Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(11):2069-2078. doi:10.7150/jca.19143 This issue Cite
Research Paper
Opposite Effects of SET7/9 on Apoptosis of Human Acute Myeloid Leukemia Cells and Lung Cancer Cells
Ye Gu1, Yuan Wang1, Xinling Wang1, Lili Gao1, Weiping Yu1 ![]() , Wei-Feng Dong2
, Wei-Feng Dong2 ![]()
1. Department of Pathophysiology, Medical school of Southeast University, Nanjing, Jiangsu, China, 210009.
2. Department of Laboratory Medicine, Cross Cancer Institute, University of Alberta, Edmonton, Alberta, Canada
Received 2017-1-11; Accepted 2017-4-1; Published 2017-7-5
Citation:
Gu Y, Wang Y, Wang X, Gao L, Yu W, Dong WF. Opposite Effects of SET7/9 on Apoptosis of Human Acute Myeloid Leukemia Cells and Lung Cancer Cells. J Cancer 2017; 8(11):2069-2078. doi:10.7150/jca.19143. https://www.jcancer.org/v08p2069.htm
Other stylesAbstract
SET7/9 is a protein lysine methyltransferases (PLMTs or PKMTs) which methylates both histone H3K4 and non-histone proteins including transcriptional factors, tumor suppressors, and membrane-associated receptors. Methylation of these proteins alters protein activity and leads to changes in cellular behavior and a series of biological processes. This study aims to investigate the role of SET7/9 in human acute myeloid leukemia (AML) and non-small-cell lung cancer (NSCLC). We examined the expression of SET7/9 in AML cells and NSCLC cells and detected the methylation status of the SET7/9 promoter region. To evaluate the effect of SET7/9 expression changes on cell apoptosis, cell apoptosis rates were determined after SET7/9 overexpression or down-regulation. Our results showed that SET7/9 induces apoptosis of AML cells and inhibits apoptosis of NSCLC cells, suggesting differential effects of SET7/9 on cellular apoptosis and carcinogenesis depending on different cancer types and genetic contexts. Furthermore, we also demonstrated that SET7/9 suppresses cell apoptosis via modulation of E2F1 under circumstance of p53 deficiency in NSCLC cells.
Keywords: SET7/9, AML cell, NSCLC cell, apoptosis, p53, E2F1.
Introduction
Epigenetic regulation of DNA methylation patterns and chromatin structure plays important role in carcinogenesis [1]. Aberrant DNA or histone methylation at CpG islands in promoter regions of tumor suppressor genes can affect tumor progression and has been frequently observed in various cancer types [2, 3]. Post-translational modifications of histones by methylation have profound influence on a series of biological processes in the context of development and cellular responses [4]. At present, histone H3 lysine 4 (H3K4), H3K9, H3K27, H3K36, H3K79 and H4K20 are among the most extensively studied histone methylation sites [4]. Methylation of different lysine residues at histone tails is critical for the regulation of gene transcription and protein function. Sequence mutations or altered expression of lysine methyl modifiers and methyl-binding proteins have been reported to be correlated with high recurrence or poor survival in prostate cancer, breast cancer, and lymphomas [5-9].
SET7/9, also known as SET7, SET9, and SETD7, is a SET domain-containing methyltransferase belonging to the protein lysine methyltransferase (PLMTs) family [10, 11]. SET7/9 methylates both lysine 4 of histone 3 (H3K4) and lysine (s) of more than 30 non-histone proteins including tumor suppressors, membrane-associated receptors and a number of transcription factors, such as K372 of p53 [12-14]. Involvement of SET7/9 in cancer development has been reported in several studies. For example, the expression of SET7/9 was associated with gastric cancer progression. Overexpression of SET7/9 inhibited oncogenic activities through regulation of Gli-1 expression in breast cancer [1, 15]. However, the role of SET7/9 in cancer development is still controversial, which may be partially due to its promiscuous targeting of different substrates [16].
