Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(12):2238-2246. doi:10.7150/jca.19295 This issue Cite
Review
Central Role of CEMIP in Tumorigenesis and Its Potential as Therapeutic Target
Li Li1 ![]() , Lin-Hai Yan2
, Lin-Hai Yan2 ![]() , Shwetha Manoj3, Ying Li4, Lu Lu5
, Shwetha Manoj3, Ying Li4, Lu Lu5
1. Department of Pharmacy, The People's Hospital of Guangxi Zhuang Autonomous Region, Nanning 530021, Guangxi Zhuang Autonomous Region, People's Republic of China
2. Department of Gastrointestinal Surgery, Affiliated Tumor Hospital of Guangxi Medical University, Nanning 530021, Guangxi Zhuang Autonomous Region, People's Republic of China
3. Quality Assurance Department, Bristol Laboratories Limited, 5 Traynor Way, Whitehouse Business Park, Peterlee, County Durham, SR8 2RU, United Kingdom
4. Department of Pharmacy, Guangxi Bone Hospital, Nanning, 530000, Guangxi Zhuang Autonomous Region, People's Republic of China
5. Department of Research, Nanning Children Rehabilitation Center, Nanning, 530003, Guangxi Zhuang Autonomous Region, People's Republic of China
Received 2017-1-22; Accepted 2017-4-25; Published 2017-7-20
Abstract
CEMIP (KIAA1199) was identified as migratory indicator protein which had been crudely studied in the last decade. Firstly its mutation site was reported to cause hearing loss due to the folding change of protein structure, meanwhile the over-expression of CEMIP referred to dreadful invasion and uncontrolled proliferation of tumor with distant metastasis, dedifferentiation, and limited survival opportunity of patients. Especially, over-expressed CEMIP also protected malignant tumor from strict microenvironment in hypoxia, low glucose and cracked barrier, leading to enhanced adaptability of tumor by stimulating the Wnt, EGFR, FGFR pathway. Here, we intend to elaborate the clinical function and dysregulation of CEMIP under the tumorous circumstance since CEMIP plays an important role in cytokine pathway and its over-expression in tumors provide a novel target for individual therapy. Targeting CEMIP would thereby dysregulate the cytokine pathway which would in turn, decide the growth and death of the vicious tumour cells.
Keywords: CEMIP/KIAA1199, Cancer, Cytokine Signaling pathway, Hyaluronic acid.
Introduction
KIAA1199, defined as cell migration inducing protein currently (CEMIP, https://www.ncbi.nlm.nih.gov/gene/57214), is located in chromosome 15q25.1, which appears in the nucleus and cytoplasm [1-3]. This protein contains a special G8 domain, associated with two nonhomologous GG domain and four pbH1 domain [4, 5]. The first report about CEMIP discussed the mutation site of protein sequence leading to asymptomatic loss of hearing i.e. non-syndromic hearing loss, wherein a histidine is mutated to a tyrosine in N-terminal of GG domain [6]. This result confirmed that the above specified domain had major involvement in the cause of the disease. Generally, G8-containing protein was probably positioned intra- or trans-membrane [4, 7]. However, transmembrane domain prediction program for CEMIP protein and a cytoplasmic localization of EGFP-CEMIP fusion protein experiment were conducted and the cytoplasm location of mature CEMIP was verified [8]. An even distribution was observed through electron microscope after being treated with postembedding immunogold staining in Corti, indicating that the favorite location of CEMIP was cytoplasm [9]. Later, other researches revealed that the subcellular location of CEMIP was confined to perinuclear space which was endoplasmic reticulum (ER) through immunofluorescence staining [10]. Identification of the possible property of CEMIP revealed that it was a secreted protein rather than a transmembrane protein, the protein with 153kDa was tested out in supernate media when using C-terminal V5 tag, and further confirmation showed the full-length CEMIP emerged in immunoprecipitant of colon cancer cell in its serum-free supernate by using anti-CEMIP monoclonal antibodies [11].
Particular to the gene fragment, there were two disease-required up-regulation sections roughly around pro-1.4 in CEMIP sequence, which were specific to -1425 to -1135 and -125 to +27, at where the fourth NK-κB (-1345 to -1333), AP-1 (-48 to -45) were positioned. Differently, effective CpG of CEMIP island lied in the first intron. The level of methylation of this region inversely correlated to CEMIP expression making it a potential therapy site. Lower the base sequence methylation in CpG island, the more aggressive the cancer progression would be as being proofed by the demethylation in aggressive breast cancer sample [12]. Except that, C-terminal also involved a B-domain which can pose a suitable binding site for binding immunoglobulin protein (BiP), leading to its excretion and correct-folding of CEMIP through ER [13]. When BiP bond to B-domain of CEMIP, calcium leakage would happen in ER, resulting in the rise of cytosolic Ca2+ concentration and the release of CEMIP [13]. On the other hand, N-terminal contained a 30 signal peptide sequence functioning in immobilized CEMIP in ER. On the knockdown of the 30 amino acids, protein secretion was enhanced and therefore, an intra cytoplasmic acceleration of mature CEMIP from ER occurred through Golgi apparatus [5]. Furthermore, fraction surviving with strongly cytoplasmic expression of CEMIP was a quarter less than the strongly nuclear-expressed one after four years post-surgery [3]. A status of nuclear confinement for CEMIP illustrated a relatively safe situation for cancer patient. It also might be the evidence for early medical intervention or diagnostic indicator when patients suffered aggressive tumor [14, 15]. Though the expression of CEMIP, placed in the nucleus, was discovered in colon adenomas regardless the UICC stage, the localization of CEMIP in the nucleus and its over-expression might be linked to the presence of β-catenin in nucleus [3]. A comparison between CEMIP-containing colorectal cells and its knock-down counterpart suggested CEMIP belonged to Wnt signaling pathway due to the same regulated tendency of the change of eight Wnt-axis genes including β-catenin expression. Basically, CEMIP assisted the catabolism of HA thereby stabilizing a suitable microenvironment for cancer growth. CEMIP has been reported another role in glycolysis and gluconeogenesis for generating nutrition in tumor cell [10]. In normal human tissues, CEMIP mRNA expressed in terms of amounts, the maximum quantity was in the brain, followed by lung, placenta and testis [8]. To be specific, the abundant expression of CEMIP in brain was found in hippocampus which supports memory function [16]. On the other hand, over-expression of CEMIP emerged in the special type of cancer cells which easily diffused to their surrounding tissues, such as gastric, colorectal, cervix, kidney and lung cancer [3, 17]. Those researches prevalently agreed the over-expression of CEMIP was associated with a severe and easily diffusible situation when patient suffered malignant tumor also a poor prognosis.
