Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(12):2263-2268. doi:10.7150/jca.19582 This issue Cite
Research Paper
MAP2K1 Mutation in Colorectal Cancer Patients: Therapeutic Challenge Using Patient-Derived Tumor Cell Lines
J. E. Kim1*, K.K. Kim2*, S. Y. Kim1, J. Lee1, S. H. Park1, J. O. Park1, Y. S. Park1, H. Y. Lim1, W. K. Kang1, S. T. Kim1 ![]()
1. Division of Hematology-Oncology, Department of Medicine, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea
2. Department of Molecular Cell Biology, Institute of Basic Science, Samsung Biomedical Research Institute, Sungkyunkwan University School of Medicine, Suwon 440-746, Korea
* Equally contributed
Received 2017-2-9; Accepted 2017-5-18; Published 2017-7-20
Abstract
BACKGROUND: The MAP2K1 K57T mutation is known to be a potential mechanism of primary and secondary resistance to EGFR inhibitors in metastatic colorectal cancer (CRC) and has also been reported to promote resistance to BRAF and MEK inhibitors. It is important to overcome therapeutic resistance to EGFR inhibitors to improve the treatment outcomes of metastatic CRC.
METHODS: We established patient-derived tumor cells (PDCs) from metastatic lesions that newly appeared during treatment with a BRAF inhibitor (LGX-818) plus an EGFR inhibitor (cetuximab) in a patient with BRAF-mutant CRC. To investigate therapeutic options to overcome acquired resistance due to MAP2K1 mutation in BRAF-mutant CRC, we performed cell viability assays using the PDCs.
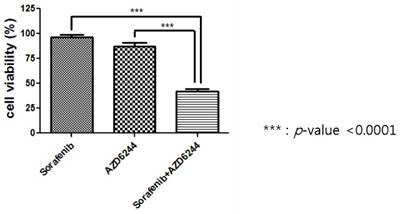
RESULTS: We tested whether the PDCs were resistant to an EGFR inhibitor (cetuximab) and a BRAF inhibitor (sorafenib) as these cells were established at the time of resistance to the EGFR plus BRAF inhibitors. Moreover, the anti-tumor effect of AZD6244 (MEK inhibitor) was evaluated because PDCs harbored a MAP2K1 mutation at the time of resistance to the EGFR plus BRAF inhibitors. MTT proliferation assays showed that monotherapy with cetuximab, sorafenib, or AZD6244 did not suppress cell viability. We next tested viability of the PDCs to combination treatment with cetuximab plus AZD6244 and sorafenib plus AZD6244. Proliferation of PDCs was significantly inhibited by sorafenib and AZD6244, but not by cetuximab plus AZD6244. Investigation of the combined effect of sorafenib and AZD6244 using the calculated combination index (CI) showed synergistic effects of sorafenib and AZD6244 in combination therapy applied to PDCs with the MAP2K1 K57T mutation.
CONCLUSION: Our results suggest that combination treatment with BRAF and MEK inhibitors might be a novel treatment strategy for MAP2K1 K57T-mutant CRC. This finding will be helpful to guide treatment of patients with CRC that is resistant to EGFR inhibitors.
Keywords: MAP2K1 mutation, MEK inhibitor, RAF inhibitor
Introduction
An improved understanding of the underlying molecular pathology of colorectal cancer (CRC) has enabled tailored treatment regimens and helped to optimize outcomes. There have been recent and rapid advances in the development of agents targeting components of receptor tyrosine kinase signaling cascades for use in cancer therapy [1-3].