During the process of carcinogenesis, SET7/9 exerts its functions by regulating p53 stability, which activates expression of its downstream transcription targets involving in induction of cell cycle arrest, cellular apoptosis, and DNA repair [17]. Loss of apoptosis pathways caused by SET7/9 mutation results in failed acetylation of p53 and may promote the development of tumors by contributing to survival of cells in inappropriate physiologic situations [17, 18]. On the contrary, SET7/9 has pro-proliferative and anti-apoptotic functions in the prostate cancer cell line LNCaP by enhancing androgen receptor (AR) activity [19]. Since p53 deficiency exists in approximately 50% of human cancers [20], the effect of SET7/9 on the apoptosis of p53-deficient malignant cells and the underlying mechanisms has largely remains unknown.
In this study, we examined the expression of SET7/9 in human acute myeloid leukemia (AML) cells and p53-deficient non-small cell lung cancer (NSCLC) cells and examined the apoptotic rates of AML cells and NSCLC cells after SET7/9 overexpression or down-regulation. We found possible correlation between decreased SET7/9 expression and methylation of its promoter region. We also present evidence of possible involvement of E2F transcription factor 1 (E2F1) in the apoptotic pathway induced by altered SET7/9 expression.
Materials and methods
Cell lines and patient specimens
Six human acute myeloid leukemia (AML) cell lines (NB4, HEL, HL-60, KG-1, KG-1a, and THP-1) and two human non-small cell lung cancer (NSCLC) cell lines (H1299 and A549) were purchased from the German Collection of Microorganisms and Cell Cultures (DSMZ, Braunschweig, Germany). The NB4, HEL, THP-1, H1299, and A549 cell lines were grown in RPMI 1640 supplemented with 10% FBS (and 1 mm sodium pyruvate: HEL and THP-1). The HL-60, KG1, and KG1a cell lines were cultured in Iscove's modified Dulbecco's medium (BioWhittaker, Verviers, Belgium) supplemented with 20% foetal bovine serum (FBS). AML patient and control samples were obtained from an established bank of marrow aspirate and peripheral blood specimens at the University of Saskatchewan. Sample collection, banking and study were performed according to guidelines approved by the University of Saskatchewan ethics review board and the principles of Declaration of Helsinki (http://www.wma.net/en/30publications/10policies/b3/). Banked patient samples were marrow aspirate (n = 22) or peripheral blood (n = 13) from 35 adult AML patients (ages 22-86) at diagnosis, including the following French-American-British subtypes: M0 (n = 4), M1 (n = 7), M2 (n = 12), M3 (n = 7), M4 (n = 2) and M5 (n = 3) as assessed by light microscopy, cytochemical stains, and multiparametric flow cytometry. The control samples were bone marrow mononuclear cells from healthy donors.
RNA extraction and reverse transcription polymerase chain reaction (RT-PCR) analysis Total RNA were isolated from AML and NSCLC cell lines as well as patient specimens using the Trizol reagent (GibcoBRL, Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocols. RNA pellets were dissolved in nuclease-free sterile water and quantified spectrophotometrically. One microgram aliquots of total RNA were reverse-transcribed by 50 U M-MLV reverse transcriptase (Promega, Medison, WI, USA) using random hexamer oligonucleotides as primers. The following primers were designed by the Primer Premier 5.0 software. SET7/9: 5′-GCAGCAGCTGAAGGCCAAGGAA-3′ (forward), 5′-ATTCCTGGGCTGGA CCGTTC-3′ (reverse) (yielding 479 bp), located between positions 80 and 559 of SET7/9 cDNA sequence (GenBank accession No. AF448510). The housekeeping gene β-actin: 5′-AGCGGGAAATCGTGCGTG-3′ (forward), 5′-CAGGGTACATGGTGGTGCC-3′ (reverse) (yielding 297 bp) (Table 1). Polymerase chain reaction (PCR) was performed in a GeneAmp PCR System 2400 (Applied Biosystems) using the Red Taq polymerase (Sigma Company, St. Louis, MO, USA) for β-actin and the HotStar Taq polymerase (Qiagen Inc., Mississauga, ON, Canada) for SET7/9. Thirty-five amplification cycles were used for detection of both the SET7/9 and β-actin expression. Annealing temperature was set at 60 ℃ for SET7/9 and 61 ℃ for β-actin. The PCR products were analyzed by 2% agarose gel electrophoresis. The expression levels were quantified using a gray-scale value converted from resulting images of RT-PCR.