The relationship between non-tumor disease and CEMIP
The majority of researches focused on the different expression level of CEMIP within tumor tissue associating with severity of cancer progression and prognosis. In physiological status, the presence of CEMIP expression was observed during early proliferating and late senescent stages in mortal cells, renal carcinoma cell was transferred by chromosome 3 (RCC23+3), whereas no detectable amount was investigated in their immortal counterparts. Therefore, the CEMIP was reported as a mediator of cellular mortality [8, 11, 18, 19]. Under the circumstance which allowed protein mutation, structural change of CEMIP lead to functional loss in cochlea. Although the 187th amino acid mutation occurred in hearing loss of human, establishing the same through in vivo animal experiments required strict criteria in murine [6]. The rats or mice are not the suitable models for detecting CEMIP expression once the gene deficiency happened at birth or later, since the rodents possessed no hearing function when they are born, and this gene is hard to be found in their hair cell and outer suclus cells whereas it can be observed in human cochlea [20]. But the mature health murine could be modeled because of their high homology to human [21]. Therefore, compared with rodents, experiments on marmoset which contained higher homology were successfully carried out. The area of CEMIP expression were accordantly observed as in human organs or tissues, suggesting a novel and preferred animal model for hearing loss [20]. In order to expose the functional change of CEMIP, three mutation points of CEMIP were identified wherein two were irrelevant but one became highly relevant site which was shown as H783Y. In this position, the mutation of histidine to tyrosine led to loss of positive charge [6]. After the mutation, the hydrophilic property of protein dramatically decreased while the aromatic ring increased its lipophilicity which could pose as a reason for its accumulation in cytoplasm via nuclear membrane. On the contrary, the extensive expression of CEMIP in potassium transportation in murine cochlea supported the assumption that expression of CEMIP protein might induce electrolyte disturbance in Corti as in human [6]. Therefore an early genetic detection needs to be instigated especially for the patients with family history [22].
Due to the regulation of hyaluronic acid (HA) by CEMIP, over-expression of CEMIP could possibly be associated with occurrence of disease in HA rich organs. HA is widely distributed in human tissues and it is a main element of forming extracellular matrix, adjusting adhesion, migration, and proliferation [23]. As for the modulation in rheumatoid arthritis (RA), the over-expression of CEMIP was investigated in synovial tissues, which has been verified by the comparison of samples from RA patients and healthy control, including serum concentration, synovial and synovial tissues. According to the ROC curve, the concentration of CEMIP in the index could be presented as a clinical diagnostic parameter. The expression of CEMIP was, then, detected through IHC and FISH where its over-expression preferred to locate in vascular endothelium [14]. Another observation in osteoarthritis (OA) and RA cells was the positive-regulation of CEMIP expression in HA degradation activity, wherein the higher expression of CEMIP existed in synovial lining of OA [24]. The similar mechanism was found in Crohn's disease wherein secreted CEMIP tended to gather in HA-rich fibroblast, and the over-expressed CEMIP was induced by IL-6 leading to activation of inflammatory pathway which extensively infected colon mucosa [25]. Additionally, suppressing the expression of CEMIP by COX-2 inhibitor supported that CEMIP also bridged inflammatory pathways and cancerous progression [26].
The other research found expression of CEMIP in physiological level related to formation of benign neoplasm, and it might accelerate canceration once its expression had been increased. Validation had been conducted through the mRNA isolation and quantification from biopsy samples. CEMIP expression level of normal duodenal mucosa remained low. Between familial adenomatous polyposis (FAP) and non-FAP groups, over-expression of CEMIP had no clear relationship. This result further indicated the high expression of CEMIP might particularly appear in the proliferation and canceration of colorectal and gastric cancer cells [15].
Summary of over-expressed CEMIP and related cancer diseases.