As many human cancers, including CRC, are associated with abnormal expression of epidermal growth factor receptor (EGFR), which is implicated in the development and prognosis of malignancy, EGFR is a potential target for cancer therapy [4]. However, benefit from anti-EGFR therapy using cetuximab and panitumumab was observed in only a selected subset of patients (10-20%), highlighting a distinct need for individualized treatment [5-8]. RAS/RAF/MAPK signaling is considered the key modulator of sensitivity and resistance to anti-EGFR therapy in CRC [9-13]. BRAF mutations are present in approximately 8% to 10% of patients with metastatic CRC and are associated with poor survival [10, 14]. BRAF encodes a protein directly downstream from RAS in the RAS/RAF/MAPK signaling pathway. Patients with metastatic BRAF-mutated CRC do not benefit from anti-EGFR antibodies in the chemotherapy refractory setting [15]. In BRAF-mutant CRC, blockade of BRAF generates a reflexive EGFR activation, which can bypass BRAF and promote tumor progression through MAPK signaling [16, 17]. Preclinical and early clinical studies reported that treatment strategies co-targeting BRAF and EGFR can suppress feedback reactivation of MAPK signaling, leading to more robust signaling and improved efficacy in BRAF-mutant CRC [16, 18, 19]. However, despite the value of combination therapy with BRAF and EGFR inhibitors in BRAF-mutant CRC, patients who derive initial benefit from treatment ultimately experience disease progression due to acquired resistance [20, 21].
Mitogen activated protein kinase 1 (MAP2K1), also called MEK1, is a protein kinase that is a known downstream target of RAF and is upstream of ERK [22]. Mutations of MAP2K1 are present in 1.5% of CRCs and most mutations cause constitutive activation of MAP2K1 kinase [23, 24]. MAP2K1 mutations also participate in the mechanisms of acquired resistance to combination treatment with BRAF and EGFR inhibitors [25].
We established patient-derived cells (PDC) from a BRAF-mutant CRC tumor that had acquired a MAP2K1 mutation at the time of resistance to combination therapy with BRAF and EGFR inhibitors. We used these PDCs to investigate therapeutic options to overcome the acquired resistance to BRAF and EGFR inhibitors caused by the MAP2K1 mutation in BRAF-mutant CRC.
Patients and Methods
Patient-Derived Tumor Cells
With informed consent, tumor samples were obtained from newly appeared hepatic metastatic lesions of a patient with BRAF-mutant metastatic CRC who had received combination treatment with BRAF (LGX-818) and EGFR inhibitors (Cetuximab). Collected tissue was minced and dissociated by enzymatic methods. The patient-derived cells (PDCs) from hepatic metastatic CRC were cultured in RPMI 1640 supplemented with 10% fetal bovine serum (FBS; Gibco BRL, Paisley, UK) and 1% antibiotic/antimycotic solution (Gibco BRL). The medium was changed every 3 days, and cells were maintained at 37°C in a humidified 5% CO2 incubator. PDCs were passaged using TrypLE Express (Gibco BRL) to detach cells when they reached 80-90% confluence.
Targeted Gene Sequencing
We conducted genomic analysis of a tumor biopsy from the patient's newly appeared metastatic hepatic lesion. Formalin-fixed paraffin embedded (FFPE) samples containing >40% tumor cellularity were dissected under a microscope from 4-μm thick unstained sections (10 to 20 slides) or from fresh frozen tissues by comparison with a hematoxylin and eosin-stained slide. Briefly, DNA was extracted using standard procedures (Qiagen) and extracted genomic DNA was sheared to 150-200 bp using Covaris S220 (Covaris, Woburn, MA, USA). Targeted genes were captured using a custom panel capture library (Agilent Technologies, Santa Clara, CA, USA) covering 2.5 Mb of exonic regions for the Illumina Paired-End Sequencing Library kit. We performed DNA sequencing of 100- or 101-bp paired-end reads using the Illumina HiSeq 2,500 sequencer (Illumina, San Diego, CA, USA). We aligned the sequencing reads to the human reference genome (GRCh37/hg19), excluded duplicated reads, and extracted uniquely mapped and properly paired reads with an insert size. Somatic alterations were detected by CancerSCAN and actionable variants included in this panel were selected based on publicly available databases such as My Cancer Genome® (http://www.mycancergenome.org/).
DNA Extraction
Cultured cells (passage 1 to 2) were harvested with TrypLE Express. Genomic DNA was isolated using a QIAamp DNA Mini Kit (Qiagen, GmBH, Hilden, Germany) according to the manufacturer's instructions. The concentration of genomic DNA was measured using a NanoDrop ND-100 (Nano Drop Technologies, Wilmington, DE, USA). Genomic DNA was stored at -80°C.