Prediction of CpG islands
CpG islands in SET7/9 gene sequence were predicted using software on the following two Web sites: http://www.ebi.ac.uk/emboss/cpgblot/#andNewcpgseek [21] and http://www.urogene.org/methprimer/ [22]. A CpG island was defined as a DNA fragment with a length of at least 200 base pairs (bp), a GC content of more than 50 %, and a ratio of more than 0.6 between the observed and expected CpGs.
Table 1
Primer sequences used for RT-PCR and MSP analyses
| Analysis | Amplified Genes/Regions | Primer | Oligonucleotide | Primer Start position | Product Size |
|---|---|---|---|---|---|
| RT-PCR | SET7/9 | Forward (5'-3') | GCAGCAGCTGAAGGCCAAGGAA | 445 | 479 bp |
| Reverse (3'-5') | ATTCCTGGGCTGGACCGTTC | 924 | |||
| P53 | Forward (5'-3') | AGAATGCCAGAGGCTGCTC | 256 | 396 bp | |
| Reverse (3'-5') | CTCGGATAAGATGCTGAGGA | 652 | |||
| β-actin | Forward (5'-3') | AGCGGGAAATCGTGCGTG | 698 | 309 bp | |
| Reverse (3'-5') | CAGGGTACATGGTGGTGCC | 1007 | |||
| MSP | methylated SET7/9 promoter alleles | Forward (5'-3') | GATTCGTTATTTTGCGGAATTC | 903 | 194 bp |
| Reverse (3'-5') | AAAACGTTTCTAACGCTCTAACG | 1096 | |||
| unmethylated SET7/9 promoter alleles | Forward (5'-3') | GTGGATTTGTTATTTTGTGGAATTT | 900 | 197 bp | |
| Reverse (3'-5') | AAAACATTTCTAACACTCTAACACC | 1096 |
For MSP analyses, the primer start position refers to the start position in the whole gene sequence of SET7/9, including the promoter region. For RT-PCR analyses, the primer start position refers to the start position in the mRNA sequences.
DNA extraction and Methylation-specific PCR (MSP) analysis
DNA was isolated from AML cell lines and patient specimens by standard phenol-chloroform extraction using the Trizol method (GibcoBRL, Invitrogen, Carlsbad, CA, USA), according to protocols provided by the manufacturer. The methylation status within the CpG island of the SET7/9 promoter was determined by MSP analysis. Briefly, 10 μg of DNA was denatured in 0.3 M NaOH at 37oC for 15 min and incubated with sodium bisulfite reagent at 55 oC for 6 h. Then DNA was purified using Wizard DNA Clean-Up Columns (Promega, Madison WI, USA), incubated in 0.3 M NaOH at 37 oC for 15 min, precipitated in ammonium acetate and ethanol, washed in 70% ethanol, and re-suspended in distilled water. Polymerase chain reaction (PCR) amplification of the SET7/9 promoter region was performed using the following primer sets designed to discriminate between methylated and unmethylated promoter alleles. Methylated: 5′-GATTCGTTATTTTGCGGAATTC-3′ (forward), 5′-AAAACGTTTCTAACGCTCTAACG-3′ (reverse) (yielding 194 bp). Unmethylated: 5′-GTGGATTTGTTATTTTGTGGAATTT-3′ (forward), 5′-AAAACATTTCTAACACTCTAACACC-3′ (reverse) (yielding 197 bp) (Table 1). Amplifications were performed in 50 μl reactions containing 200 - 400 ng of bisulfite-treated DNA, 1× Qiagen PCR Buffer, 1.5 mM MgCl2, 0.4 mM of each dNTP, 0.2 µM of each primer set, and 0.2 units of HotStar Taq (Qiagen Inc., Mississauga, ON, Canada). The amplification conditions were as follows: initial denature at 95°C for 13 min followed by 35 cycles of 1 min at 95°C, 1 min at the optimized annealing temperature (60°C for methylated SET7/9 promoter alleles and 60°C for un-methylated alleles), 1 min of elongation at 72°C, ending with a 10-min extension at 72°C. Amplification products were resolved by electrophoresis in a 2% agarose gel staining with ethidium bromide. A sample of bisulfite-modified CpGenome universally methylated DNA (Chemicon, Temecula, CA, USA) was used as positive control.
