Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(12):2256-2262. doi:10.7150/jca.19566 This issue Cite
Research Paper
Development of a new analog of SGK1 inhibitor and its evaluation as a therapeutic molecule of colorectal cancer
Xuchun Liang1,#, Chunling Lan2,#, Jinzhe Zhou3,#, Wencheng Fu1, Xuesha Long1, Yu An2, Guanming Jiao1, Kejin Wang1, Yongqin Li1, Jiahong Xu4, Qi Huang3, Bin Xu2, ![]() , Junjie Xiao1,
, Junjie Xiao1, ![]()
1. Regeneration and Ageing Lab, School of Life Science, Shanghai University, Shanghai 200444, China
2. Department of Chemistry, Qianweichang College, Innovative Drug Research Center, Shanghai University, Shanghai 200444, China
3. Department of General Surgery, Tongji Hospital, Tongji University School of Medicine, Shanghai 200065, China
4. Department of Cardiology, Tongji Hospital, Tongji University School of Medicine, Shanghai 200065, China
#These authors contributed equally to this work.
Received 2017-2-8; Accepted 2017-5-2; Published 2017-7-20
Abstract
Colorectal cancer (CRC) is one of the most leading causes of cancer-related death worldwide. The serum and glucocorticoid inducible kinase SGK1 is highly expressed and involved in several tumors. GSK650394, a SGK1 inhibitor, has been proved to be effective in impeding tumor growth in vitro. In this study, we developed a novel analog of GSK650394, and evaluated its effects on CRC cells and tumor growth both in vitro and in vivo. HCT116 cells were treated with a concentration gradient of new developed compounds and cholecystokinin octapeptide (CCK-8) assay was used to calculate the IC50 value of every analog. Cell proliferation analysis was estimated from EdU staining and flow cytometry in vitro, and immunohistochemistry of Ki67 and PCNA in vivo. Cell migration analysis was examined using the transwell assay. In vivo tumor growth was determined in athymic nude mice by injecting the HCT116 cells in the subcutaneous tissue, followed by the injection of QGY-5-114-A. We found that new developed GSK650394 analog QGY-5-114-A has lower IC50 value, and treatment with QGY-5-114-A significantly inhibited CRC cell proliferation and migration in vitro. Besides that, colonic tumor growth was also dramatically restricted by QGY-5-114-A in vivo. In conclusion, pharmacological treatment with QGY-5-114-A impedes CRC tumor cell proliferation, migration and tumor growth.
Keywords: Colorectal cancer, Serum and glucocorticoid inducible kinase 1, Inhibitor, Cell proliferation, Cell migration, Colonic tumor growth
Introduction
Colorectal cancer (CRC) is the most frequently diagnosed cancer, and remains the leading cause of cancer-related death worldwide [1]. The cause of CRC is very complex and diverse, including multiple factors such as diet, genetic predisposition, lifestyle, environmental factors and the presence of long-standing inflammatory bowel diseases [2-4]. Multiple systems and pathways are involved in colorectal carcinogenesis and contribute to the tumor initiation and development. Abnormal DNA methylation, chromosomal instability pathway, inflammation, microRNA, DNA Mismatch Repair (MMR) system and consequential microsatellite instability (MSI) have been reported to be responsible for CRC [5-9]. Various therapeutic agents targeting the above-mentioned pathways have resulted in a significant reduction of mortality in early stage CRC. However, the morbidity and mortality remain high in patients with advanced stage disease [10]. Thus, the optimization of clinical treatment and the investigation of novel molecular-targeted therapeutics for CRC are critically needed.
Serum-and glucocorticoid-regulated kinase 1 (SGK1) belongs to the AGC family of serine/threonine protein kinases. SGK1 is up-regulated by insulin and growth factors through the phosphatidylinositide-3-kinase (PI3K), 3-phosphoinositide dependent kinase (PDK1) and PDK2 [11]. SGK1 promotes tumor growth by enhancing tumor cell survival, motility, invasiveness, adhesiveness and epithelial to mesenchymal transition [12-14]. Following deficiency of APC (adenoma polyposis coli), SGK1 knockout mice developed less intestinal tumors than their wild-type littermates [15]. In addition, SGK1 inhibitors (GSK650394, LY294002 and EMD638683) proved to be effective in vitro against prostate cancer cells [16], breast cancer [13] and the colorectal cancer [17]. Thus, we hypothesized that SGK1 would be an attractive target for developing novel anti-CRC therapies.
