IFN-β [131]. Recently Westcott et al. demonstrated that IFN-2α rather than IFN-β may be more effective for improving VSV selectivity in cancer cells [132]. Other interesting studies on improving therapeutic synergy have shown the oncolytic effect of WT VSV or (VSV)-MΔ51 engineered to express suicide enzyme, cytosine deaminase/uracil phosphoribosyltransferase which allow the conversion of non-toxic prodrug (5-fluorocytosine) into toxic, chemotherapeutic 5-fluorouracil (5-FU), which exhibited a considerable bystander effect at the tumor site [133, 134]. Another important problem associated with oncolytic virus therapy is generation of undesirable host immune response. For example neutralizing antibodies can be raised through prior systemic administration of VSV during therapy. Diverse techniques can be applied to protect the oncolytic virus from inactivation by pre-existing antibodies in the blood. Among such techniques one of them is combination of virotherpy with pharmacological immunosuppression to control immune responses [135, 136]. Other methods involve protecting the VSV from neutralization through virus shielding strategies such as serotype switching when the entire VSV-G glycoprotein gene sequence is replaced with that of another serotype or chemical modification [137].
5. Current Status of VSV Based Virotherapy in Ovarian Cancer Animal Models
Anticancer activity of VSV has been shown across a spectrum of malignancies in the preclinical setting [104]. Because significant progress has been made to understand the mechanisms of VSV oncolytic selectivity and activity, therefore a strong rationale exists for the use of VSV in ovarian cancer therapy. In ovarian cancer research one of the major challenges is lack of suitable animal model systems targeting different cell types that truly replicate human ovarian disease. Multiple histological subtypes of ovarian cancer are being treated with similar surgical and therapeutic approaches, even though they originate from different cells and are characterized by various genetic and genomic alterations. Most researchers utilize murine xenograft and transgenic models of ovarian carcinoma. Xenografts of human or mouse ovarian cancer cells in immunodeficient mice allow for investigation of growth, metastasis, and treatment response of ovarian tumors in a live animal. Perspectives for personalized cancer therapy has led to, an increased interest in patient-derived xenografts (PDX) engrafted into immune-compromised mice for preclinical modeling. Tumor cells together with parenchymal and stromal tumor components, can be transplanted into ovarian bursa (intrabursally, IB) or into mouse peritoneal space (intraperitoneally, IP) generating “the Avatar”, an orthotopic PDX model allowing further study of tumor development and metastases [138]. However, xenografts do not illustrate the initial transforming events leading to tumorigenesis. Apart from problems with species differences between host and graft, another major disadvantage of immunodeficient models for oncolytic virotherapy studies is their increased susceptibility to viral infection. Since an inherent neurotropism of wild type VSV poses a severe threat to immunocompromised animals, a recombinant, attenuated version of virus has become a favored approach. A recent study of Dold et al. revealed that wild type VSV caused neurotoxicity at 102 PFU per mice while doses up to 109 PFU of VSV pseudotyped with the glycoprotein of the lymphocytic choriomeningitis virus (LCMV)-VSV-GP were well tolerated even in immmunodeficient, tumor-bearing NOD/SCID mice. However, treatment of subcutaneous xenografts of human ovarian carcinoma with VSV-GP was not very effective. Authors concluded that recurrence of tumors in vivo was related to the intact antiviral mechanisms in the tumor cells and that inhibition of VSV-GP activity was mediated by IFN as application of interferon modulators improved oncolysis [139]. Since mouse xenograft models lack a functional immune system, the development of immunocompetent, genetically engineered mouse models of epithelial ovarian cancer that more accurately recapitulate metastatic ovarian carcinoma is crucial [140]. The cellular origin of ovarian cancer has remained disputable therefore a variety of transgenic animal models have been developed for the various histotypes of ovarian cancer. Genetically engineered mouse models of ovarian cancer faithfully recapitulate many aspects of human disease and have greater clinical relevance because tumor development, neovasculaturization and immune responses take place within the context of normal tissues. Consequently, cancer in transgenic mouse models may be more difficult to treat. There are some excellent mouse transgenic models of EOC [141] but only some were exploited in studies on oncolytic efficacy of VSV. In a first transgenic model of spontaneous EOC (SV40) large T antigen, including the small and large T antigen (TAg) was expressed genes under transcriptional control of the Müllerian inhibiting substance type II receptor promoter (MISIIR). Female TgMISIIR-Tag DR26 transgenic mice spontaneously develop bilateral poorly differentiated serous tumors derived from the ovarian surface epithelium (OSE) [142]. Lesions resemble high-grade serous ovarian carcinoma (HGSOC), the most common form of ovarian cancer. Expression of Simian virus 40 (SV40) large T antigen causes inactivation of p53 and pRB which is a crucial feature as HGSOC is characterized by a high frequency of TP53 mutations, observed in 96% cases. Dysregulation of Rb1 are seen in about 67% cases [143, 144]. Another model is represented by a naturally occurring mutant, white spotting variant (Wv) mice which harbor a point mutation in the kinase domain of the c-kit gene. Mutations at the mouse W/c-kit locus lead to intrinsic defects in stem cells of the melanocytic, hematopoietic, and germ cell lineages. The Wv/Wv mice develop tubular adenomas, a benign epithelial neoplasm that can become invasive in older Wv mice [145].
The results of oncolytic cancer therapy with VSV in these two mouse models are quite different. Initial testing the therapeutic efficacy of an attenuated strain VSV-Δ51 in a TgMISIIR-Tag DR26 mouse was not satisfactory [146]. Nonetheless this group reported later that replication and spread of the virus was augmented within tumors arising on the ovaries by the histone deacetylase inhibitor (HDI) treatment which weakened the innate antiviral responses [147]. Arulanandam et al. study demonstrated that treatment with VSV-Δ51-GM-CSF and microtubule-destabilizing agents (MDAs) such as colchicine both delivered i.p. led to significantly prolonged survival of TgMISIIR-TAg mice as compared with either monotherapy [148]. On the other hand, a study utilizing Wv mice, demonstrated the high activity of VSV in the monotherapy. VSV was particularly efficient in clearing tumor lesions without significant replication in any other organs and tissues [149]. Discrepancies in outcome in these models may be related to the differences within tumors. The ovarian tubular adenomas in the ovary of the Wv mice resemble rather the low-grade ovarian cancer tumors, which usually have a wild-type Trp53 and KRas or Raf-1 activating mutation while lesions in TgMISIIR-TAg animals are similar to HGSOC with inactivated p53 [150]. The differences in two models are highlighted by Cai et al. The study revealed that even the addition of an oncogenic mutation, such as Trp53 deletion or reduction of Pten gene dosage in Wv mice did not convert the benign ovarian epithelial tumors into malignant cancer resembling HGSOC [151]. Nevertheless the white spotting variant (Wv) mouse model can provide important insights into the etiology and pathogenesis of ovarian cancer and is an excellent research model for the menopause study. There are also several disadvantages of using mouse models. The researchers pointed out that some mouse models do not accurately mimic human molecular mechanisms of inflammatory responses [152]. For example unlike human ovaries, murine ovaries have a covering bursa that protects the peritoneal cavity from epithelial cell shedding in the OSE, which could inhibit metastasis. Additionally there are differences in the cycle between human and mice. Humans are a mono-ovulatory species and have menstrual cycles rather than estrous cycles [18]. All of these make the interpretation of mouse models extremely challenging.