Transfection
Overexpression and shRNA-induced down-regulation of SET7/9 were achieved using the pCMV-Tag5B vectors. Overexpression of p53 was achieved using the pIRES2-Zs1 vector. Transfection of the vectors into AML and NSCLC cells were performed using the Superfect Transfection Reagent (Qiagen, Valenca, CA) following the manufacturer's instructions. Cells transfected with empty vectors and scrambled siRNA vectors were used as negative controls. The SET7/9 and p53 overexpression and control vectors were constructed by Invitrogen Corporation (CA, USA). The overexpression and silencing efficiencies were tested using western blotting analyses.
Western blot analysis
For preparation of protein extraction, approximately 1×107 cells were collected, washed with ice-cold phosphate-buffered saline, re-suspended, and lysed on ice in 1 ml RIPA lysis buffer (sc-24948, Santa Cruz Biotechnology, Santa Cruz, CA, USA). The supernatants were quantified using the Bradford reagent (BioRad, Hercules, CA, USA). Protein lysates (30 μg) were resolved on 12% SDS polyacrylamide gel and electro-blotted onto polyvinylidene fluoride membrane (Immobilon P; Millipore, Billerica, MA, USA). The membranes were blocked with 5% non-fat dry milk in Tris-buffered saline and washed at room temperature and immunoblotted with the following primary antibodies: SET7/9 (1:1000, #2813; Cell Signaling), β-actin (1:5000, #A5441; Sigma Aldrich), p53 (1:1000, #2527; Cell Signaling), p21 (1:1000, #2947; Cell Signaling), E2F1(1:1000, #ab179445; Abcam), and p65 (1:1000, #8242; Cell Signaling). Finally, the appropriate secondary antibodies (HRP-conjugated anti-rabbit IgG, 1:5000, #7074; Cell Signaling; and HRP-conjugated anti-mouse IgG, 1:5000, #7076; Cell Signaling) were applied. Signals were detected by the enhanced chemiluminescence method (Pierce, Rockford, IL, USA).
Cell apoptosis assay
After a 48 h transfection, the transfected cells were seeded in 24-well plates at a density of 1 × 105 cells per well and incubated overnight. Cells were treated with or without adriamycin (Adr) (0.5µmol/L) for 12 h. For Hoechst 33342 staining, cells were stained with 10 µg/mL Hoechst 33342 (Sigma, St. Louis, MO) in PBS for 10 min in the dark at 37 °C and cell apoptosis was examined under a fluorescence microscope (ZEISS, NY, USA). For flow cytometry analysis, cells were collected by trypsinization, washed with ice-cold PBS, and re-suspended in binding buffer. According to the manufacturer's protocol of Annexin V-FITC Apoptosis Detection Kit (BD Biosciences PharMingen, San Diego, CA, USA), cells were stained with annexin-V-FITC for 15 min at room temperature and then were stained with propidium iodure (PI) for 15 min in dark. The cell cycle and cell apoptosis were detected by an EPICS®XL flow cytometer (Beckman Coulter, Miami, FL, USA) using the EXPO 32 ADC Software (Beckman Coulter).
Statistical analysis
All experimental data were analyzed by the SPSS 18.0 software. Student's t-test was used to compare the apoptotic rates of cells between treatment and control groups. A threshold value was set at 0.05 (two tailed).