In this study, we designed and synthesized 39 new analogs of SGK1 inhibitor GSK650394. And one of these compounds named as QGY-5-114-A was more effective than GSK650394 in vitro and in vivo against colonic tumor cells. Therefore, the present study develops a new competitive inhibitor of SGK1, which is likely to have therapeutic utility in CRC.
Methods
Preparation of Phenyl(1-phenyl-1H-pyrrolo[2,3-b]pyridin-3-yl)methanone (General Procedure) [17]: 1-phenyl-1H- pyrrolo[2,3-b]pyridine (0.39 g, 2.0 mmol) was added to a stirred suspension of AlCl3 (1.4 g, 10 mmol) in CH2Cl2 (48 mL). After the mixture was stirred at room temperature for 1 h, benzoyl chloride (1.4 g, 10 mmol) was added dropwise and the resulting mixture stirred for 21h. MeOH (10 mL) was added cautiously to quench the reaction, the solvents were removed under vacuum, and the residual solid was purified by flash chromatography on silica gel (petroleum ether/EtOAc = 10:1) to afford 3f (0.45 g, 75%) as a white solid.
Preparation of (1-(2-Chlorophenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)(phenyl)- methanone (QGY-5-114-A) (General Procedure) [17]: To a 15 mL sealed tube was added phenyl(1-phenyl-1H- pyrrolo[2,3-b]pyridin-3-yl)methanone (89.5 mg, 0.3 mmol), [RhCp*Cl2]2 (3.7 mg, 0.006 mmol), Cu(TFA)2 (174 mg, 0.6 mmol), Li2CO3 (22 mg, 0.3 mmol), t-BuNC (50 mg, 0.6 mmol) and DCE (1.5 mL). The reaction mixture was stirred at 130 oC under air atmosphere. After reacted for 46 h, the reaction was diluted by EtOAc (10 mL) and quenched with aqueous ammonia solution (3 M, 10 mL). The aqueous layer was extracted with EtOAc (3×10 mL) and the combined extract was dried over Na2SO4, filtered, and concentrated in vacuo. The given residue was purified by column chromatography on silica gel (petroleum ether/EtOAc = 20:1) to give the clorinated product QGY-5-114-A (70.9 mg, 71%) as a white solid.
Cell culture
Colorectal cancer cell lines HCT116 and HT29 were purchased from KeyGEN BioTECH. CO., LTD (Nanjing, China). HCT116 cells were cultured in Dulbecco's modified Eagle's medium (DMEM, Corning) supplemented with 10% fetal bovine serum (FBS, CellMax) and 1% PS (penicillin-streptomycin, Gibco), and HT29 cells were cultured in RPMI 1640 medium (KeyGEN) supplemented with 10% FBS (Biological Industries) and 1% PS, and maintained in a humidified atmosphere containing 5% CO2 at 37 ºC.
Cell viability assay
Cell viability was detected using the cholecystokinin octapeptide (CCK-8) assay (Bioworld). After 6-8 h starvation, HCT116 cells were plated in 96 well plate at 2×105 cells/mL per well, and grown for 24 h. Then, cells were treated with various concentrations of compounds (0, 25, 50, 75, 100 and 200 μM) for 24 h, 1 hour before the end of the experiment, CCK-8 was added and incubated in 37 ºC blocking the light following the manufacturer's instructions. Then the cell proliferation inhibition was calculated by measuring the optical density values (OD) at 450 nm using microplate reader (Bio-Rad), the half maximal inhibitory concentration (IC50) value of every compound on HCT116 cells was calculated using SPSS software through probit model by logit-transformed.