6. VSV virotherapy in ovarian cancer-future study directions
The potential oncolytic effectiveness of VSV in ovarian cancer should be more precisely evaluated in newly emerged experimental models (in vivo or in vitro) that recapitulate different aspects of human ovarian cancer. Such experimental models include mouse model of fallopian tube origins of HGSOC and mouse models with altered signaling pathways, which are not only involved in ovarian cancer development, but are also crucial for the innate immune responses. Not to mention that animal models should address a metastatic potential of ovarian cancer spheroids, presence and biology of CSCs, and their role in chemoresistance. Table 2 presents a simplified overview of the development of various mouse models addressing the issue of the origin of high grade serous ovarian cancer (HGSOC) as well as potential mechanisms of VSV oncoselectivity. In the contexts of recent findings that HGSOC is considered a müllerian origin resembling the epithelium of the fallopian tube, mogp-TAg transgenic mouse has been developed [153]. In this model the SV40 large T-antigen (TAg) is expresses under the control of the mouse müllerian-specific murine oviduct-specific glycoprotein promoter (Ovgp-1) that is highly active in the fallopian tube. Transgenic mogp-TAg mice develop neoplastic lesions in the fallopian tube resembling human serous tubal intraepithelial carcinoma (STIC) with disrupted p53 and Rb pathways [154]. Therefore mogp-TAg mice model is valuable not only for elucidating the mechanisms of serous ovarian tumorigenesis, but may also serve as more accurate model for testing the efficacy of VSV as an oncolytic virus.
New experimental mouse models of high-grade serous ovarian carcinoma (HGSOC)
| Promoter | Targeted gene | Mechanism | Tumorigenesis | Reference |
|---|---|---|---|---|
| Ovgp-1 | SV40 TAg | Inactivation of p53 and pRb | Neoplastic lesions in the fallopian tube resembling human serous tubal intraepithelial carcinoma (STIC) a potential precursor of ovarian HGSC | Miyoshi et al., 2002 [153] Sherman-Baust et al., 2014 [154] |
| MISIIR (Amhr2-Cre) | Dicer-/-& Pten-/- | Disruption of PI3K and AKT pathways (increased phosphorylation of AKT, STMN1- stathmin) and BIRC5 -survivin) | High grade serous carcinomas from the oviduct. Tumors spread to the ovary and metastasize throughout the abdominal cavity. Upregulated expression of cytokeratin 14, 17, and 8, E-cadherin, CA125 | Kim et al., 2012 [155] |
| Pax8-Cre | Brca 1 or Brca 2-/-, p53-/-, Pten-/- | Alteration in PTEN/PI3K pathway | Serous tubal intraepithelial carcinoma (precursor lesion to ovarian HGSC and peritoneal carcinomas). Upregulated expression of including cytokeratin-8, STMN1, PAX2, P53, Ki-67, and CA-125 | Perets et al., 2013 [160] |
| AdCre | Pik3ca & Pten-/- | Disruption of PI3K and AKT pathways (increased phosphorylation of AKT) | Serous papillary hyperplasia of the ovaries | Kinross et al., 2012 [156] |
| AdCre | Pten-/- & Apc-/-& p53-/- | Disruption of PI3K/AKT/mTOR pathway | High grade metastatic ovarian carcinomas CD24+ cells showed greater tumor initiation rates | Burgos-Ojeda et al., 2015 [161] |
STIC, serous tubal intraepithelial carcinoma; HGSC, high-grade serous carcinoma; MISIIR, Müllerian inhibiting substance type II receptor; PAX2 and PAX8, transcription factors; CA-125, cancer antigen 125; Ki-67, a nuclear protein expressed in proliferating mammalian cells; PI3K, phosphatidylinositol 3-kinase; AKT, serine/threonine kinase, mTOR, mammalian target of rapamycin; PTEN, phosphatase and tensin homolog; Dicer, endoribonuclease
In another transgenic model of EOC a conditional knock out of Dicer and Pten led to activation of the phosphatidylinositol 3-kinase (PI3K) and AKT pathway, and resulted in the development of high grade serous adenocarcinomas from the oviduct. Authors suggest that the lesions consist of cells of a mesenchymal lineage since they originate in the stroma of the fallopian tube. Moreover, in this model the primary tumors metastasize throughout the abdominal cavity [155]. Kinross et al. confirmed development of HGSOC in mice with yet another alteration of PI3K/AKT/mTOR pathway with deletion of Pten and mutation in PIK3CA gene enhancing PI3K activity [156]. PTEN is the tumor-suppressor phosphatase with phosphatidylinositol 3,4,5 trisphosphate (PIP3) as its primary substrate. Because PIP3 is the main cellular product of PI3K, PTEN is capable of antagonizing PI3K activity and negatively regulates the PI3K-AKT cell-survival signaling pathway [157] (Figure 1). PTEN loss and simultaneous activation of AKT and mTOR signaling is frequently observed in human ovarian carcinomas. Martins et al. revealed that PTEN not only is frequently deleted in HGSOC but reduced PTEN expression is associated with significantly worse survival [158]. Another important role of PTEN is associated with its role as the p53 regulator which is a key factor as the majority of HGSOC also harbor mutations of p53. It has been shown that PTEN and p53 interact and regulate each other at the transcription as well as protein level. PTEN regulates p53 protein levels through AKT-dependent and independent mechanisms which could be one of the switching mechanism between cell death and survival [159]. Perets et al. established serous tubal intraepithelial carcinoma as the precursor lesion to high-grade serous ovarian and peritoneal carcinomas in mouse models by targeting the Brca, Tp53, and Pten genes [160]. Burgos-Ojeda et al. utilized yet another transgenic murine model of ovarian cancer with conditional deletion of Pten additionally to Apc and Trp53 in the ovarian surface epithelium (OSE), which results in the generation of high grade metastatic ovarian carcinomas. Although these tumors have endometrioid histology, in the presence of a p53 mutation they, have a high grade metastatic phenotype similar to that seen in patients with high grade serous cancer. What is interesting is that cell lines and primary tumors derived from this model had higher expression of CD24 and CD44 markers that have been associated with cancer stem cells (CSCs). Moreover these CD24+ cells possessed higher tumorigenic ability and demonstrated a greater ability to passage to form secondary spheres. CD24+ cells had also increased pSTAT3 and expressed the stem cell genes Nanog, cyclin-D1, and c-Myc [161]. It is not surprising since Schubbert et al. demonstrated recently that PTEN loss leads the development of CSCs that share properties with somatic stem cells, including the capacity for self-renewal and multi-lineage differentiation [162]. Moreover another study revealed that PTEN is also crucial for stem cell maintenance as PTEN loss can lead to the emergence and proliferation of cancer stem cell (CSC) clones [163]. Therefore it would be worth testing oncolytic efficacy of VSV in ovarian cancer mouse model based on fallopian tube transformation or with deletion of PTEN in particular because tumor suppressor PTEN has a critical role in antiviral innate immunity. Recently, Li et al. demonstrated that PTEN is involved in IRF3-mediated, innate immunity and can be responsible for the inhibition of VSV replication. VSV-induced induction of type I interferon was severely impaired in PTEN-mutant mice and this effect was accompanied with higher VSV titers in the livers of PTEN-mutant mice and higher VSV-triggered mortality than in PTEN-wild-type mice.
A schematic overview of the molecular network involved in VSV oncoselectivity. Binding of growth factors to the receptor tyrosine kinase (RTK) activates the receptor complex, which in turn recruits and activates PI3K. Activated PI3K converts PIP2 to PIP3, which subsequently mediates the phosphorylation of AKT. Tumor suppressor PTEN negatively regulates the pathway by removing the 3-phosphate from PIP3, converting it back to PIP2. PTEN is crucial for the activation of IRF3, its import into the nucleus and production of IFN-β. Loss of PTEN leads to over-activation of AKT and subsequently mTOR, which is associated with uncontrolled cell growth, proliferation, and survival. PTEN loss leads also to the development of cancer stem cells (CSCs) and an impaired cellular responses to viral infection. Up-regulation of LDLR in ovarian carcinomas enables VSV to enter cells with altered PTEN function through LDLR. Abbreviations: PI3K, phosphatidylinositol 3-kinase; PIP2, phosphatidylinositol (4,5)-bisphosphate); PIP3, phosphatidylinositol (3,4,5)-trisphosphate); AKT, serine/threonine kinase; IRF3, interferon regulatory factor-3; mTOR, mammalian target of rapamycin; LDLR, low-density lipoprotein receptor. See text for further details.