Cell Treatment and Viability Assay
After pathologic confirmation, cells were seeded at a density of 1-2 × 106 cells/10-mm dish for immunoblot analysis or 5,000 cells/well in 96-well plates for cell proliferation assays and treated for 3-5 days with various concentrations of drugs as indicated. Inhibition of cell proliferation was determined using Cell Titer Glo (Promega, Madison, WI, USA) according to the manufacturer's protocol. Interactions between drugs were presented as the combination index (CI), calculated by dividing the expected growth inhibition rate by the observed growth inhibition rate: CI <1.0 indicates antagonistic cytotoxicity; CI=1.0 is additive cytotoxicity; and CI >1.0 is synergistic cytotoxicity.
Immunoblot Analysis
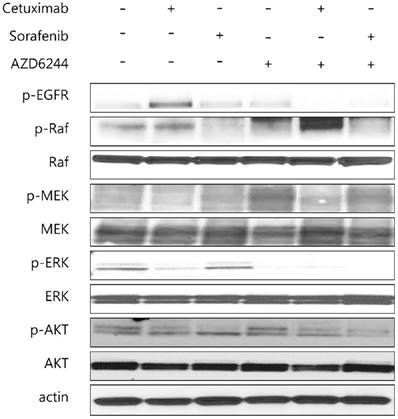
Total proteins from PDCs were isolated using RIPA buffer (Sigma-Aldrich, St. Louis, MO, USA) containing a protease inhibitor cocktail (Roche, Mannheim, Germany) and phosphatase inhibitor cocktail (Roche), and protein concentration was determined using a Quick Start Bradford Protein Assay (Bio-Rad, Hercules, CA, USA). Aliquots containing 30 μg of protein were subjected to 10% SDS-polyacrylamide gel electrophoresis and electrotransferred to nitrocellulose membranes. The membranes were blocked with 5% nonfat dry milk in Tris-buffered saline containing 0.1% v/v Tween 20 and probed overnight at 4°C with specific antibodies against the following proteins: p-EGFR, p-RAF, RAF, p-MEK, MEK, p-ERK, ERK, p-Rb1, Rb1, P-AKT, AKT (Cell Signaling Technology, Beverly, MA, USA), and beta actin (Sigma Aldrich). Horseradish peroxidase-conjugated anti-rabbit or anti-mouse IgG (Vector, Burlingame, CA, USA) was used as a secondary antibody, and signals were detected by chemiluminescence using ECL Western Blotting Substrate (Thermo Scientific, Rockford, IL, USA) and visualized using LAS-4000 (Fujifilm, Tokyo, Japan).
Droplet Digital PCR Analysis for MAP2K1 Mutation (K56T)
MEK1 K57T mutation in PDCs was analyzed by droplet digital PCR (ddPCR) using a Raindrop system (Rain Dance Technologies, Lexington, MA). The sequences of PCR primers used for MEK1 K57T mutant detection were as follows: forward primer 5'-GCGCCTTGAGGCCTTTCTTA -3'; reverse primer 5'-CAAAGTCGTCATCCTTCAGTTCTC-3'. The probes were 5'-[FAM]CACCTTCTGCGTCTGG[MGB]-3' for wild type and 5'-[VIC]CCACCTTCTGCTTCTGG[MGB]-3' for mutant type. For Droplet Digital PCR, the sample DNA was mixed with TaqMan genotyping Master Mix (Thermo Fisher Scientific Inc., Waltham, MA, USA) and aqueous droplets in oil were amplified using the C1000 Touch Thermal cycler (Bio-Rad, Pleasanton, CA). PCR conditions were as follows: 95°C for 10 min; 45 cycles of 95°C for 15 s and 60°C for 60 s; 98°C for 10 min; and 4°C hold. After the reaction, the PCR plate was read and individual sample droplets were analyzed using RainDrop Analyst II software (Rain Dance Technologies).