Flow cytometry
HCT116 and HT29 cells were plated in 12 well culture plastic dish at 2× 105 cells per well. Briefly, cells were treated with vehicle DMSO or 50 μM compound for 24 h. After cold ethanol fixation overnight, cells were resuspended in 100 μl PBS. Subsequently, these samples were assessed using propidium iodide (PI, Sigma) staining followed by flow cytometry (Beckman).
EdU proliferation assay
Detection of compounds effect on HCT116 and HT29 cell proliferation was performed according to the manual of Cell-Light™ EdU Apollo®488 Cell Tracking Kit (Ribobio). Two hours before the end of cell treatment by compound, 50 μM EdU labeling medium was added to the cell culture to finish co-incubation. Afterwards, cells were washed with PBS and fixed with 4% paraformaldehyde overnight and neutralized with glycine for 5 min. After washed with PBS and 0.5% TritonX-100 for 10 min, anti-EdU working solution was added to stain at room temperature for 30 min. Subsequently, the DNA contents were stained with 5 μg/ml Hoechst 33342 for 30 min, and then observed under a fluorescent microscope (Leica Microsystems). The quantity of cell nuclear was calculated with Cell Profiler software.
Migration
The migration ability of HCT116 cells was examined in 6.5 mm transwell chambers with 8 μm pores (Costar). Briefly, 600 μl DMEM supplemented with 10% FBS was added to the bottom chamber. 200 μl resuspended cells (2×106/ml) treated with compound in serum-free DMEM were added to the top chamber. After migration for 48 h, cleaned cells upper surface of the membrane with a cotton swab and washed with PBS. Then the chamber was stained with crystal violet (Bioworld) for 5 min. The quantity of cells on the bottom of the membrane was performed under a microscope (Leica Microsystems).
Mice and treatment
Athymic nude mice aged 6 weeks were purchased from the Cavens lab (Changzhou, China). Under SPF conditions, the mice were bred in 12 h light/12 h dark cycle in an airy room at constant temperature about 21-23 °C. All animal experiments were conducted in accordance with institutional guidelines. Transplanted tumors were generated through implanting subcutaneously in 8-week old mice. HCT116 cells were suspended in 10% Matrigel and injected in the subcutaneous tissue of nude mice at a density of 3×106 cells/200 μL per mouse. After two weeks, compound (50 mM, 1:5 dilution, 40 μl) was injected around the subcutaneous every other day. Transplanted tumors were dissected after xenograft implantation 8 days, and then measured, fixed in 4% PFA and frozen in liquid nitrogen until use.
Immunohistochemistry on tissue section
Tissues from the xenograft tumor were embedded in paraffin and sliced to 5-μm thickness. Tissue sections were maintained in a 65 °C incubator 2 h for drying. After dewaxing and dehydration, antigens were unmasked by pressure cooking for 10 min in citrate buffer according to antibody specification. Then sections were immersed in 0.3% hydrogen peroxide for 10 min to destruct the endogenous peroxidase activity. The next steps were performed avoiding exposing to light. 5% BSA used to block non-specific binding for 5 min, subsequently covered each slice with 50 μL primary antibodies against Ki67 (Abcam, 1:300, v/v, dilution), PCNA (Proteintech, 1:300, v/v, dilution), incubation overnight at 4 °C. Then covered with appropriate secondary antibodies for 1 h at room temperature and added each slice with fresh prepared 3,3-diaminobenzidine tetrahydrochloride (DAB) solution and hematoxylin counterstain. After staining, sections were dehydrated through increasing concentrations of ethanol and xylene. A total of 15 fields were randomly selected for each section in the viable region of the tumor under the microscope after mounted coverslip with neutral resin containing xylene.
Statistical analysis
All statistics of experiments were analyzed by GraphPad Prism 5.0 or SPSS20.0 software. All data were shown as mean ± SE. A Student's t test was performed to determine difference between the control and treated group. For all of the tests, P < 0.05 was considered statistically significant.