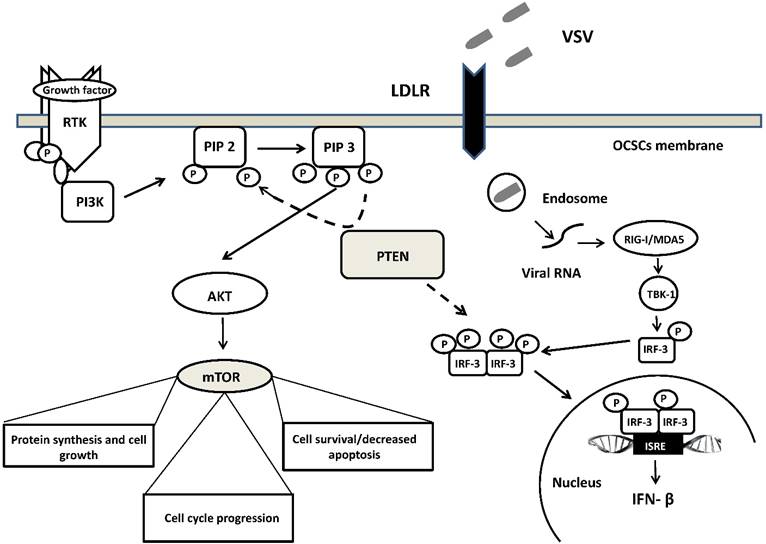
Moreover, deficiency in PTEN and its phosphatase activity increased the replication of VSV in human prostate cancer cells PC-3. Enhancement of VSV replication and induction of type I interferon was independent of the PI3K/Akt signaling pathway and was related rather to the impairment import of IRF3 (a master transcription factor responsible for IFN-β production) into the nucleus. [164] (Figure 1). However, it is worth mentioning that regardless of the suppressive effects on the antiviral response of acute deletion of PTEN, its loss does not always account for the difference in susceptibility to VSV infection. Recently, Yu et al. study revealed that deletion of Pten in murine prostate epithelial progenitor (MPE) caused poor response to interferon and increased susceptibility to VSV infection. On the other hand, tumor-derived Pten-/- cells were resistant to VSV infection. Authors speculate that sensitivity to VSV infection observed in early tumor development, following Pten deletion is lost during tumor evolution due to other mutations or heterogeneity in the tumor microenvironment [165]. The role of PTEN in participating in VSV oncoselectivity should then be further elucidated.
One of the reasons that EOC is difficult to treat is due to the unique mechanism of ovarian cancer metastasis. EOC dissemination involves local invasion of pelvic and abdominal organs through aggregates of malignant cells (spheroids). Intra-peritoneal metastatic lesions are initiated during proteinase-mediated shedding of single cells and spheroids that subsequently adhere to peritoneal mesothelial cells [166]. In vitro cultured spheroids possess high tumorigenic ability. Pease et al. revealed that spheroids can be formed by budding directly as adherent clusters from a monolayer of ovarian cancer cell lines. This phenomenon was accompanied by expression of vimentin and lack of cortical E-cadherin suggesting epithelial to mesenchymal transition (EMT). Furthermore, cells grown as spheroids acquired progressive resistance to the chemotherapy drugs paclitaxel and cisplatin in comparison to their adherent counterparts [167]. The main problem associated with the current developments of new antitumor agents is that they do not address the anchorage- and vascular-independent ovarian cancer spheroids. Therefore studying ovarian cancer development in a three-dimensional (3D) system has many advantages. Currently there is no stable ovarian CSC model, nevertheless, hanging drop culture method, 3D cultures with three-dimensional matrices or co-culture of ovarian cancer cells with mesothelial cells or multiple cell types provide for now an excellent platform to study adhesion, invasion, as well as tumor proliferation and differentiation [168, 169]. These techniques require highly specified spheroid culture conditions with media containing growth factors such as LIF (leukaemia inhibitory factor), FGF (fibroblast growth factor), EGF (epidermal growth factor) and Insulin. 3-dimensional spheroids rather than monolayer cultures should be used to screen for anticancer drugs or oncolytic efficacy of viruses. In an in vitro model of epithelial ovarian cancer metastasis Tong et al. demonstrated different kinetics of Maraba virus (MRBV)-mediated killing of epithelial ovarian cancer (EOC) between monolayer and tumor spheroids [98]. Both VSV and Maraba virus are classified as vesiculoviruses and they require low-density lipoprotein receptor (LDLR) for binding and entry of host cells [170]. MRBV entry into EOC spheroid cells was significantly reduced in comparison to adherent cells and this effect was associated with decreased expression of LDLR protein in spheroids. However, after initial delay MRBV replicated within spheroids generating significant cell killing effects [97] Pampalakis et al. has found up-regulated of LDLR in ovarian carcinomas [171] that implies that VSV might targets ovarian cancer spheroids as well.
Concluding remarks
EOC metastasis occurs via the shedding of malignant cells from the primary tumor into the peritoneal cavity. Ovarian cancer stem cells (CSCs) within spheroids, commonly present in cancer associated ascites are believed to be a source of both chemotherapy resistance and metastases. Cancer cells multicellular spheroids are less sensitive to apoptosis. Therefore virotherapy with oncolytic virus penetrating deep into the hypoxic and acidic region of the 3D spheroids seems a quite appealing idea. Vesicular stomatitis virus (VSV) is a is an attractive oncolytic agent for cancer virotherapy. Since VSV is able to replicate in healthy cells, several approaches have been used to engineer a VSV that replicates preferentially in tumor cells. The potential of VSV to eradicate ovarian CSCs is not confirmed however more and more studies point out the key elements of antiviral response and signaling pathways that may improve VSV oncolysis. Therefore employing newly emerged, genetically engineered mouse models of human serous epithelial ovarian cancer (HGSOC), or with alteration of PI3K/AKT/mTOR pathway, and 3D tumor spheroid models for screening VSV oncolytic potential is necessary in order to be able to determine its real role as a promising approach to ovarian cancer therapy.
Abbreviations
EOC: epithelial ovarian cancer CSCs: cancer stem cells CICs: cancer initiating cells VSV: vesicular stomatitis virus HGSOC: high grade serous ovarian cancer STIC: serous tubal intraepithelial carcinoma Bmi1: B cell-specific Moloney murine leukemia virus integration site 1 EMT: epithelial to mesenchymal transition MISIIR: Müllerian inhibiting substance type II receptor promoter SV40 Tag: Simian virus 40 large T antigen.
Acknowledgements
The authors thank Ms. Izabella Orzechowska, LSHTM, London, UK for English-language editing
Funding
This work was supported by Wroclaw Centre of Biotechnology, programme: The Leading National Research Centre (KNOW) for years 2014-2018.
Statement of author contributions
O. B. U. conceived and wrote the manuscript, J. M. wrote the section concerning ovarian cancer detection and treatment, M. R gave conceptual advice, Z. K. revised the manuscript critically for important intellectual content.
All authors were involved in reviewing the paper and had final approval of the submitted and published versions.