Results
Patient
A 45-year-old man initially presented in 2013 with stage IV, KRAS wild type and BRAF mutant rectosigmoid colon cancer. He underwent lower anterior resection for the primary lesion and hepatic sectionectomy for a metastatic liver lesion. Recurrence with hepatic and intra-abdominal lymph node metastases occurred after eight cycles of postoperative XELOX (capecitabine and oxaliplatin) chemotherapy. At the time of recurrence he was enrolled in a clinical trial of combination therapy with BRAF (LGX-818) and EGFR inhibitors (cetuximab) for BRAF-mutant CRC. The combination treatment with LGX-818 and cetuximab initially stabilized the disease; however, a computed tomography (CT) scan 3.5 months after treatment showed newly appeared hepatic metastasis and aggravation of the pre-existing hepatic lesion. At this time, core biopsy from the newly appeared hepatic lesion was performed and after tumor confirmation we generated PDCs from the patient. The biopsied tumor sample was analyzed by target sequencing and the tumor was found to harbor the BRAF V6003 and MAP2K1 K57T mutations. Genomic profiling of previous surgical samples taken at the time of diagnosis using targeted sequencing did not show the MAP2K1 K57T mutation. We also confirmed the MAP2K1 K57T mutation in PDCs by ddPCR.
The combination effect of Sorafenib and AZD6244.
| Treatment A | Treatment B | Combination Treatment (1:1) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Drug | Conc. | *MGI | †P value | Drug | Conc. | MGI | †P value | ‡Expected | §Observed | †P value | *Index | |||
| Sorafenib | 0.1 | 0.96 | 0.0201 | AZD6244 | 0.1 | 1.13 | 0.0024 | 1.09 | 0.92 | 0.0308 | 1.19 | |||
| (mM) | 0.3 | 0.97 | 0.3060 | (mM) | 0.3 | 1.07 | 0.1163 | 1.04 | 0.77 | 0.0097 | 1.35 | |||
| 1.0 | 0.96 | 0.2882 | 1.0 | 0.87 | 0.0141 | 0.84 | 0.42 | >0.0001 | 1.99 | |||||
| 3.0 | 0.99 | 0.0629 | 3.0 | 0.74 | 0.0002 | 0.73 | 0.31 | >0.0001 | 2.33 | |||||
| 10 | 0.73 | >0.0001 | 10 | 0.55 | >0.0001 | 0.40 | 0.28 | >0.0001 | 1.42 | |||||
*MGI is mean growth inhibition rate and calculated by dividing the expected growth inhibition rate by the observed growth inhibition rate. > 1 indicates synergistic effect, ≈ 1 indicates additive effect, and < 1 indicates antagonistic effect.
†P value was calculated by paired t test compared with no treatment. ‡Growth inhibition rate of treatment A x growth inhibition rate of treatment B. §Growth inhibition rate of combination on treatments A and B.
Anti-proliferation assay in K57T mutated PDC. A) Cells were exposed to indicated drug in increasing dose for 3 days. The effects were determined using Cell Titer glo method according to the manufacturer's manual. B) The table represents the genetic feature of PDC and sensitivity to Cetuximab, Sorafenib and AZD6244 alone or combination.
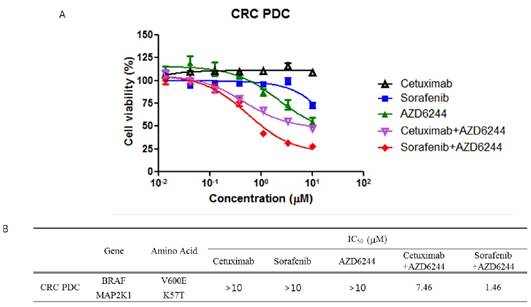
The effect of Sorafenib and AZD6244 combination.
The proliferative signaling related proteins were subject to western blotting analysis, including p-Raf, Raf, p-MEK, MEK, p-ERK, ERK, p-AKT and AKT.