| Subcellular location | Study models | Pathology | Origin | Function | Ref. |
|---|---|---|---|---|---|
| cytoplasm | RCC23+3 | N/M | Immortal cell | Physiological distribution, Immortal proliferation | [8] |
| cytoplasm | PDAC tissues biopsies | pancreatic ductal adenocarcinoma | carcinoma | Migration, shorter survival | [29] |
| blood | Pdx1-Cre:K-RasLSLG12D mouse models | Predictor for tumorigenesis | [30] | ||
| N/M | BxPC-3, PANC-1 | Increase of light molecule weight of HA, Malignant change Inflammatory pathway target | [26] | ||
| N/M | TU-KATOIII, SNU16 | Gastric cancer | Invasion, migration, Glycogen breakdown | [16] | |
| cytoplasm | Dissection from patient | Poor differentiation, Distant metastasis, Peritoneal dissemination, Late stage | [33] | ||
| N/M | MDAMB453 | breast cancer | Drug resistance related | [39] | |
| cytoplasm | HPV-positive CaSki | Stabilize EGFR, Phenotype change | [37] | ||
| cytoplasm | MDA-MB-231 | Invasion Proliferation | [28] | ||
| N/M | microarray | colorectal adenomas | Linker of Wnt signaling pathway | [35] | |
| N/M | SW480 DLD-1 | Degradation of HA, Phenotype imply poor survival, serological marker | [11] | ||
| N/M | SW480, HeLa | Migration from hypoxia | [42] | ||
| cytoplasm | Sample from patient | Invasion depth Lymph node metastasis | [18] | ||
| endoplasmic reticulum | HEK293 | Secreted ability, Assumed by Golgi apparatus to vesicles | [5] | ||
| perinuclear space | SW480 | Wnt signal target, Ca2+ amplification | [10] | ||
| cytoplasm | Sample from patients | Proliferation motility and adhesion | [3] | ||
| nucleus | Better prognosis | [3] | |||
| N/M | Sample from patients Cell lines: HCT116, SW480, HT29, LOVO and SW620 | Tumor survive, invasion, metastasis, Poor prognosis, Downstream of miRNA-126a | [27] | ||
| N/M | Sample from patient | oral squamous cell carcinoma | Potential indicator for cancer progression | [32] | |
| N/M | U87-MG | glioblastoma multiformae | blastoma | senescence | [34] |
| N/M | array-CGH | uterine leiomyosarcoma | sarcoma | Located at 15q25.1 Related to uterine leiomyosarcoma | [1] |
N/M: not mention; Ref.: reference.
Note: This table briefly listed the cancer-related researches which were included in this paper.
Tendency of high expression of CEMIP referred to severe situation in cancer patients
High measurement result of CEMIP mRNA expression in colorectal tissues indicated a statistically significant difference existing between adjacent normal tissues and the former except in some cases where it was a confirmation of malignant transformation, severe invasion or metastasis thereby ending up in a poor prognosis [27]. Inter-constrain between CEMIP and its upstream gene miRNA-126a revealed they were directly interdependent targeted sites. Furthermore, univariate Cox regression analysis demonstrated high CEMIP expression positively correlated with low differentiation, late TNM stage, tumor invasion depth, lymph node metastasis and high CEA concentration [19]. Despite the majority trend of over-expression of CEMIP observed in many cancer, a series of experiments in breast cancer reversely provided evidence through inhibition of tumor proliferation, adhesion, tumor angiogenesis and migration meanwhile accelerated apoptosis happened when CEMIP was absent [28]. In pancreatic ductal adenocarcinoma, over-expressed CEMIP alerted an aggressive tumor progression and early mortality [26, 29, 30]. Due to the secretion ability of CEMIP, high CEMIP level could be detected in cancer patients' blood, and the blood CEMIP positively correlated to grade of tumor malignancy. According to the report, detection of CEMIP could avoid the false positive result which was caused by carbohydrate antigen marker detection [30]. Among variety of cancer tissues, over-expression of CEMIP was commonly observed existed, especially in the late stage accompanying with dissemination. CEMIP was raised as a potential predictor for skin cancer after it was screened out through the cell lines which was extracting out of the mice with myeloma culture [31]. There were evidences supporting the high expression among many cancer cells. Through large scale microarrays, CEMIP gene and its protein were investigated and a 14.66 fold higher expression among in vitro breast cancer cells was observed compared with the control cells [28]. Other report, according to the mass spectrometry, showed the protein level of CEMIP was significantly higher in late stage oral squamous cell carcinoma (OSCC), with 9.85 fold change. Following real time quantitative real-time reverse qRT-PCR validated the over-expression of CEMIP in OSCC cells [32].
According to a survival study [11], the low expression of CEMIP stabilized at above 70% and 80% survival rate whereas the survival rate of the high expression group continually decreased to below 20% and approximately 30% respectively, both in stage III and II. In gastric cancer, the over-expressed-trend of CEMIP has also reflected in the distant metastasis, late TNM stage and poor prognosis which has been shown through 5-years survival rate. There was an obvious drop of gap between low- and high-expression group wherein the proportion of low-expression group presented 60% above. However, there was no parallel candidate and their CEMIP expression with at least the same TNM stage were stated in the comparison. Also, IHC results were absent, to support the patient who received postoperative chemotherapy which limited the scope of prognosis [33]. It was also worth to note that the suppression of tumor invasion and growth appeared in situ among the exact CEMIP-knockdown samples [28].
Although there is strong relationship between expression of CEMIP and tumourigenesis as it has been emphasised in uterine leiomyosarcomas, prostate cancer and glioblastoma multiforme [1, 8, 34], no further evidence showed whether the over-expression of CEMIP could be an impetus of cell carcinogenesis. However, over-expressed CEMIP, indeed, promoted the transformation of tumor phenotype. IHC provided the clues that high proportion of up-regulated CEMIP expression was observed in submucosa where was intruded by the cancer cell. Then the different expression level of CEMIP among phenotype change, epithelial-to-mesenchymal (EMT), was suggested by detecting intensity of E-cadherin. The intensive E-cadherin of adjacent normal tissue and the sparse intensity of invasive front indicated EMT process was stimulated by over-expressed CEMIP. The similar outcomes were also proofed through observation of breast carcinoma with a decrease of E-cadherin and cytokerain -8/-18 and the increase of Twist-1 [13].