Results
IC50 and kinetic analysis of QGY-5-114-A
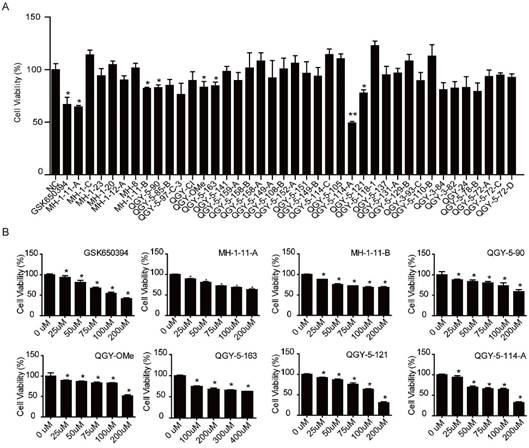
We first designed and synthesized new 39 analogs of SGK1 inhibitor GSK650394 (Fig. 1). We then preliminarily examined their inhibitory activities with CCK-8 assay. The results showed that those analogs, numbered as MH-1-11-A, MH1-11-B, QGY-5-90, QGY-OMe, QGY-5-163, QGY-5-114-A and QGY-5-121, could inhibit the cell viability in colonic tumor cell line HCT116 (Fig. 2A). To compare the inhibitory potency of these 7 analogs, we calculated their IC50 values by determining the concentration needed to inhibit half of the maximum cell viability of these compounds. Using CCK-8 assay, we showed that GSK650394 inhibited HCT116 cell viability with an IC50 value of 135.5 μM. And only compound QGY-5-114-A showed a significantly lower IC50 value of 122.9 μM, indicating the better inhibitory potency than commercial inhibitor GSK650394 (Fig. 2B and Table 1).
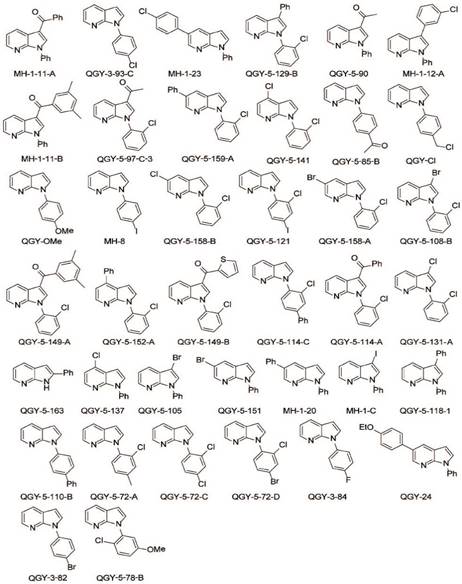
Chemical structure of GSK650394 and 39 new developed analogs.
Seven compounds of the 39 newly developed GSK650394 analogs suppress cell viability. (A) Cell viability analysis of HT116 cells treated with 50 μM GSK650394 and 39 new analogs, as determined by CCK-8 assay. (B) Cell viability analysis of HT116 cells treated with various concentrations of compounds.
The IC50 value of SGK650394 and new developed chemical compounds
| Number | IC50 |
|---|---|
| GSK650394 | 135.5 μM |
| MH-1-11-A | 346.4 μM |
| MH-1-11-B | 473.0 μM |
| QGY-5-90 | 340.8 μM |
| QGY-OMe | 266.9 μM |
| QGY-5-163 | 1528.7 μM |
| QGY-5-114-A | 122.9 μM |
| QGY-5-121 | 133.9 μM |
QGY-5-114-A restrains colonic tumor cell proliferation
Several studies have showed that SGK1 regulates survival, proliferation, migration and invasion of cancer cells. In this study, we first evaluated the proliferation of HCT116 cells treated with QGY-5-114-A. The results showed that QGY-5-114-A inhibited HCT116 cell proliferation, revealed by a significant decrease in EdU staining (Fig. 3A). This conclusion was further confirmed by cell cycle analysis via flow cytometry (Fig. 3B).
QGY-5-114-A inhibits HCT116 cell proliferation. (A) EdU staining of HCT116 cells and EdU-positive cell proportion. Scale bar, 100 μm; n = 4; ***, P<0.001; (B) HCT116 cell cycle analysis by flow cytometry. ***, P<0.001.