Competing Interests
All authors declare no conflicts of interest.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7-30
2. McCluggage WG. Morphological subtypes of ovarian carcinoma: a review with emphasis on new developments and pathogenesis. Pathology (Phila). 2011;43:420-32
3. Bapat SA. Human ovarian cancer stem cells. Reprod Camb Engl. 2010;140:33-41
4. Ahmed N, Abubaker K, Findlay JK. Ovarian cancer stem cells: Molecular concepts and relevance as therapeutic targets. Mol Aspects Med. 2014;39:110-25
5. Chen X, Zhang J, Zhang Z, Li H, Cheng W, Liu J. Cancer stem cells, epithelial-mesenchymal transition, and drug resistance in high-grade ovarian serous carcinoma. Hum Pathol. 2013;44:2373-84
6. Cripe TP, Wang P-Y, Marcato P, Mahller YY, Lee PW. Targeting Cancer-initiating Cells With Oncolytic Viruses. Mol Ther. 2009;17:1677-82
7. Smith TT, Roth JC, Friedman GK, Gillespie GY. Oncolytic viral therapy: targeting cancer stem cells. Oncolytic Virotherapy. 2014;3:21-33
8. Tong Y, Qian W. Targeting cancer stem cells with oncolytic virus. Stem Cell Investig. 2014;1:20
9. Balachandran S, Barber GN. Defective translational control facilitates vesicular stomatitis virus oncolysis. Cancer Cell. 2004;5:51-65
10. Simovic B, Walsh SR, Wan Y. Mechanistic insights into the oncolytic activity of vesicular stomatitis virus in cancer immunotherapy. Oncolytic Virotherapy. 2015;4:157-67
11. Hasan N, Ohman AW, Dinulescu DM. The promise and challenge of ovarian cancer models. Transl Cancer Res. 2015;4:14-28
12. Brown DL, Andreotti RF, Lee SI, Dejesus Allison SO, Bennett GL, Dubinsky T. et al. ACR appropriateness criteria© ovarian cancer screening. Ultrasound Q. 2010;26:219-23
13. Zeng J, Yin J, Song X, Jin Y, Li Y, Pan L. Reduction of CA125 Levels During Neoadjuvant Chemotherapy Can Predict Cytoreduction to No Visible Residual Disease in Patients with Advanced Epithelial Ovarian Cancer, Primary Carcinoma of Fallopian tube and Peritoneal Carcinoma. J Cancer. 2016;7:2327-32
14. Kadija S, Stefanovic A, Jeremic K, Radojevic MM, Nikolic L, Markovic I. et al. The utility of human epididymal protein 4, cancer antigen 125, and risk for malignancy algorithm in ovarian cancer and endometriosis. Int J Gynecol Cancer Off J Int Gynecol Cancer Soc. 2012;22:238-44
15. Nolen BM, Lokshin AE. Biomarker Testing for Ovarian Cancer: Clinical Utility of Multiplex Assays. Mol Diagn Ther. 2013;17:139-46
16. Fan L, Yin M, Ke C, Ge T, Zhang G, Zhang W. et al. Use of Plasma Metabolomics to Identify Diagnostic Biomarkers for Early Stage Epithelial Ovarian Cancer. J Cancer. 2016;7:1265-72
17. Bookman MA, Brady MF, McGuire WP, Harper PG, Alberts DS, Friedlander M. et al. Evaluation of new platinum-based treatment regimens in advanced-stage ovarian cancer: a Phase III Trial of the Gynecologic Cancer Intergroup. J Clin Oncol Off J Am Soc Clin Oncol. 2009;27:1419-25
18. Vaughan S, Coward JI, Bast RC, Berchuck A, Berek JS, Brenton JD. et al. Rethinking ovarian cancer: recommendations for improving outcomes. Nat Rev Cancer. 2011;11:719-25
19. Aghajanian C, Finkler NJ, Rutherford T, Smith DA, Yi J, Parmar H. et al. OCEANS: A randomized, double-blinded, placebo-controlled phase III trial of chemotherapy with or without bevacizumab (BEV) in patients with platinum-sensitive recurrent epithelial ovarian (EOC), primary peritoneal (PPC), or fallopian tube cancer (FTC). J Clin Oncol Off J Am Soc Clin Oncol. 2011;29:LBA5007
20. Perren TJ, Swart AM, Pfisterer J, Ledermann JA, Pujade-Lauraine E, Kristensen G. et al. A phase 3 trial of bevacizumab in ovarian cancer. N Engl J Med. 2011;365:2484-96
21. Teoh D, Secord AA. Antiangiogenic agents in combination with chemotherapy for the treatment of epithelial ovarian cancer. Int J Gynecol Cancer Off J Int Gynecol Cancer Soc. 2012;22:348-59
22. Ansaloni L, Coccolini F, Morosi L, Ballerini A, Ceresoli M, Grosso G. et al. Pharmacokinetics of concomitant cisplatin and paclitaxel administered by hyperthermic intraperitoneal chemotherapy to patients with peritoneal carcinomatosis from epithelial ovarian cancer. Br J Cancer. 2015;112:306-12
23. Sp S, G P, Jaka R, Rauthan A, Hs M, Karanth S. Secondary Cytoreductive Surgery and Hyperthermic Intraperitoneal Chemotherapy (CRS+HIPEC) for Recurrent Epithelial Ovarian Cancer (EOC): Indian Experience. J Mol Genet Med. 2016:1-7
24. Chung C, Lee R. An update on current and emerging therapies for epithelial ovarian cancer: Focus on poly(adenosine diphosphate-ribose) polymerase inhibition and antiangiogenesis. J Oncol Pharm Pract. 2016 [Epub ahead of print]
25. Virchow Von Rudolf. Art. XIV.-Die Cellular Pathologie, in ihrer Begrundung auf physiologische und pathologische Gewebelehre. Am J Med Sci. 1861;1:465-79
26. Wicha MS, Liu S, Dontu G. Cancer Stem Cells: An Old Idea—A Paradigm Shift. Cancer Res. 2006;66:1883-90
27. Greaves M, Maley CC. Clonal evolution in cancer. Nature. 2012;481:306-13
28. Beck B, Blanpain C. Unravelling cancer stem cell potential. Nat Rev Cancer. 2013;13:727-38
29. Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CHM, Jones DL. et al. Cancer stem cells-perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006;66:9339-44
30. Nguyen LV, Vanner R, Dirks P, Eaves CJ. Cancer stem cells: an evolving concept. Nat Rev Cancer. 2012;12:133-43
31. Xia P. Surface markers of cancer stem cells in solid tumors. Curr Stem Cell Res Ther. 2014;9:102-11
32. Tirino V, Desiderio V, Paino F, Rosa AD, Papaccio F, Noce ML. et al. Cancer stem cells in solid tumors: an overview and new approaches for their isolation and characterization. FASEB J. 2013;27:13-24
33. Bapat SA, Mali AM, Koppikar CB, Kurrey NK. Stem and progenitor-like cells contribute to the aggressive behavior of human epithelial ovarian cancer. Cancer Res. 2005;65:3025-9
34. Zhan Q, Wang C, Ngai S. Ovarian Cancer Stem Cells: A New Target for Cancer Therapy. BioMed Res Int. 2013;2013:916819. doi: 10.1155/2013/916819
35. Gao M-Q, Choi Y-P, Kang S, Youn JH, Cho N-H. CD24+ cells from hierarchically organized ovarian cancer are enriched in cancer stem cells. Oncogene. 2010;29:2672-80
36. Meng E, Long B, Sullivan P, McClellan S, Finan MA, Reed E. et al. CD44+/CD24- ovarian cancer cells demonstrate cancer stem cell properties and correlate to survival. Clin Exp Metastasis. 2012;29:939-48
37. Long H, Xie R, Xiang T, Zhao Z, Lin S, Liang Z. et al. Autocrine CCL5 Signaling Promotes Invasion and Migration of CD133+ Ovarian Cancer Stem-Like Cells via NF-κB-Mediated MMP-9 Upregulation. STEM CELLS. 2012;30:2309-19