MTT Assay and Immunoblot Assay Using Patient-Derived Cells
To investigate therapeutic options to overcome the acquired resistance associated with the MAP2K1 mutation in BRAF-mutant CRC that is resistant to combination treatment with BRAF and EGFR inhibitors, we performed a cell viability assay using the MAP2K1- and BRAF-mutant CRC PDC line. We tested whether the PDCs were resistant to EGFR (cetuximab) and BRAF inhibitors (sorafenib) as these cells were established at the time of tumor resistance to EGFR plus BRAF inhibitors. Moreover, the antitumor effect of AZD6244 (MEK inhibitor) was evaluated because the PDCs harbored a MAP2K1 mutation at the time of resistance to EGFR plus BRAF inhibitors. MTT proliferation assays showed that cetuximab, sorafenib, and AZD6244 monotherapies did not suppress cell viability.
Discussion
The MAP2K1 K57T mutation is known to be a potential mechanism of primary and secondary resistance to EGFR inhibitors in metastatic CRC [26, 27] and has also been reported to promote resistance to BRAF and MEK inhibitors [25]. For successful treatment of metastatic CRC it is necessary to overcome therapeutic resistance to EGFR inhibitors. In the present study, MAP2K1 K57T-mutant PDCs were sensitive to combination treatment with BRAF and MEK inhibitors, with downregulation of the downstream pathway involving ERK phosphorylation. Moreover, the calculated combination index (CI) showed synergistic effects of the combination of BRAF and MEK inhibitors in PDCs with the MAP2K1 K57T mutation. These findings suggested that combination treatment with BRAF and MEK inhibitors might be a novel treatment strategy for patients with MAP2K1 K57T-mutant CRC.
The progressive lesion in our patient that newly appeared after combination therapy with the EGFR inhibitor (cetuximab) plus a RAF (LGX-818) inhibitor was biopsied and analyzed by targeted sequencing. The post-EGFR/RAF therapy progression sample retained the original BRAF V600E mutation and harbored a new MAP2K1 K57T mutation compared with the pre-EGFR/RAF therapy tumor sample. The MAP2K1 mutation is known to be one of the potential candidates for primary and/or secondary resistance to EGFR inhibitors in CRC [25-27]. Also, alterations in the MAPK signaling pathway are important drivers of acquired resistance in BRAF-mutant cancer. MAP2K1 is a component of the oncogenic RAS-MAPK pathway [13, 28] and MAP2K1 mutations that activate this pathway have been observed in melanoma, CRC, gastric cancer, Langerhans cell histiocytosis, and hairy cell leukemia [29-31]. MAP2K1 mutation was associated the resistance of EGFR inhibitor of ALK inhibitor in non-small cell lung cancer [32,33]. Previous studies reported that MAP2K1 mutations in cancer cells led to downstream ERK phosphorylation and increased colony formation that was inhibited with the MEK inhibitor AZD6244 [34]. In this study, established MAP2K1-mutant PDCs were insensitive to AZD 6244 alone, inconsistent with findings from previous studies. This discrepancy may be caused by co-existing genomic alterations. The PDCs used in this study originated from a BRAF V600E and MAP2K1 K57T mutant tumor sample. The BRAF V600E mutation would affect the findings for cytotoxicity of AZD6244 alone.
Accurate prediction of anti-tumor effects of molecularly targeted agents before clinical trial design and implementation in cancer patients is essential to realize the goal of precision medicine. Ideal preclinical models should closely resemble the actual tumors in terms of genomic profiles and drug response. Recently, patient-derived tumor cells have been suggested as an alternative preclinical model for use as a prediction system for preclinical drug testing [35]. Our group previously demonstrated the usefulness of our PDC system as a promising model for preclinical experiments in various cancer types including CRC [36]. In the present study, we successfully established PDCs from a metastatic lesion that newly appeared in a patient with BRAF-mutant CRC during treatment with BRAF (LGX-818) plus EGFR inhibitors (cetuximab). Although MAP2K1 K57T-mutant CRC is very rare, it is important to establish a precise treatment strategy based on the genomic profile and to study mechanisms for overcoming resistance to EGFR inhibitors. From this perspective, our successfully established PDCs from a patient with BRAF V600E and MAP2K1 K57T mutant CRC that was resistant to combination treatment with BRAF and MEK inhibitors could be considered important preparation for preclinical research.