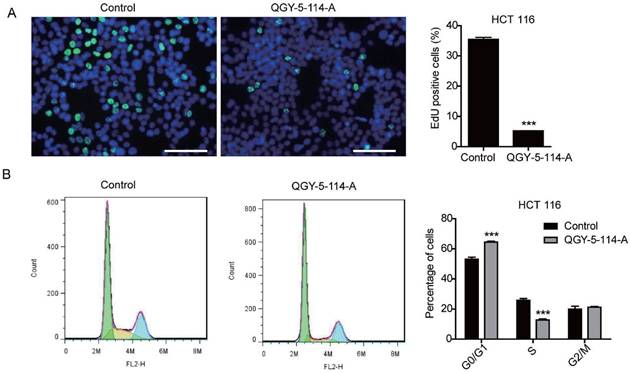
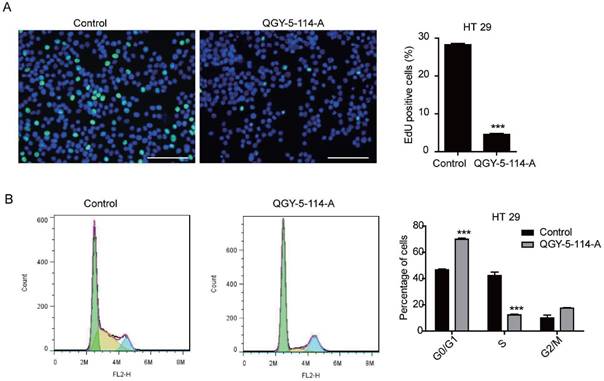
The cell cycle distribution of HCT116 cells was affected by QGY-5-114-A, with a significant increase in G0/G1 population and a decrease in S phase cells. In order to investigate whether QGY-5-114-A could affect other colonic tumor cells, we also performed EdU staining and cell cycle analysis in HT29 cells. The same results were obtained (Fig. 4A and Fig. 4B). Taken together, our results revealed that QGY-5-114-A could suppress the proliferation of colonic tumor cells.
QGY-5-114-A inhibits HT29 cell proliferation. (A) EdU staining of HT29 cells and EdU-positive cell proportion. Scale bar, 100 μm; n=4; ***, P<0.001; (B) Cell cycle analysis of HT29 cells by flow cytometry. ***, P<0.001.
QGY-5-114-A impedes colonic tumor cell migration in vitro
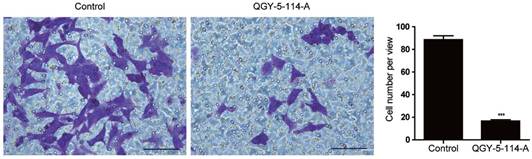
CRC is one of the most lethal malignancies, with the high rates of metastasis. To explore the biological functions of QGY-5-114-A in CRC migration ability, we first performed transwell migration assay in HCT116 cells. Cells treated with QGY-5-114-A displayed impaired migration (Fig. 5A), and the percentage of cells on the bottom of membrane was notably lower than control cells (Fig. 5B). These results indicated that QGY-5-114-A restrains CRC metastasis in vitro.
QGY-5-114-A suppresses the migration of HCT116 cells. Decrease in HCT116 cell numbers after migration for 48 hours by QGY-5-116, n=4; Scale bars, 100 μm; ***, P<0.001.