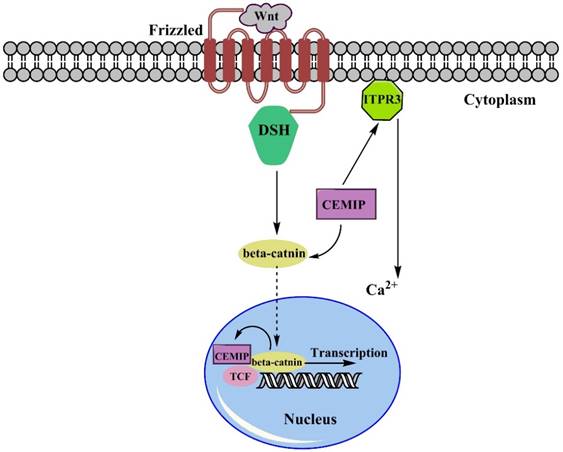
There is much evidence to support that the over-expression of CEMIP facilitates tumor progression. To be more specific on explaining the dynamic process here we choose some of the main regulatory pathways to state how over-expression of CEMIP resulted in tumor malignancy. Remarkable up-regulation of CEMIP was observed in all transcriptional profile of adenomas sample with an average increase of 5.8-fold increase and were filtered, and almost half of the Wnt targets was positively corelated to expression of CEMIP [35]. As been well described, the dual-regulated function of CEMIP in regulating Wnt/β-catenin pathway is mainly through Ca2+ [10, 18]. Based on the current studies (Fig. 1), transmembrane receptor Frizzled (Fz) firstly received Wnt signals, then passed it to DSH to form a complex, activating β-catenin with association of CEMIP flowed towards nucleus. After reaching in nucleus, β-catenin trigger TCF. However, since the expression of CEMIP affects the expression of β-catenin [3], the CEMIP would run to cytoplasm due to its self-trigger [13]. Then CEMIP would deliver Wnt signal to activate TCF, which would further initiate the duplication and transcript small secreted proteins of Wnt. Another sub-pathway stated that Ca2+ was regulated by the activation of ITPR3, releasing Ca2+, thereby breaking intracellular homeostasis through Wnt signaling pathway [36]. In colon cancer pattern where TCF1 or TCF4 are deactivated, CEMIP showed its down-regulation that indicated two suppressors of CEMIP/Wnt pathway. The similar result appeared when LS174 cell underwent a knockdown or inhibition of TCF4 to suppress Wnt pathway [35].
The Simple Wnt/β-catenin pathway involving CEMIP. The Wnt signals will be captured by Frizzled receptors, then they are passed to β-catenin after forming complex with DSH. Over-expression of CEMIP will emphasize this formation and stimulate movement of DSH-Wnt complex towards nucleus. Also, the expression of nucleus-settled β-catenin positively induce the expression of CEMIP. Once bond to TCF complex, the transcription of small secreted protein would be started. Besides, CEMIP also stimulated the ITR3 leading the increase of Ca2+, which was also one of the Wnt signaling factor leading to intracellular ions disordered.
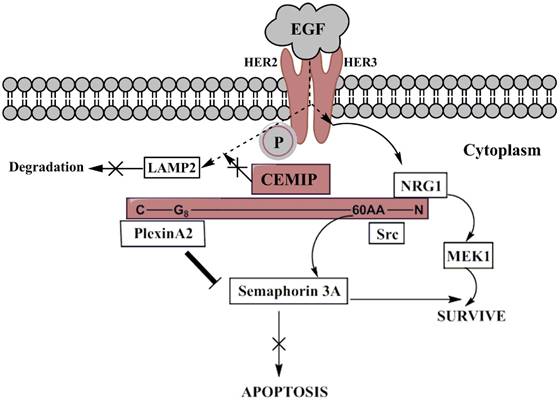
On the EGFR signaling pathway (Fig. 2), around 100 amino acids of N-terminal including G8 domain of CEMIP possessed the binding ability to Plexin A2 so that Plexin A2 signal would not further trigger the Semaphorin 3A-apoptosis [37]. On activating the pathway, cancer cell would be able to escape from apoptosis. Meanwhile, the same region of N-Terminal of CEMIP also bond to Semaphorin 3A/EGFR survival pathway. Based on similar decreased trend of Src, c-Cbl, MEK1 were shown among the deletion of CEMIP in cervical cancer-derived cells. However, CEMIP did not affect the dimerization, instead, transmitted the EGF signal after phosphorylation of HER2 and HER3 followed by MEK1 pathway, leading to the survival of tumor cell. Also, CEMIP inhibited the EGFR which tended to settle in LAMP2, resulting in lowering the degradation of EGF in lysosomes [37, 38]. Besides, the accordant trend of up-regulated CEMIP and other differentially expressed genes was discovered in glioblastoma tissues through gene analysis. However, further verified experiments have not been produced. The possibility of another gene, EPHb which was related to the tumor invasion was later reported. It was affected by CEMIP through cascading CCL2/Mcp-1, further enhancing PI3K and ERK1/2 to keep tumor cells alive [34].
The EGFR signaling pathway involving CEMIP mediation. When EGF signals firstly attach HER2 receptor, heterodimerization and phosphorylation of HER2 and HER3 will be automatically processed leading to enablement of EGFR signaling pathway. The signal will head into two branches, descending activation and degradation where over-expressed CEMIP acts as connected junction. The light purple color represents CEMIP and its simple protein structure with specific binding site, wherein survival signal Src targeted 60AA near N-terminal, and NRG1 bond to N-terminal. Src branch towards Semaphorin-3A-meidated survive and NRG1 lead to proliferation through activation of MEK1. Plexin A2 bond to G8 domain of C-terminal initiating suppression of apoptosis. The dashed arrows describe the EGF signaling routine, so do the curved arrows. Thick line from Plexin A2 to Semaphorin 3A means it cuts off the delivery of apoptotic signal to Semaphorin 3A. The participation of over-expressed CEMIP also prevent EGF signals from consumption of lysosomal enzyme (LAMP2).
Recently, FGFR pathway also caught our attention due to the over-expression of CEMIP strongly accumulated FGFR-apoptotic suppression and cross-linked to EGFR tumor pathway. When searching the signaling axis of anti-breast cancer drug, Phenylbutyrate, ZEB1/ESRP1/CEMIP axis was revealed in cancer phenotype variation. ESRP1 was an independent indicator for EMT process, the negative regulation of it was maintained by ZEB1 expression, while ESRP1 was upstream suppressor for CEMIP. Besides, both ESRP1 and ZEB1 was involved in FGFR signaling pathway which suggested CEMIP could be a cross-linker in this pathway [39].