Although generalization of our results is limited because they are based on a single case, this study suggests that the combination of BRAF and MEK inhibitors might be a novel treatment strategy for MAP2K1 K57T-mutant CRC. Furthermore, in terms of the MAP2K1 K57T mutation, which has been linked to mechanisms of primary and secondary resistance to EGFR inhibitor in metastatic CRC, our findings might guide the treatment of patients with CRC that is resistant to EGFR inhibitors.
Acknowledgements
This work was supported by a grant from the Korean Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea (HI14C3418, HI16C1990). Support was also provided by grants (GF01140111) and (SMX1170421) of Samsung Medical Center.
Competing Interests
None declared.
References
1. Cunningham D, Humblet Y, Siena S. et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med. 2004;351:337-345
2. Hurwitz H, Fehrenbacher L, Novotny W. et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350:2335-2342
3. Mayer RJ. Targeted therapy for advanced colorectal cancer-more is not always better. N Engl J Med. 2009;360:623-625
4. Scartozzi M, Bearzi I, Berardi R. et al. Epidermal growth factor receptor (EGFR) status in primary colorectal tumors does not correlate with EGFR expression in related metastatic sites: implications for treatment with EGFR-targeted monoclonal antibodies. J Clin Oncol. 2004;22:4772-4778
5. Bokemeyer C, Bondarenko I, Makhson A. et al. Fluorouracil, leucovorin, and oxaliplatin with and without cetuximab in the first-line treatment of metastatic colorectal cancer. J Clin Oncol. 2009;27:663-671
6. Van Cutsem E, Kohne CH, Hitre E. et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med. 2009;360:1408-1417
7. Douillard JY, Siena S, Cassidy J. et al. Randomized, phase III trial of panitumumab with infusional fluorouracil, leucovorin, and oxaliplatin (FOLFOX4) versus FOLFOX4 alone as first-line treatment in patients with previously untreated metastatic colorectal cancer: the PRIME study. J Clin Oncol. 2010;28:4697-4705
8. Sobrero AF, Maurel J, Fehrenbacher L. et al. EPIC: phase III trial of cetuximab plus irinotecan after fluoropyrimidine and oxaliplatin failure in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26:2311-2319
9. Benvenuti S, Sartore-Bianchi A, Di Nicolantonio F. et al. Oncogenic activation of the RAS/RAF signaling pathway impairs the response of metastatic colorectal cancers to anti-epidermal growth factor receptor antibody therapies. Cancer Res. 2007;67:2643-2648
10. Van Cutsem E, Kohne CH, Lang I. et al. Cetuximab plus irinotecan, fluorouracil, and leucovorin as first-line treatment for metastatic colorectal cancer: updated analysis of overall survival according to tumor KRAS and BRAF mutation status. J Clin Oncol. 2011;29:2011-2019
11. Bardelli A, Siena S. Molecular mechanisms of resistance to cetuximab and panitumumab in colorectal cancer. J Clin Oncol. 2010;28:1254-1261
12. Rajagopalan H, Bardelli A, Lengauer C. et al. Tumorigenesis: RAF/RAS oncogenes and mismatch-repair status. Nature. 2002;418:934
13. Normanno N, Rachiglio AM, Lambiase M. et al. Heterogeneity of KRAS, NRAS, BRAF and PIK3CA mutations in metastatic colorectal cancer and potential effects on therapy in the CAPRI GOIM trial. Ann Oncol. 2015;26:1710-1714
14. Yokota T, Ura T, Shibata N. et al. BRAF mutation is a powerful prognostic factor in advanced and recurrent colorectal cancer. Br J Cancer. 2011;104:856-862
15. Di Nicolantonio F, Martini M, Molinari F. et al. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J Clin Oncol. 2008;26:5705-5712
16. Prahallad A, Sun C, Huang S. et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 2012;483:100-103
17. Corcoran RB, Ebi H, Turke AB. et al. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov. 2012;2:227-235