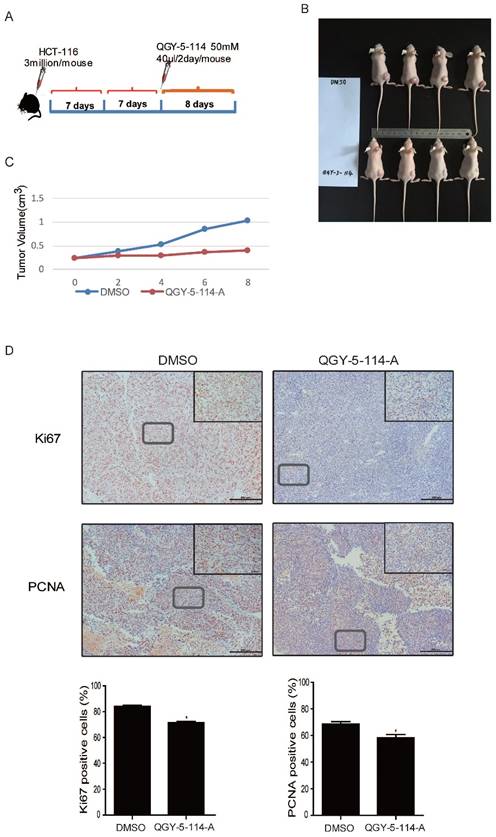
QGY-5-114-A inhibits colonic tumor growth and cell proliferation in vivo
We explored the therapeutic effect of QGY-5-114-A on CRC growth by administering it into nude mice. CRC implant tumors were induced by subcutaneous injection of HCT116 cells, followed by treatment with the compound QGY-5-114-A (Fig. 6A). The results showed that QGY-5-114-A prominently reduced xenograft tumor size and volume until the mice were dissected on day 8 (Fig. 6B and 6C). In addition, we detected the xenograft tumor tissues by immunohistochemical analysis for proliferation markers including Ki67 and PCNA (Fig. 6D). In xenograft tumors treated with QGY-5-114-A, PCNA- and Ki67-positive cell populations were dramatically reduced. Collectively, these results suggest that QGY-5-114-A is impactful to depress the proliferation of CRC cells and tumor growth during the CRC development.
QGY-5-114-A restrains tumor growth and cell proliferation in subcutaneous xenograft mice model. (A) The procedure of establishing the xenotransplant mice model. (B) Transplanted tumor size of control and mice injected with QGY-5-114-A. n=4; (C) Transplanted tumor volume. n=4; (D) Reduction in Ki67- and PCNA-positive cell populations in skin tissues treated with QGY-5-114-A. Scale bars, 200 μm; *, P<0.05;
Discussion
The present study describes the development of a novel compound, QGY-5-114-A, that was a new analog of SGK1 inhibitor GSK650394. We also examined the effects of QGY-5-114-A on CRC tumor cell proliferation, migration and tumor growth. GSK650394, a commercial inhibitor of SGK1, has previously been shown to antagonize SGK1 activity and inhibit prostate cancer cell growth [16]. Our results showed that, compared with GSK650394, this novel compound QGY-5-114-A has lower molecular weight, simpler chemical structure and lower IC50 value (Fig. 1, 2 and Table 1), indicating that QGY-5-114-A has a promising application prospect as SGK1 inhibitor.
Previous studies reported that GSK650394 inhibits the enzymatic activity of both SGK1 and other members of the SGK protein family [16], and another SGK1 commercial inhibitor EMD638683 also inhibits the other two SGK1 isoforms with similar potency [18]. Therefore, extensive studies are needed to determine the specificity and effectiveness of QGY-5-114-A in inhibiting the enzymatic activity of SGK1.
The inhibition of colonic tumor growth by QGY-5-114-A is fully consistent with the previous reports that SGK1 mutation reduces spontaneous intestinal tumor number in APC-deficient mice [15] and SGK1 inhibition counteracts colonic tumor growth in vivo [17]. Several mechanisms, such as mTOR, FOXO3a, β-catenin, p53 and NF-kB, have been reported or predicted to be involved in SGK1-dependent tumor cell proliferation and tumor growth [19-25]. However, the explicit molecular mechanism of SGK1 in regulating colorectal cancer is still not clear. Accordingly, QGY-5-114-A will facilitate the identification of pathways regulated by SGK1 in CRC, which may develop more effective CRC treatments. Besides that, the identification and characterization of novel SGK1 antagonist by performing optimization on the basis of known compounds could be emulated by the identification of other small molecule inhibitors with ideal pharmacokinetic characteristics.