Tumor resistance in harsh microenvironment due to over-expression of CEMIP
Generally, tumor cells suffer the strict microenvironment due to its rapid uncontrolled growth and high requirement of nutrient supplement. Hypoxia was an apparent condition in tumor growth, wherein the abnormal formation of vessels and heterogeneity were generally observed. As for angiogenesis, several related factors such as VGF, PDGF-B and TGF were promoted, so that the existed blood vessels would generate a new branch to feed tumor cell. Together, hypoxia provided the dedifferentiation and aggressiveness happened in cancer progression [40, 41]. Moreover, research into hypoxia stated that hypoxia could induce expression of CEMIP in colon cancer cells, CEMIP hypoxia promoter region was triggered by the related factors, such as hypoxia-inducible-factor-2α (HIF-2α), resulting in an increase in CEMIP's expression. Researchers also analyzed a span of HRE gene located in CEMIP, however, same increased level of activity could not be seen in the knockout HRE of CEMIP through luciferase assay, which implied there were co-factors for regulating expression of CEMIP. After hydroxylation, mutated HIF-2α was screened, SW480 cells showed the migrated ability via the over expression of HIF2a mutant and a drop of migration in mutant HIF-2α appeared in the sliced CEMIP models. Its increased promoter role in CEMIP up-regulation later was confirmed with the association with cell migration, helping tumor to get rid of hypoxia microenvironment. Another fragment of CEMIP promoter called H3K4me3 which was regarded as a transcription activator was abundant even in the less aggressive cell line models SW480 after anoxic culture, the results indicated hypoxic stress, indeed, enhanced the up-regulation of CEMIP [42].
The tumor suffered from the hypoxia environment since its fast proliferation and vascular formation. To adjust this situation, the energy supplement inside tumor was mainly produced through glycolysis. CEMIP has been confirmed in its function in depolymerization of HA, and it can boost the gluconeogenesis providing energy. Besides, a hypothesis was raised that CEMIP might affect the tumor incidence via stimulating its metabolism, wherein the preferred glycolysis tended to be relevance with elevated expression of CEMIP [28]. As for the glucose supplement, CEMIP found its ability in glycogen breakdown after binding to glycogen phosphorylase brain form (PYGB) in gastric cancer tissues forming a glucose-1-phosphate which also was the important intermediate for glycolysis and gluconeogenesis. Growth of the cell and the simultaneous decrease in glucose level were investigated in the meantime, the fraction of glycogen enhanced the energy supplement while the CEMIP activated the cell survival signaling pathway, leading to the survival of tumor cell [17, 37, 38].
HA degradation
Recently CEMIP was found to be involved in HA catabolism in many diseases. HA takes part in formation of tissue matrix and contributes to structure modeling, degradation of HA is initiated through binding of HA to the linking domain of CD44 which is present at the cell surface thereby triggering the encoding of a single pass transmembrane protein [43]. CD44 was also one of the upstream gene of CEMIP whose high expression reflected the poor prognosis. It had been reported that its over expression contributed to the aggressive capacity in Ewing sarcoma tissues. The experiments specifically showed a faster adhesion of binding HA to EW7 (Ewing sarcoma cell line) and a more effective attachment of EW7 and immobilized HA was observed to prove its invasion ability [44]. HA depolymerisation phenomenon had been duplicated in vitro by using [3H]HA labeled medium. Since the particular 30 amino acids of N-terminal was predicted to be a signal trigger for HA degradation, CEMIP-V5 which accepted the selected 30AA of human CEMIP (hCEMIP) and its 30AA-knockdown counterpart (CEMIP Δ30aa-v5) were used for distinguishing whether the HA underwent degradation or not. On the other hand, this sequence initiated the maturation process from ER, then forced the protein secreted extracellularly out of perinuclear leading to cytoplasm accumulation [5]. Among the cultured skin fibroblast, the depolymerization of HA did not rely on CD44 and the related enzymes, which was confirmed by the zero effect on HA degradation after incubating siRNA with knockdown counterpart. Followed that the elimination of HA associated with CEMIP which was screened after the histamine and TGF-β1 treatment criteria, also, the HA catabolism due to the quantity of CEMIP according to the rule of michaelis-menten equation [45].