38. Chau WK, Ip CK, Mak ASC, Lai H-C, Wong AST. c-Kit mediates chemoresistance and tumor-initiating capacity of ovarian cancer cells through activation of Wnt/β-catenin-ATP-binding cassette G2 signaling. Oncogene. 2013;32:2767-81
39. Zhang S, Balch C, Chan MW, Lai H-C, Matei D, Schilder JM. et al. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res. 2008;68:4311-20
40. Walters Haygood CL, Arend RC, Straughn JM, Buchsbaum DJ. Ovarian cancer stem cells: Can targeted therapy lead to improved progression-free survival? World J Stem Cells. 2014;6:441-7
41. Steg AD, Bevis KS, Katre AA, Ziebarth A, Dobbin ZC, Alvarez RD. et al. Stem cell pathways contribute to clinical chemoresistance in ovarian cancer. Clin Cancer Res Off J Am Assoc Cancer Res. 2012;18:869-81
42. Abubaker K, Latifi A, Luwor R, Nazaretian S, Zhu H, Quinn MA. et al. Short-term single treatment of chemotherapy results in the enrichment of ovarian cancer stem cell-like cells leading to an increased tumor burden. Mol Cancer. 2013;12:24
43. Yang G, Lu X, Fu H, Jin L, Yao L, Lu Z. Chemotherapy not only enriches but also induces cancer stem cells. Biosci Hypotheses. 2009;2:393-5
44. Donnenberg VS, Meyer EM, Donnenberg AD. Measurement of multiple drug resistance transporter activity in putative cancer stem/progenitor cells. Methods Mol Biol Clifton NJ. 2009;568:261-79
45. Ween MP, Armstrong MA, Oehler MK, Ricciardelli C. The role of ABC transporters in ovarian cancer progression and chemoresistance. Crit Rev Oncol Hematol. 2015Nov;96(2):220-56
46. Chen HHW, Kuo MT. Role of Glutathione in the Regulation of Cisplatin Resistance in Cancer Chemotherapy. Met-Based Drugs. 2010:2010 doi: 10.1155/2010/430939
47. Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, Kulp AN. et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458(7239):780-3
48. Landen CN, Goodman B, Katre AA, Steg AD, Nick AM, Stone RL. et al. Targeting aldehyde dehydrogenase cancer stem cells in ovarian cancer. Mol Cancer Ther. 2010;9:3186-99
49. Bodnar L, Stanczak A, Cierniak S, Smoter M, Cichowicz M, Kozlowski W. et al. Wnt/β-catenin pathway as a potential prognostic and predictive marker in patients with advanced ovarian cancer. J Ovarian Res. 2014;7:16
50. Lessard J, Sauvageau G. Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature. 2003;423:255-60
51. Yang M-H, Hsu DS-S, Wang H-W, Wang H-J, Lan H-Y, Yang W-H. et al. Bmi1 is essential in Twist1-induced epithelial-mesenchymal transition. Nat Cell Biol. 2010;12:982-92
52. Paranjape AN, Balaji SA, Mandal T, Krushik EV, Nagaraj P, Mukherjee G. et al. Bmi1 regulates self-renewal and epithelial to mesenchymal transition in breast cancer cells through Nanog. BMC Cancer. 2014;14:785
53. Chiu W-T, Huang Y-F, Tsai H-Y, Chen C-C, Chang C-H, Huang S-C. et al. FOXM1 confers to epithelial-mesenchymal transition, stemness and chemoresistance in epithelial ovarian carcinoma cells. Oncotarget. 2014;6:2349-65
54. Ip CKM, Li S-S, Tang MYH, Sy SKH, Ren Y, Shum HC. et al. Stemness and chemoresistance in epithelial ovarian carcinoma cells under shear stress. Sci Rep. 2016Jun1;6:26788
55. Alvero AB. Recent insights into the role of NF-kappaB in ovarian carcinogenesis. Genome Med. 2010;2:56
56. Patel N, Morris A, Patel R, Carter G, Cooper D, Murr M. PKCδVIII and Bcl2 Increase Ovarian Cancer Survival via LncRNA NEAT1 Secreted by Obese Adipose Derived Stem Cells. FASEB J. 2015;29:578.7
57. Yemelyanova A, Vang R, Kshirsagar M, Lu D, Marks MA, Shih IM. et al. Immunohistochemical staining patterns of p53 can serve as a surrogate marker for TP53 mutations in ovarian carcinoma: an immunohistochemical and nucleotide sequencing analysis. Mod Pathol Off J U S Can Acad Pathol Inc. 2011;24:1248-53
58. Yang-Hartwich Y, Soteras MG, Lin ZP, Holmberg J, Sumi N, Craveiro V. et al. p53 protein aggregation promotes platinum resistance in ovarian cancer. Oncogene. 2015;34:3605-16
59. Park JT, Li M, Nakayama K, Mao T-L, Davidson B, Zhang Z. et al. Notch3 Gene Amplification in Ovarian Cancer. Cancer Res. 2006;66:6312-8
60. McAuliffe SM, Morgan SL, Wyant GA, Tran LT, Muto KW, Chen YS. et al. Targeting Notch, a key pathway for ovarian cancer stem cells, sensitizes tumors to platinum therapy. Proc Natl Acad Sci U S A. 2012;109:E2939-2948
61. Zeng C, Chen T, Zhang Y, Chen Q. Hedgehog signaling pathway regulates ovarian cancer invasion and migration via adhesion molecule CD24. J Cancer. 2017;8:786-92
62. Liao J, Qian F, Tchabo N, Mhawech-Fauceglia P, Beck A, Qian Z. et al. Ovarian Cancer Spheroid Cells with Stem Cell-Like Properties Contribute to Tumor Generation, Metastasis and Chemotherapy Resistance through Hypoxia-Resistant Metabolism. PLOS ONE. 2014;9:e84941
63. Wang YC, Juric D, Francisco B, Yu RX, Duran GE, Chen GK. et al. Regional activation of chromosomal arm 7q with and without gene amplification in taxane-selected human ovarian cancer cell lines. Genes Chromosomes Cancer. 2006;45:365-74
64. Srivastava AK, Han C, Zhao R, Cui T, Dai Y, Mao C. et al. Enhanced expression of DNA polymerase eta contributes to cisplatin resistance of ovarian cancer stem cells. Proc Natl Acad Sci. 2015;112:4411-6
65. Ahn HJ, Kim YS, Kim J-U, Han SM, Shin JW, Yang HO. Mechanism of taxol-induced apoptosis in human SKOV3 ovarian carcinoma cells. J Cell Biochem. 2004;91:1043-52
66. Jia L, Zhang S, Ye Y, Li X, Mercado-Uribe I, Bast RC. et al. Paclitaxel inhibits ovarian tumor growth by inducing epithelial cancer cells to benign fibroblast-like cells. Cancer Lett. 2012;326:176-82
67. Li S, Tong J, Rahman MM, Shepherd TG, McFadden G. Oncolytic virotherapy for ovarian cancer. Oncolytic Virotherapy. 2012;1:1-21
68. Hartkopf AD, Fehm T, Wallwiener D, Lauer U. Oncolytic virotherapy of gynecologic malignancies. Gynecol Oncol. 2011;120:302-10
69. Search of: NCT02364713 - List Results - ClinicalTrials.gov. https://clinicaltrials.gov/ct2/results?term=NCT02364713&Search=Search
70. Search of: NCT00408590 - List Results - ClinicalTrials.gov. https://clinicaltrials.gov/ct2/results?term=NCT00408590&Search=Search
71. Search of: NCT02068794 - List Results - ClinicalTrials.gov. https://clinicaltrials.gov/ct2/results?term=NCT02068794&Search=Search
72. Breitbach CJ, De Silva NS, Falls TJ, Aladl U, Evgin L, Paterson J. et al. Targeting tumor vasculature with an oncolytic virus. Mol Ther J Am Soc Gene Ther. 2011;19:886-94
73. Search of: NCT02759588 - List Results - ClinicalTrials.gov. https://clinicaltrials.gov/ct2/results?term=NCT02759588&Search=Search