In conclusion, we developed 39 novel analogs of SGK1 inhibitor GSK650394 and one of them, QGY-5-114-A, has lower molecular weight and IC50 value. Treatment with QGY-5-114-A significantly restrains colonic tumor cell proliferation, migration and tumor growth, indicating QGY-5-114-A will be a promising therapeutic inhibitor in the treatment of CRC and other malignancies.
Abbreviations
CRC, colorectal cancer; MMR, mismatch repair; SGK1, serum-and glucocorticoid-regulated kinase 1; PI3K, phosphatidylinositide-3-kinase; PDK1, 3-phosphoinositide dependent kinase; APC, adenoma polyposis coli.
Acknowledgements
This work was supported by the grants from National Natural Science Foundation of China (81470515 to JH Xu and 21672136 to B Xu), Shanghai Medical Guide Project from Shanghai Science and Technology Committee (134119a3000 to JH Xu) and the development fund for Shanghai talents (to JJ Xiao).
Competing Interests
The authors declare no competing financial interests.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7-30
2. Burn J, Gerdes AM, Macrae F, Mecklin JP, Moeslein G, Olschwang S. et al. Long-term effect of aspirin on cancer risk in carriers of hereditary colorectal cancer: an analysis from the CAPP2 randomised controlled trial. Lancet. 2011;378:2081-7
3. Cappellani A, Zanghi A, Di Vita M, Cavallaro A, Piccolo G, Veroux P. et al. Strong correlation between diet and development of colorectal cancer. Front Biosci (Landmark Ed). 2013;18:190-8
4. Abu-Remaileh M, Bender S, Raddatz G, Ansari I, Cohen D, Gutekunst J. et al. Chronic inflammation induces a novel epigenetic program that is conserved in intestinal adenomas and in colorectal cancer. Cancer Res. 2015;75:2120-30
5. Wheeler JM, Bodmer WF, Mortensen NJ. DNA mismatch repair genes and colorectal cancer. Gut. 2000;47:148-53
6. Gyparaki MT, Basdra EK, Papavassiliou AG. DNA methylation biomarkers as diagnostic and prognostic tools in colorectal cancer. J Mol Med (Berl). 2013;91:1249-56
7. Garcia-Anguita A, Kakourou A, Tsilidis KK. Biomarkers of Inflammation and Immune Function and Risk of Colorectal Cancer. Curr Colorectal Cancer Rep. 2015;11:250-8
8. Amirkhah R, Schmitz U, Linnebacher M, Wolkenhauer O, Farazmand A. MicroRNA-mRNA interactions in colorectal cancer and their role in tumor progression. Genes Chromosomes Cancer. 2015;54:129-41
9. Gupta S, Ashfaq R, Kapur P, Afonso BB, Nguyen TP, Ansari F. et al. Microsatellite instability among individuals of Hispanic origin with colorectal cancer. Cancer. 2010;116:4965-72
10. Schlussel AT, Gagliano RA Jr, Seto-Donlon S, Eggerding F, Donlon T, Berenberg J. et al. The evolution of colorectal cancer genetics-Part 1: from discovery to practice. J Gastrointest Oncol. 2014;5:326-35
11. Kobayashi T, Cohen P. Activation of serum- and glucocorticoid-regulated protein kinase by agonists that activate phosphatidylinositide 3-kinase is mediated by 3-phosphoinositide-dependent protein kinase-1 (PDK1) and PDK2. Biochem J. 1999;339(Pt 2):319-28
12. Wu W, Chaudhuri S, Brickley DR, Pang D, Karrison T, Conzen SD. Microarray analysis reveals glucocorticoid-regulated survival genes that are associated with inhibition of apoptosis in breast epithelial cells. Cancer Res. 2004;64:1757-64
13. Tangir J, Bonafe N, Gilmore-Hebert M, Henegariu O, Chambers SK. SGK1, a potential regulator of c-fms related breast cancer aggressiveness. Clin Exp Metastasis. 2004;21:477-83