18. Corcoran RB, Dias-Santagata D, Bergethon K. et al. BRAF gene amplification can promote acquired resistance to MEK inhibitors in cancer cells harboring the BRAF V600E mutation. Sci Signal. 2010;3:ra84
19. Montero-Conde C, Ruiz-Llorente S, Dominguez JM. et al. Relief of feedback inhibition of HER3 transcription by RAF and MEK inhibitors attenuates their antitumor effects in BRAF-mutant thyroid carcinomas. Cancer Discov. 2013;3:520-533
20. Yaeger R, Cercek A, O'Reilly EM. et al. Pilot trial of combined BRAF and EGFR inhibition in BRAF-mutant metastatic colorectal cancer patients. Clin Cancer Res. 2015;21:1313-1320
21. Hyman DM, Puzanov I, Subbiah V. et al. Vemurafenib in Multiple Nonmelanoma Cancers with BRAF V600 Mutations. N Engl J Med. 2015;373:726-736
22. Avizienyte E, Roth S, Loukola A. et al. Somatic mutations in LKB1 are rare in sporadic colorectal and testicular tumors. Cancer Res. 1998;58:2087-2090
23. Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330-337
24. Cerami E, Gao J, Dogrusoz U. et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401-404
25. Ahronian LG, Sennott EM, Van Allen EM. et al. Clinical Acquired Resistance to RAF Inhibitor Combinations in BRAF-Mutant Colorectal Cancer through MAPK Pathway Alterations. Cancer Discov. 2015;5:358-367
26. Bertotti A, Papp E, Jones S. et al. The genomic landscape of response to EGFR blockade in colorectal cancer. Nature. 2015;526:263-267
27. Siravegna G, Mussolin B, Buscarino M. et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat Med. 2015;21:795-801
28. Wong NA, Gonzalez D, Salto-Tellez M. et al. RAS testing of colorectal carcinoma-a guidance document from the Association of Clinical Pathologists Molecular Pathology and Diagnostics Group. J Clin Pathol. 2014;67:751-757
29. Marks JL, Gong Y, Chitale D. et al. Novel MEK1 mutation identified by mutational analysis of epidermal growth factor receptor signaling pathway genes in lung adenocarcinoma. Cancer Res. 2008;68:5524-5528
30. Murugan AK, Dong J, Xie J. et al. MEK1 mutations, but not ERK2 mutations, occur in melanomas and colon carcinomas, but none in thyroid carcinomas. Cell Cycle. 2009;8:2122-2124
31. Wagle N, Emery C, Berger MF. et al. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J Clin Oncol. 2011;29:3085-3096
32. Marks JL, Gong Y, Chitale D. et al. Novel MEK1 mutation identified by mutational analysis of epidermal growth factor receptor signaling pathway genes in lung adenocarcinoma. J Clin Oncol. 2008;68:5524-5528
33. Crystal AS, Shaw AT, Sequist LV. et al. Patient-derived models of acquired resistance can identify effective drug combinations for cancer. Science. 2014;346:1480-1486
34. Yoon J, Koo KH, Choi KY. MEK1/2 inhibitors AS703026 and AZD6244 may be potential therapies for KRAS mutated colorectal cancer that is resistant to EGFR monoclonal antibody therapy. Cancer Res. 2011;71:445-453
35. Lee JY, Kim SY, Park C. et al. Patient-derived cell models as preclinical tools for genome-directed targeted therapy. Oncotarget. 2015;6:25619-25630
36. Song HN, Lee C, Kim ST. et al. Molecular characterization of colorectal cancer patients and concomitant patient-derived tumor cell establishment. Oncotarget. 2016;7:19610-19619
Author contact
![]() Corresponding author: Dr. Seung Tae Kim, Division of Hematology/Oncology, Department of Medicine, Samsung Medical Center, Sungkyunkwan University School of Medicine, 81 Irwon-ro, Gangnam-gu, Seoul 06351, Korea. Tel: +82-2-3410-1779; Fax: +82-2-3410-1754; E-mail: seungtae1.kimcom
Corresponding author: Dr. Seung Tae Kim, Division of Hematology/Oncology, Department of Medicine, Samsung Medical Center, Sungkyunkwan University School of Medicine, 81 Irwon-ro, Gangnam-gu, Seoul 06351, Korea. Tel: +82-2-3410-1779; Fax: +82-2-3410-1754; E-mail: seungtae1.kimcom