74. Search of: NCT02275039 - List Results - ClinicalTrials.gov. https://clinicaltrials.gov/ct2/results?term=NCT02275039&Search=Search
75. Search of: NCT02028117 - List Results - ClinicalTrials.gov. https://clinicaltrials.gov/ct2/results?term=NCT02028117&Search=Search
76. Kim KH, Dmitriev I, O'Malley JP, Wang M, Saddekni S, You Z. et al. A phase I clinical trial of Ad5.SSTR/TK.RGD, a novel infectivity-enhanced bicistronic adenovirus, in patients with recurrent gynecologic cancer. Clin Cancer Res Off J Am Assoc Cancer Res. 2012;18:3440-51
77. Search of: NCT00562003 - List Results - ClinicalTrials.gov. https://clinicaltrials.gov/ct2/results?term=NCT00562003&Search=Search
78. Chen G, Zhang S, He X, Liu S, Ma C, Zou X-P. Clinical utility of recombinant adenoviral human p53 gene therapy: current perspectives. OncoTargets Ther. 2014;7:1901-9
79. Search of: NCT02435186 - List Results - ClinicalTrials.gov. https://clinicaltrials.gov/ct2/results?term=NCT02435186&Search=Search
80. Search of: NCT02140996 - List Results - ClinicalTrials.gov. https://clinicaltrials.gov/ct2/results?term=NCT02140996&Search=Search
81. Search of: NCT01199263 - List Results - ClinicalTrials.gov. https://clinicaltrials.gov/ct2/results?term=NCT01199263&Search=Search
82. Clements D, Helson E, Gujar SA, Lee PW. Reovirus in cancer therapy: an evidence-based review. Oncolytic Virotherapy. 2014;3:69-82
83. Zhang X, Meng S, Zhang R, Ma B, Liu T, Yang Y. et al. GP73-regulated oncolytic adenoviruses possess potent killing effect on human liver cancer stem-like cells. Oncotarget. 2016;7:29346-58
84. Yano S, Tazawa H, Hashimoto Y, Shirakawa Y, Kuroda S, Nishizaki M. et al. A genetically engineered oncolytic adenovirus decoys and lethally traps quiescent cancer stem-like cells in S/G2/M phases. Clin Cancer Res Off J Am Assoc Cancer Res. 2013;19:6495-505
85. Lv S, Ye Z, Liu P, Huang Y, Li L, Liu H. et al. 11R-P53 and GM-CSF Expressing Oncolytic Adenovirus Target Cancer Stem Cells with Enhanced Synergistic Activity. J Cancer. 2017;8:199-206
86. Li J, Zeng W, Huang Y, Zhang Q, Hu P, Rabkin SD. et al. Treatment of breast cancer stem cells with oncolytic herpes simplex virus. Cancer Gene Ther. 2012;19:707-14
87. Mahller YY, Williams JP, Baird WH, Mitton B, Grossheim J, Saeki Y. et al. Neuroblastoma cell lines contain pluripotent tumor initiating cells that are susceptible to a targeted oncolytic virus. PloS One. 2009;4:e4235
88. Yoo SY, Bang SY, Jeong S-N, Kang DH, Heo J. A cancer-favoring oncolytic vaccinia virus shows enhanced suppression of stem-cell like colon cancer. Oncotarget. 2016;7:16479-89
89. Wang H, Chen NG, Minev BR, Szalay AA. Oncolytic vaccinia virus GLV-1h68 strain shows enhanced replication in human breast cancer stem-like cells in comparison to breast cancer cells. J Transl Med. 2012;10:167
90. Redding N, Zhou H-Y, Lun X, Senger D, Forsyth P, Robbins S. et al. The utility of oncolytic viruses against neuroblastoma. In Banff, In The 5th International Meeting on Replicating Oncolytic Virus Therapeutics. Alberta, Canada. 2009
91. Zemp FJ, Lun X, McKenzie BA, Zhou H, Maxwell L, Sun B. et al. Treating brain tumor-initiating cells using a combination of myxoma virus and rapamycin. Neuro-Oncol. 2013;15:904-20
92. Rahman MM, Madlambayan GJ, Cogle CR, McFadden G. Oncolytic viral purging of leukemic hematopoietic stem and progenitor cells with Myxoma virus. Cytokine Growth Factor Rev. 2010;21:169-75
93. Marcato P, Dean CA, Giacomantonio CA, Lee PWK. Oncolytic reovirus effectively targets breast cancer stem cells. Mol Ther J Am Soc Gene Ther. 2009;17:972-9
94. Dautzenberg IJC, van den Wollenberg DJM, van den Hengel SK, Limpens RWA, Bárcena M, Koster AJ. et al. Mammalian orthoreovirus T3D infects U-118 MG cell spheroids independent of junction adhesion molecule-A. Gene Ther. 2014;21:609-17
95. van den Hengel SK, Balvers RK, Dautzenberg IJC, van den Wollenberg DJM, Kloezeman JJ, Lamfers ML. et al. Heterogeneous reovirus susceptibility in human glioblastoma stem-like cell cultures. Cancer Gene Ther. 2013;20:507-13
96. Bach P, Abel T, Hoffmann C, Gal Z, Braun G, Voelker I. et al. Specific elimination of CD133+ tumor cells with targeted oncolytic measles virus. Cancer Res. 2013Jan15;73(2):865-74
97. Brun J, McManus D, Lefebvre C, Hu K, Falls T, Atkins H. et al. Identification of genetically modified Maraba virus as an oncolytic rhabdovirus. Mol Ther J Am Soc Gene Ther. 2010;18:1440-9
98. Tong JG, Valdes YR, Barrett JW, Bell JC, Stojdl D, McFadden G. et al. Evidence for differential viral oncolytic efficacy in an in vitro model of epithelial ovarian cancer metastasis. Mol Ther Oncolytics. 2015;2:15013. doi: 10.1038/mto.2015.13
99. Barber GN. VSV-tumor selective replication and protein translation. Oncogene. 2005;24:7710-9
100. Ge P, Tsao J, Schein S, Green TJ, Luo M, Zhou ZH. CryoEM Model of the Bullet-Shaped Vesicular Stomatitis Virus. Science. 2010;327:689-93
101. LETCHWORTH GJ, RODRIGUEZ LL, DEL CBARRERA J. Vesicular Stomatitis. Vet J. 1999;157:239-60
102. Lichty BD, Power AT, Stojdl DF, Bell JC. Vesicular stomatitis virus: re-inventing the bullet. Trends Mol Med. 2004;10:210-6
103. Vesicular Stomatitis,Sore Mouth of Cattle and Horses, Indiana Fever 2016 Jan. Iowa State University, College of Veterinary Medicine. www.cfsph.iastate.edu/IICAB
104. Hastie E, Grdzelishvili VZ. Vesicular stomatitis virus as a flexible platform for oncolytic virotherapy against cancer. J Gen Virol. 2012;93:2529-45
105. Yasmeen A, Zhang L, Al Moustafa A-E. Does the vesicular stomatitis virus really have a selective oncolytic effect in human cancer? Int J Cancer J Int Cancer. 2010;126:2509-10
106. Orzechowska B, Antoszków Z, Błach-Olszewska Z. Individual differentiation of innate antiviral immunity in humans; the role of endogenous interferons and tumor necrosis factor in the immunity of leukocytes. Arch Immunol Ther Exp (Warsz). 2003;51:51-60
107. Orzechowska B, Antoszkow Z, Siemieniec I, Lorenc M, Jatczak B, Blach-Olszewska Z. Cytokine production by human leukocytes with different expressions of natural antiviral immunity and the effect of antibodies against interferons and TNF-? Arch Immunol Ther Exp. 2007;55:111-7
108. Rybka K, Orzechowska B, Siemieniec I, Leszek J, Zaczynska E, Pajak J. et al. Age related natural antiviral non-specific immunity of human leukocytes. Med Sci Monit Int Med J Exp Clin Res. 2003;9:BR413-417