Feasibility of murine CEMIP (mCEMIP) associated with HA catabolism due to its high homology to hCEMIP with 91% and 81% identical to coding sequence and G8 domain respectively was verified via glycosamineglycan-binding assay. Note that the less homology of mCEMIP G8 domain caused extracellular binding which might be the reason of detection of different size of HA fragement [21]. Additionally, transmembrane protein 2 (TMEM2) of zebrafish was found to be an analogue of CEMIP which could potentially contain a better therapeutic effect in HA completed catabolism [46]. When HEK293 cells was transfected with CEMIP, it was endowed with a specific function of degenerating HA at the site of β-endo-N-acetylglucosamine bond. Later the fragment immunoperticitated assay illustrated that CEMIP with its particular HA binding segment validated this depolymerized site [24]. CEMIP had been reported for its highly relationship between extracellular matrix formation and the degradation of HA which was the basic structural materials. Another upstream regulator miRNA-188-5p was rendered with the capacity of directly suppressing CEMIP via binding 3'UTR site. Being measured by qRT-PCR, CEMIP was decreased in accordance with cleavage of glycosaminoglycan resulting in aggravating synovial fibroblasts in rheumatoid arthritis (RSAF) by structure degradation of cartilage and increasing migration potential [47]. To attain more specific details of rodent pattern, two age group were compared using two-months-old (2M) mice and six-months-old (6M) mice in order to investigate the HA catabolism with its related genes under the mechanical overload situation in mandibular joint. Indeed the results indicated the increase of HYAL2 in both and CEMIP existed in the 6M experimental and control mice samples [48]. The increase of HYAL2 coupling with over-expression of CEMIP coincidently presented that the HA degradation by HYAL2 might be affected by CEMIP. CEMIP could be suppressed by TGF-β1 resulting in acceleration of HA synthesis in synovial fluids, affecting intermediate-sized HA synthesis. This result also confirmed that the regulation of HYAL2 due to over-expression of CEMIP, lead to partially broken HA, an intermediate-sized fragment [25, 29, 49]. Another reverse testified evidence illustrated the increased average molecular weight of HA, as well as the total amount of HA in hippocampus of the CEMIP-knock-out mice, however their HYAL1 and HYAL2 remained unaffected [16]. The same observation in heavy chain HA and over-expression of CEMIP had been shown in TGF-β1 co-culture porcine aortic valves which heightened expressed CEMIP both at young stages and calcified tissue due to the lowered HA level [49]. This phenomenon enhanced the migratory association of CEMIP since the low molecular weight HA assisted tumor migration [26, 29]. Since E-cadherin was used as an indicator of the cellular transition of EMT, it was then discovered the inverse correlation between HYALI intensity and the progress of cancer. After immunohistochemical, E-cadherin staining showed higher level in endometrium, the HYAL1 showed low intensity whereas the HYAL2 remained negligible change. In general, HYAL1 and HYAL2 corporately catalyzed the decomposition of HA, wherein HYAL2 started the process by degrading HA to 20kDa molecule followed by HYAL1 taking care of the rest of the process [50]. HYAL2 has been reported that was facilitated by the CEMIP activation. An inconsistent with two related enzymes provided evidence that mediation of CEMIP assisted function of HYAL2 [51]. High concentration of HA was normally observed in tumor, previous researches addressed its balance of production and metabolism influenced invasiveness, angiogenesis and cancer progression [52]. According to the reports of CEMIP-induced HA degradation, it turns to be helpful that over expression of CEMIP stimulated HA degradation [48, 49, 51]. However, an extremely high level of HA could also suppress cancer progression. Besides, no related reports so far have showed that at accurate level of HA has the over-expression of CEMIP has been found in tumor cell. There were evidences which illustrated over-expression of CEMIP activated HYAL2 to initiate HA depolymerization, but the specific concentration of HA would this process start was not stated, as well as at what level of HA the activation of HYAL2 may or may not be stopped.
Future assumable therapeutic sites of cancer aiming at CEMIP
Although over-expression of CEMIP was generally validated in adenocarcinoma, which might be due to its secreted ability, the participation of down-regulated miRNA-126a in OSCC explained this excepted type of tumor [27]. Thereby pervasiveness of detecting the plasmic level of CEMIP might be a novel index for predicting tumor progress and prognosis of patient, if the quantified relationship of over-expressed CEMIP and TNM stage could be verified.
Based on the reported data, it was no doubt over-expressed CEMIP impaired health condition and accelerated malignancy and progression of tumor. Unfortunately, no therapy is existed aiming at CEMIP mediation. Interaction among CEMIP, inflammatory pathway and cytokine signaling pathway highlight that CEMIP is a connection site in various signaling cross-talk. Previous studies have provided precious evidence for further discovery of accurate mechanism at cytokines level. In our research team, our unpublished results indicate over-expression of CEMIP activate multidrug resistance gene 1 (MDR1). Therefore we assume either drug treatment or gene mediation towards limiting expression of CEMIP would aid in better and faster release for cancer patients.
Conclusion
In summary, CEMIP with its broadened and potential function in adjusting tumor microenvironment attracted researchers' attention recently. Besides, many reports have stated that over-expression of CEMIP lead to a horrible prognosis, and dangerous tumor progression. To our knowledge, CEMIP is a hardcore component in bridging various cytokines signaling pathways, thereby transmitting or even amplifying signals in stabilizing tumor malignancy, but still, the more completed branch of cytokine signaling pathway remains unknown. Evidence on why over-expressed CEMIP could be an indicator for cancerization has been stated with detailed gene sequence. Hence the secreted level of CEMIP reflected in its concentration in blood, and through the detection of CEMIP by running blood test, cancer stage could be accurately predicted. Despite the major tendency of over-expressed CEMIP highly associated with tumor malignancy, many researches have not thrown light into how we can suppress CEMIP thereby precisely improving individual chemotherapy. As silencing gene experiments showed a decrease of CEMIP expression, this finding might be an index for knockdown or silencing of CEMIP when necessary. Based on our assumption, the accurate methods of suppressing CEMIP could be helpful for adjuvant therapy in preventing chemo-drug resistance, discovering novel chemo-agent or targeted gene treatment.
Acknowledgements
This study was funded by grants from the National Science Foundation for Young Scholars of China (No. 81502120), the Science Foundation for Young Scholars of Guangxi Medical University (No. GXMUYSF201404) and the Guangxi Medical University Training Program for Distinguished Young Scholars. Authorship contributions: All authors collected data; Li Li and Lin-Hai Yan wrote the paper; LH Yan provided technical support; and all authors discussed and commented on the manuscript.
Competing Interests
None declared.