14. Cheng J, Truong LD, Wu X, Kuhl D, Lang F, Du J. Serum- and glucocorticoid-regulated kinase 1 is upregulated following unilateral ureteral obstruction causing epithelial-mesenchymal transition. Kidney Int. 2010;78:668-78
15. Wang K, Gu S, Nasir O, Foller M, Ackermann TF, Klingel K. et al. SGK1-dependent intestinal tumor growth in APC-deficient mice. Cell Physiol Biochem. 2010;25:271-8
16. Sherk AB, Frigo DE, Schnackenberg CG, Bray JD, Laping NJ, Trizna W. et al. Development of a small-molecule serum- and glucocorticoid-regulated kinase-1 antagonist and its evaluation as a prostate cancer therapeutic. Cancer Res. 2008;68:7475-83
17. Towhid ST, Liu GL, Ackermann TF, Beier N, Scholz W, Fuchss T. et al. Inhibition of colonic tumor growth by the selective SGK inhibitor EMD638683. Cell Physiol Biochem. 2013;32:838-48
18. Ackermann TF, Boini KM, Beier N, Scholz W, Fuchss T, Lang F. EMD638683, a novel SGK inhibitor with antihypertensive potency. Cell Physiol Biochem. 2011;28:137-46
19. Feng Z, Liu L, Zhang C, Zheng T, Wang J, Lin M. et al. Chronic restraint stress attenuates p53 function and promotes tumorigenesis. Proc Natl Acad Sci U S A. 2012;109:7013-8
20. Amato R, D'Antona L, Porciatti G, Agosti V, Menniti M, Rinaldo C. et al. Sgk1 activates MDM2-dependent p53 degradation and affects cell proliferation, survival, and differentiation. J Mol Med (Berl). 2009;87:1221-39
21. You H, Jang Y, You-Ten AI, Okada H, Liepa J, Wakeham A. et al. p53-dependent inhibition of FKHRL1 in response to DNA damage through protein kinase SGK1. Proc Natl Acad Sci U S A. 2004;101:14057-62
22. Mikosz CA, Brickley DR, Sharkey MS, Moran TW, Conzen SD. Glucocorticoid receptor-mediated protection from apoptosis is associated with induction of the serine/threonine survival kinase gene, sgk-1. J Biol Chem. 2001;276:16649-54
23. Zhang L, Cui R, Cheng X, Du J. Antiapoptotic effect of serum and glucocorticoid-inducible protein kinase is mediated by novel mechanism activating I{kappa}B kinase. Cancer Res. 2005;65:457-64
24. Avruch J, Hara K, Lin Y, Liu M, Long X, Ortiz-Vega S. et al. Insulin and amino-acid regulation of mTOR signaling and kinase activity through the Rheb GTPase. Oncogene. 2006;25:6361-72
25. Xu Y, Chen SY, Ross KN, Balk SP. Androgens induce prostate cancer cell proliferation through mammalian target of rapamycin activation and post-transcriptional increases in cyclin D proteins. Cancer Res. 2006;66:7783-92
Author contact
![]() Corresponding authors: Dr. Junjie Xiao Regeneration and Ageing Lab, School of Life Science, Shanghai University, 333 Nan Chen Road, Shanghai 200444, China; Tel: 0086-21-66138131; Fax: 0086-21-66138131; E-mail: junjiexiaoedu.cn. And Dr. Bin Xu Department of Chemistry, Innovative Drug Research Center, 99 Shang Da Road, Shanghai 200444, China; Tel: 0086-21-66132830; Fax: 0086-21-66132830; E-mail: xubinedu.cn
Corresponding authors: Dr. Junjie Xiao Regeneration and Ageing Lab, School of Life Science, Shanghai University, 333 Nan Chen Road, Shanghai 200444, China; Tel: 0086-21-66138131; Fax: 0086-21-66138131; E-mail: junjiexiaoedu.cn. And Dr. Bin Xu Department of Chemistry, Innovative Drug Research Center, 99 Shang Da Road, Shanghai 200444, China; Tel: 0086-21-66132830; Fax: 0086-21-66132830; E-mail: xubinedu.cn