109. Orzechowska B, Chaber R, Wiśniewska A, Pajtasz-Piasecka E, Jatczak B, Siemieniec I. et al. Baicalin from the extract of Scutellaria baicalensis affects the innate immunity and apoptosis in leukocytes of children with acute lymphocytic leukemia. Int Immunopharmacol. 2014;23:558-67
110. Jatczak B, Leszek J, Siemieniec I, Sochocka M, Wiśniewska A, Tarkowski R. et al. Age- and disease-related innate immunity of human leukocytes ex vivo. Exp Gerontol. 2012;47:8-13
111. Piasecki E, Knysz B, Zwolińska K, Gąsiorowski J, Lorenc M, Zalewska M. et al. Inhibition of vesicular stomatitis virus replication in the course of HIV infection in patients with different stages of immunodeficiency. Viral Immunol. 2010;23:567-76
112. Johnson JE, Nasar F, Coleman JW, Price RE, Javadian A, Draper K. et al. Neurovirulence properties of recombinant vesicular stomatitis virus vectors in non-human primates. Virology. 2007;360:36-49
113. Carey BL, Ahmed M, Puckett S, Lyles DS. Early Steps of the Virus Replication Cycle Are Inhibited in Prostate Cancer Cells Resistant to Oncolytic Vesicular Stomatitis Virus. J Virol. 2008;82:12104-15
114. Oliere S, Arguello M, Mesplede T, Tumilasci V, Nakhaei P, Stojdl D. et al. Vesicular Stomatitis Virus Oncolysis of T Lymphocytes Requires Cell Cycle Entry and Translation Initiation. J Virol. 2008;82:5735-49
115. Lyles DS, Mckenzie MO, Ahmed M, Woolwine SC. Potency of Wild-Type and Temperature-Sensitive Vesicular Stomatitis Virus Matrix Protein in the Inhibition of Host-Directed Gene Expression. Virology. 1996;225:172-80
116. Rajani KR, Pettit Kneller EL, McKenzie MO, Horita DA, Chou JW, Lyles DS. Complexes of Vesicular Stomatitis Virus Matrix Protein with Host Rae1 and Nup98 Involved in Inhibition of Host Transcription. PLoS Pathog. 2012;8:e1002929
117. Castelló A, Izquierdo JM, Welnowska E, Carrasco L. RNA nuclear export is blocked by poliovirus 2A protease and is concomitant with nucleoporin cleavage. J Cell Sci. 2009;122:3799-809
118. Ayala-Breton C, Barber GN, Russell SJ, Peng K-W. Retargeting vesicular stomatitis virus using measles virus envelope glycoproteins. Hum Gene Ther. 2012;23:484-91
119. Muik A, Kneiske I, Werbizki M, Wilflingseder D, Giroglou T, Ebert O. et al. Pseudotyping vesicular stomatitis virus with lymphocytic choriomeningitis virus glycoproteins enhances infectivity for glioma cells and minimizes neurotropism. J Virol. 2011;85:5679-84
120. Wollmann G, Drokhlyansky E, Davis JN, Cepko C, van den Pol AN. Lassa-vesicular stomatitis chimeric virus safely destroys brain tumors. J Virol. 2015;89:6711-24
121. Betancourt D, Ramos JC, Barber GN. Retargeting Oncolytic Vesicular Stomatitis Virus to Human T-Cell Lymphotropic Virus Type 1-Associated Adult T-Cell Leukemia. J Virol. 2015;89:11786-800
122. Kelly EJ, Nace R, Barber GN, Russell SJ. Attenuation of Vesicular Stomatitis Virus Encephalitis through MicroRNA Targeting. J Virol. 2010;84:1550-62
123. Ammayappan A, Nace R, Peng K-W, Russell SJ. Neuroattenuation of Vesicular Stomatitis Virus through Picornaviral Internal Ribosome Entry Sites. J Virol. 2013;87:3217-28
124. Wongthida P, Jengarn J, Narkpuk J, Koonyosying P, Srisutthisamphan K, Wanitchang A. et al. In Vitro and In Vivo Attenuation of Vesicular Stomatitis Virus (VSV) by Phosphoprotein Deletion. PLoS ONE. 2016;11:e0157287. doi: 10.1371/journal.pone.0157287
125. Muik A, Dold C, Geiß Y, Volk A, Werbizki M, Dietrich U. et al. Semireplication-competent vesicular stomatitis virus as a novel platform for oncolytic virotherapy. J Mol Med Berl Ger. 2012;90:959-70
126. Wollmann G, Rogulin V, Simon I, Rose JK, van den Pol AN. Some attenuated variants of vesicular stomatitis virus show enhanced oncolytic activity against human glioblastoma cells relative to normal brain cells. J Virol. 2010;84:1563-73
127. Obuchi M, Fernandez M, Barber GN. Development of Recombinant Vesicular Stomatitis Viruses That Exploit Defects in Host Defense To Augment Specific Oncolytic Activity. J Virol. 2003;77:8843-56
128. Jenks N, Myers R, Greiner SM, Thompson J, Mader EK, Greenslade A. et al. Safety Studies on Intrahepatic or Intratumoral Injection of Oncolytic Vesicular Stomatitis Virus Expressing Interferon-β in Rodents and Nonhuman Primates. Hum Gene Ther. 2010;21:451-62
129. LeBlanc AK, Naik S, Galyon GD, Jenks N, Steele M, Peng K-W. et al. Safety studies on intravenous administration of oncolytic recombinant vesicular stomatitis virus in purpose-bred beagle dogs. Hum Gene Ther Clin Dev. 2013;24:174-81
130. Zhang L, Steele MB, Jenks N, Grell J, Suksanpaisan L, Naik S. et al. Safety Studies in Tumor and Non-Tumor-Bearing Mice in Support of Clinical Trials Using Oncolytic VSV-IFNβ-NIS. Hum Gene Ther Clin Dev. 2016;27:111-22
131. Viral Therapy in Treating Patient With Liver Cancer - Full Text View - ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT01628640?term=VSV&rank=11
132. Westcott MM, Liu J, Rajani K, D'Agostino R, Lyles DS, Porosnicu M. Interferon Beta and Interferon Alpha 2a Differentially Protect Head and Neck Cancer Cells from Vesicular Stomatitis Virus-Induced Oncolysis. J Virol. 2015;89:7944-54
133. Porosnicu M, Mian A, Barber GN. The oncolytic effect of recombinant vesicular stomatitis virus is enhanced by expression of the fusion cytosine deaminase/uracil phosphoribosyltransferase suicide gene. Cancer Res. 2003;63:8366-76
134. Leveille S, Samuel S, Goulet M-L, Hiscott J. Enhancing VSV oncolytic activity with an improved cytosine deaminase suicide gene strategy. Cancer Gene Ther. 2011;18:435-43
135. Altomonte J, Wu L, Chen L, Meseck M, Ebert O, García-Sastre A. et al. Exponential enhancement of oncolytic vesicular stomatitis virus potency by vector-mediated suppression of inflammatory responses in vivo. Mol Ther J Am Soc Gene Ther. 2008;16:146-53
136. Jha BK, Dong B, Nguyen CT, Polyakova I, Silverman RH. Suppression of Antiviral Innate Immunity by Sunitinib Enhances Oncolytic Virotherapy. Mol Ther J Am Soc Gene Ther. 2013;21:1749-57
137. Tesfay MZ, Kirk AC, Hadac EM, Griesmann GE, Federspiel MJ, Barber GN. et al. PEGylation of vesicular stomatitis virus (VSV) extends virus persistence in blood circulation of passively immunized mice. J Virol. 2013;87:3752-9
138. Zayed AA, Mandrekar SJ, Haluska P. Molecular and clinical implementations of ovarian cancer mouse avatar models. Chin Clin Oncol. 2015;4:30
139. Dold C, Urbiola CR, Wollmann G, Egerer L, Muik A, Bellmann L. et al. Application of interferon modulators to overcome partial resistance of human ovarian cancers to VSV-GP oncolytic viral therapy. Mol Ther Oncolytics. 2016;3:16021