References
1. Raish M, Khurshid M, Ansari MA. et al. Analysis of molecular cytogenetic alterations in uterine leiomyosarcoma by array-based comparative genomic hybridization. J Cancer Res Clin Oncol. 2012;138:1173-1186
2. Dash DP, Silvestri G, Hughes AE. Fine mapping of the keratoconus with cataract locus on chromosome 15q and candidate gene analysis. Mol Vis. 2006;12:499-505
3. Birkenkamp-Demtroder K, Maghnouj A, Mansilla F. et al. Repression of KIAA1199 attenuates wnt-signalling and decreases the proliferation of colon cancer cells. Br J Cancer. 2011;105:552-561
4. He QY, Liu XH, Li Q. et al. G8: A novel domain associated with polycystic kidney disease and non-syndromic hearing loss. Bioinformatics. 2006;22:2189-2191
5. Yoshida H, Nagaoka A, Nakamura S. et al. N-terminal signal sequence is required for cellular trafficking and hyaluronan-depolymerization of KIAA1199. FEBS Lett. 2014;588:111-116
6. Abe S, Usami S, Nakamura Y. Mutations in the gene encoding KIAA1199 protein, an inner-ear protein expressed in deiters' cells and the fibrocytes, as the cause of nonsyndromic hearing loss. J Hum Genet. 2003;48:564-570
7. Guo J, Cheng H, Zhao S. et al. Gg: A domain involved in phage LTF apparatus and implicated in human MEB and non-syndromic hearing loss diseases. FEBS Lett. 2006;580:581-584
8. Michishita E, Garces G, Barrett JC. et al. Upregulation of the KIAA1199 gene is associated with cellular mortality. Cancer Lett. 2006;239:71-77
9. Usami S, Takumi Y, Suzuki N. et al. The localization of proteins encoded by CRYM, KIAA1199, UBA52, COL9A3, and COL9A1, genes highly expressed in the cochlea. Neuroscience. 2008;154:22-28
10. Tiwari A, Schneider M, Fiorino A. et al. Early insights into the function of KIAA1199, a markedly overexpressed protein in human colorectal tumors. PloS One. 2013;8:e69473
11. Fink SP, Myeroff LL, Kariv R. et al. Induction of KIAA1199/CEMIP is associated with colon cancer phenotype and poor patient survival. Oncotarget. 2015;6:30500-30515
12. Kuscu C, Evensen N, Kim D. et al. Transcriptional and epigenetic regulation of KIAA1199 gene expression in human breast cancer. PloS One. 2012;7:e44661
13. Evensen NA, Kuscu C, Nguyen HL. et al. Unraveling the role of KIAA1199, a novel endoplasmic reticulum protein, in cancer cell migration. J Natl Cancer Inst. 2013;105:1402-1416
14. Yang X, Qiu P, Chen B. et al. KIAA1199 as a potential diagnostic biomarker of rheumatoid arthritis related to angiogenesis. Arthritis Res Ther. 2015;17:140
15. van Heumen BW, Roelofs HM, te Morsche RH. et al. Duodenal mucosal risk markers in patients with familial adenomatous polyposis: Effects of celecoxib/ursodeoxycholic acid co-treatment and comparison with patient controls. Orphanet J Rare Dis. 2013;8:181
16. Yoshino Y, Ishisaka M, Tsuruma K. et al. Distribution and function of hyaluronan binding protein involved in hyaluronan depolymerization (HYBID, KIAA1199) in the mouse central nervous system. Neuroscience. 2017;347:1-10
17. Terashima M, Fujita Y, Togashi Y. et al. KIAA1199 interacts with glycogen phosphorylase kinase beta-subunit (PHKB) to promote glycogen breakdown and cancer cell survival. Oncotarget. 2014;5:7040-7050
18. Zhang Y, Jia S, Jiang WG. KIAA1199 and its biological role in human cancer and cancer cells (review). Oncol Rep. 2014;31:1503-1508
19. Xu J, Liu Y, Wang X. et al. Association between KIAA1199 overexpression and tumor invasion, TNM stage, and poor prognosis in colorectal cancer. Int J Clin Exp Pathol. 2015;8:2909-2918
20. Hosoya M, Fujioka M. Distinct expression pattern of a deafness gene, KIAA1199, in a primate cochlea. Biomed Res Int. 2016;2016:1781894
21. Yoshida H, Nagaoka A, Nakamura S. et al. Murine homologue of the human KIAA1199 is implicated in hyaluronan binding and depolymerization. FEBS Open Bio. 2013;3:352-356
22. Usami S, Wagatsuma M, Fukuoka H. et al. The responsible genes in Japanese deafness patients and clinical application using invader assay. Acta Otolaryngol. 2008;128:446-454
23. Cheng XB, Kohi S, Koga A. et al. Hyaluronan stimulates pancreatic cancer cell motility. Oncotarget. 2016;7:4829-4840
24. Yoshida H, Nagaoka A, Kusaka-Kikushima A. et al. KIAA1199, a deafness gene of unknown function, is a new hyaluronan binding protein involved in hyaluronan depolymerization. Proc Natl Acad Sci U S A. 2013;110:5612-5617
25. Soroosh A, Albeiroti S, West GA. et al. Crohn's disease fibroblasts overproduce the novel protein KIAA1199 to create proinflammatory hyaluronan fragments. Cell Mol Gastroenterol Hepatol. 2016;2:358-368.e354
26. Kohi S, Sato N, Koga A. et al. KIAA1199 is induced by inflammation and enhances malignant phenotype in pancreatic cancer. Oncotarget. 2017;8:17156-17163
27. Zhang D, Zhao L, Shen Q. et al. Down-regulation of KIAA1199/CEMIP by miR-216a suppresses tumor invasion and metastasis in colorectal cancer. Int J Cancer. 2017;140:2298-2309
28. Jami MS, Hou J, Liu M. et al. Functional proteomic analysis reveals the involvement of KIAA1199 in breast cancer growth, motility and invasiveness. BMC cancer. 2014;14:194
29. Koga A, Sato N, Kohi S. et al. KIAA1199/CEMIP/HYBID overexpression predicts poor prognosis in pancreatic ductal adenocarcinoma. Pancreatology. 2017;17:115-22
30. Suh HN, Jun S, Oh AY. et al. Identification of KIAA1199 as a biomarker for pancreatic intraepithelial neoplasia. Sci Rep. 2016;6:38273
31. Koh Y, Kim D, Jung WJ. et al. Revealing genomic profile that underlies tropism of myeloma cells using whole exome sequencing. Int J Genomics. 2015;2015:675379
32. Chanthammachat P, Promwikorn W, Pruegsanusak K. et al. Comparative proteomic analysis of oral squamous cell carcinoma and adjacent non-tumour tissue from thailand. Arch Oral Biol. 2013;58:1677-1685
33. Matsuzaki S, Tanaka F, Mimori K. et al. Clinicopathologic significance of KIAA1199 overexpression in human gastric cancer. Ann Surg Oncol. 2009;16:2042-2051
34. Motaln H, Gruden K, Hren M. et al. Human mesenchymal stem cells exploit the immune response mediating chemokines to impact the phenotype of glioblastoma. Cell Transplant. 2012;21:1529-1545
35. Sabates-Bellver J, Van der Flier LG, de Palo M. et al. Transcriptome profile of human colorectal adenomas. Mol Cancer Res. 2007;5:1263-1275
36. Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006;127:469-480
37. Shostak K, Zhang X, Hubert P. et al. NF-κB-induced KIAA1199 promotes survival through EGFR signalling. Nat Commun. 2014;5:5232
38. Shostak K, Chariot A. EGFR and NF-κB: Partners in cancer. Trends Mol Med. 2015;21:385-393
39. Kikuchi M, Yamashita K, Waraya M. et al. Epigenetic regulation of ZEB1-RAB25/ESRP1 axis plays a critical role in phenylbutyrate treatment-resistant breast cancer. Oncotarget. 2016;7:1741-1753
40. Axelson H, Fredlund E, Ovenberger M. et al. Hypoxia-induced dedifferentiation of tumor cells-a mechanism behind heterogeneity and aggressiveness of solid tumors. Semin Cell Dev Biol. 2005;16:554-563
41. Vaupel P. Hypoxia and aggressive tumor phenotype: Implications for therapy and prognosis. Oncologist. 2008;13(Suppl 3):21-26
42. Evensen NA, Li Y, Kuscu C. et al. Hypoxia promotes colon cancer dissemination through up-regulation of cell migration-inducing protein (CEMIP). Oncotarget. 2015;6:20723-20739
43. Liang J, Jiang D, Noble PW. Hyaluronan as a therapeutic target in human diseases. Adv Drug Deliv Rev. 2016;97:186-203
44. Paulis YW, Huijbers EJ, van der Schaft DW. et al. CD44 enhances tumor aggressiveness by promoting tumor cell plasticity. Oncotarget. 2015;6:19634-19646
45. Nagaoka A, Yoshida H, Nakamura S. et al. Regulation of hyaluronan (HA) metabolism mediated by HYBID (hyaluronan-binding protein involved in HA depolymerization, KIAA1199) and HA synthases in growth factor-stimulated fibroblasts. J Biol Chem. 2015;290:30910-30923
46. Yamamoto H, Tobisawa Y, Inubushi T. et al. A mammalian homolog of the Zebrafish Transmembrane Protein 2 (TMEM2) is the long-sought-after cell surface hyaluronidase. J Biol Chem. 2017;292:7304-7313
47. Ruedel A, Dietrich P, Schubert T. et al. Expression and function of microrna-188-5p in activated rheumatoid arthritis synovial fibroblasts. Int J Clin Exp Pathol. 2015;8:4953-4962
48. Shinohara T, Izawa T, Mino-Oka A. et al. Hyaluronan metabolism in overloaded temporomandibular joint. J Oral Rehabil. 2016;43:921-928
49. Krishnamurthy VK, Stout AJ, Sapp MC. et al. Dysregulation of hyaluronan homeostasis during aortic valve disease. Matrix Biol. 2016 [Epub ahead of print]
50. Schmaus A, Bauer J, Sleeman JP. Sugars in the microenvironment: The sticky problem of HA turnover in tumors. Cancer Metastasis Rev. 2014;33:1059-1079
51. Nykopp TK, Pasonen-Seppanen S, Tammi MI. et al. Decreased hyaluronidase 1 expression is associated with early disease recurrence in human endometrial cancer. Gynecol Oncol. 2015;137:152-159
52. Toole BP. Hyaluronan: From extracellular glue to pericellular cue. Nat Rev Cancer. 2004;4:528-539
Author contact
![]() Corresponding authors: Li Li, Department of Pharmacy, The People's Hospital of Guangxi Zhuang Autonomous Region, No.6 Taoyuan Road, Nanning 530021, Guangxi Zhuang Autonomous Region, People's Republic of China; Tel: +86-771-2186160; E-mail: snailwlilycom (Li L); And Dr. Lin-Hai Yan, Department of Gastrointestinal Surgery, Affiliated Tumor Hospital of Guangxi Medical University, No. 71, Hedi Road, Nanning 530021, Guangxi Zhuang Autonomous Region, People's Republic of China. Tel: +86-771-5344230; E-mail: lhyanedu.cn (Yan LH).
Corresponding authors: Li Li, Department of Pharmacy, The People's Hospital of Guangxi Zhuang Autonomous Region, No.6 Taoyuan Road, Nanning 530021, Guangxi Zhuang Autonomous Region, People's Republic of China; Tel: +86-771-2186160; E-mail: snailwlilycom (Li L); And Dr. Lin-Hai Yan, Department of Gastrointestinal Surgery, Affiliated Tumor Hospital of Guangxi Medical University, No. 71, Hedi Road, Nanning 530021, Guangxi Zhuang Autonomous Region, People's Republic of China. Tel: +86-771-5344230; E-mail: lhyanedu.cn (Yan LH).