140. Šale S, Orsulic S. Models of ovarian cancer metastasis: Murine models. Drug Discov Today Dis Models. 2006;3:149-54
141. Garson K, Gamwell LF, Pitre EM, Vanderhyden BC. Technical challenges and limitations of current mouse models of ovarian cancer. J Ovarian Res. 2012;5:39
142. Connolly DC, Hensley HH. Xenograft and Transgenic Mouse Models of Epithelial Ovarian Cancer and Non Invasive Imaging Modalities to Monitor Ovarian Tumor Growth In situ -Applications in Evaluating Novel Therapeutic Agents. Curr Protoc Pharmacol Editor Board SJ Enna Ed-Chief Al. 2009;45:14.12.1-14.12.26
143. Brachova P, Thiel KW, Leslie KK. The consequence of oncomorphic TP53 mutations in ovarian cancer. Int J Mol Sci. 2013;14:19257-75
144. Mittempergher L. Genomic Characterization of High-Grade Serous Ovarian Cancer: Dissecting Its Molecular Heterogeneity as a Road Towards Effective Therapeutic Strategies. Curr Oncol Rep. 2016;18:44
145. Reith AD, Rottapel R, Giddens E, Brady C, Forrester L, Bernstein A. W mutant mice with mild or severe developmental defects contain distinct point mutations in the kinase domain of the c-kit receptor. Genes Dev. 1990;4:390-400
146. Testing the therapeutic efficacy of vesicular stomatitis virus as a single agent or in combination with the histone deacetylase inhibitor, MS-275, in a mouse model of ovarian cancer Depatrment of Cellular and Molecular Medicine; Ottawa, Canada. 2009 Valerie Snoulton. https://www.ruor.uottawa.ca/handle/10393/28069
147. Nguyên TL-A, Abdelbary H, Arguello M, Breitbach C, Leveille S, Diallo J-S. et al. Chemical targeting of the innate antiviral response by histone deacetylase inhibitors renders refractory cancers sensitive to viral oncolysis. Proc Natl Acad Sci U S A. 2008;105:14981-6
148. Arulanandam R, Batenchuk C, Varette O, Zakaria C, Garcia V, Forbes NE. et al. Microtubule disruption synergizes with oncolytic virotherapy by inhibiting interferon translation and potentiating bystander killing. Nat Commun. 2015;6:6410
149. Capo-chichi CD, Yeasky TM, Heiber JF, Wang Y, Barber GN, Xu X-X. Explicit targeting of transformed cells by VSV in ovarian epithelial tumor-bearing Wv mouse models. Gynecol Oncol. 2010;116:269
150. Shih I-M, Kurman RJ. Ovarian tumorigenesis: a proposed model based on morphological and molecular genetic analysis. Am J Pathol. 2004;164:1511-8
151. Cai KQ, Wang Y, Smith ER, Smedberg JL, Yang D-H, Yang W-L. et al. Global deletion of Trp53 reverts ovarian tumor phenotype of the germ cell-deficient white spotting variant (Wv) mice. Neoplasia N Y N. 2015;17:89-100
152. Vandamme TF. Use of rodents as models of human diseases. J Pharm Bioallied Sci. 2014;6:2-9
153. Miyoshi I, Takahashi K, Kon Y, Okamura T, Mototani Y, Araki Y. et al. Mouse transgenic for murine oviduct-specific glycoprotein promoter-driven simian virus 40 large T-antigen: tumor formation and its hormonal regulation. Mol Reprod Dev. 2002;63:168-76
154. Sherman-Baust CA, Kuhn E, Valle BL, Shih I-M, Kurman RJ, Wang T-L. et al. A genetically engineered ovarian cancer mouse model based on fallopian tube transformation mimics human high-grade serous carcinoma development. J Pathol. 2014;233:228-37
155. Kim J, Coffey DM, Creighton CJ, Yu Z, Hawkins SM, Matzuk MM. High-grade serous ovarian cancer arises from fallopian tube in a mouse model. Proc Natl Acad Sci U S A. 2012;109:3921-6
156. Kinross KM, Montgomery KG, Kleinschmidt M, Waring P, Ivetac I, Tikoo A. et al. An activating Pik3ca mutation coupled with Pten loss is sufficient to initiate ovarian tumorigenesis in mice. J Clin Invest. 2012;122:553-7
157. Cully M, You H, Levine AJ, Mak TW. Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat Rev Cancer. 2006;6:184-92
158. Martins FC, Santiago I de, Trinh A, Xian J, Guo A, Sayal K. et al. Combined image and genomic analysis of high-grade serous ovarian cancer reveals PTEN loss as a common driver event and prognostic classifier. Genome Biol. 2014;15:526
159. Nakanishi A, Kitagishi Y, Ogura Y, Matsuda S. The tumor suppressor PTEN interacts with p53 in hereditary cancer (Review). Int J Oncol. 2014;44:1813-9
160. Perets R, Wyant GA, Muto KW, Bijron JG, Poole BB, Chin KT. et al. Transformation of the Fallopian Tube Secretory Epithelium Leads to High-grade Serous Ovarian Cancer in Brca;Tp53;Pten Models. Cancer Cell. 2013;24:751-65
161. Burgos-Ojeda D, Wu R, McLean K, Chen Y-C, Talpaz M, Yoon E. et al. CD24+ Ovarian Cancer Cells Are Enriched for Cancer-Initiating Cells and Dependent on JAK2 Signaling for Growth and Metastasis. Mol Cancer Ther. 2015;14:1717-27
162. Schubbert S, Jiao J, Ruscetti M, Nakashima J, Wu S, Lei H. et al. Methods for PTEN in Stem Cells and Cancer Stem Cells. Methods Mol Biol Clifton NJ. 2016;1388:233-85
163. Ciuffreda L, Falcone I, Incani UC, Del Curatolo A, Conciatori F, Matteoni S. et al. PTEN expression and function in adult cancer stem cells and prospects for therapeutic targeting. Adv Biol Regul. 2014;56:66-80
164. Li S, Zhu M, Pan R, Fang T, Cao Y-Y, Chen S. et al. The tumor suppressor PTEN has a critical role in antiviral innate immunity. Nat Immunol. 2016;17:241-9
165. Yu N, Puckett S, Antinozzi PA, Cramer SD, Lyles DS. Changes in Susceptibility to Oncolytic Vesicular Stomatitis Virus during Progression of Prostate Cancer. J Virol. 2015;89:5250-63
166. Sawada K, Mitra AK, Radjabi AR, Bhaskar V, Kistner EO, Tretiakova M. et al. Loss of E-cadherin promotes ovarian cancer metastasis via alpha 5-integrin, which is a therapeutic target. Cancer Res. 2008;68:2329-39
167. Pease JC, Brewer M, Tirnauer JS. Spontaneous spheroid budding from monolayers: a potential contribution to ovarian cancer dissemination. Biol Open. 2012;1:622-8
168. Lengyel E, Burdette J, Kenny H, Matei D, Pilrose J, Haluska P. et al. Epithelial Ovarian Cancer Experimental Models. Oncogene. 2014;33:3619-33
169. Ffrench B, Gasch C, O'Leary JJ, Gallagher MF. Developing ovarian cancer stem cell models: laying the pipeline from discovery to clinical intervention. Mol Cancer. 2014;13:262
170. Finkelshtein D, Werman A, Novick D, Barak S, Rubinstein M. LDL receptor and its family members serve as the cellular receptors for vesicular stomatitis virus. Proc Natl Acad Sci U S A. 2013;110:7306-11
171. Pampalakis G, Politi A-L, Papanastasiou A, Sotiropoulou G. Distinct cholesterogenic and lipidogenic gene expression patterns in ovarian cancer - a new pool of biomarkers. Genes Cancer. 2015;6:472-9
Author contact
![]() Corresponding author: orzechowpan.wroc.pl.
Corresponding author: orzechowpan.wroc.pl.