Figure 1
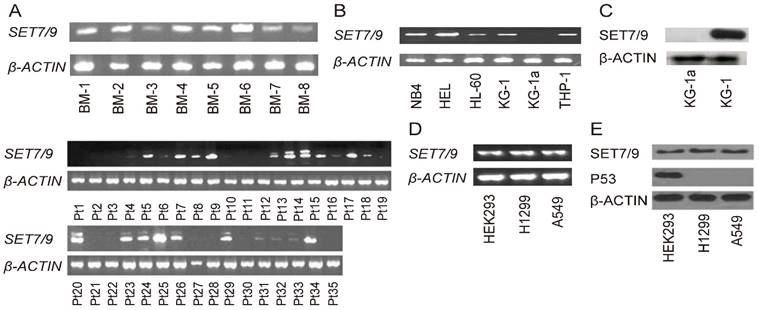
Expression analyses of SET7/9 in AML and NSCLC. (A) Expression of SET7/9 mRNA in eight normal bone marrow samples and 35 AML samples. (B) Expression of SET7/9 mRNA in the six AML cell lines. (C) Expression of SET7/9 protein in the KG-1a and KG-1 AML cells. (D) Expression of SET7/9 mRNA in the normal human embryonic cell line, HEK293, and two NSCLC cell lines, H1299 and A549. (E) Expression of SET7/9 and p53 protein in the normal human embryonic cell line, HEK293, and two NSCLC cell lines, H1299 and A549. Expression of β-actin at both the mRNA and protein levels was used as internal control. BM, normal bone marrow samples; Pt, clinical sample from AML patient.
Results
Expression of SET7/9 in AML and NSCLC
We examined the expression of SET7/9 in 35 bone marrow samples of AML patients and eight bone marrow samples from normal person. Expression of SET7/9 mRNA was detected in all the eight normal samples and 17 AML samples (48.6%), but was weakly detected or undetectable in the other 18 AML samples (51.4%) (Figure 1A). In the six AML cell lines NB4, HEL, HL-60, THP-1, KG-1a, and KG-1, expression of SET7/9 mRNA was detected in the NB4, HEL, HL-60, THP-1, and KG-1 cells, but was not detected in the KG-1a cells (Figure 1B). Consistently, western blot analyses revealed expression of SET7/9 protein in the KG-1 cells but not in the KG-1a cells (Figure 1C). Furthermore, expression of SET7/9 at both the mRNA and protein levels was detected in two NSCLC cell lines, H1299 and A549, as well as in normal human embryonic cells HEK293 (Figure 1D, E). However, expression of the p53 protein, which regulates cell apoptosis, was not detectable in either of the two NSCLC cell lines (Figure 1E).
Promoter methylation associated with SET7/9 expression in AML cells
One CpG island, containing 486 CpG sites, was identified in the 5'-flanking of SET7/9 gene, covering the region -195 to +1005 relative to the transcription start site. Using MSP analysis, we found that SET7/9 promoter was methylated in the KG-1a cell line with undetectable SET7/9 expression, but was un-methylated in AML cell lines HEL, HL-60, KG-1, and THP-1 with evident SET7/9 mRNA and protein expression (Figure 2A). We also randomly selected three SET7/9 positive AML samples (Pt7, Pt9, and Pt23) and one SET7/9 negative AML samples (Pt19) and examined the methylation status of SET7/9 promoter. Our results showed that all the three SET7/9 positive samples were un-methylated, while the SET7/9 negative sample was methylated (Figure 2B).
Figure 2
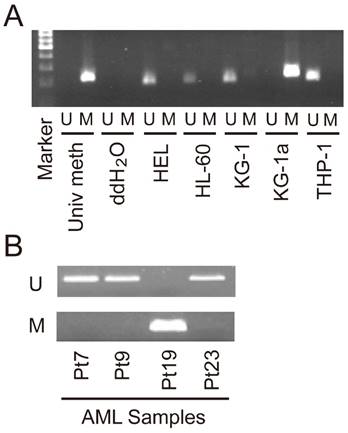
Representational methylation status of SET7/9 promoter in (A) AML cell lines and (B) clinical samples. Pt, clinical sample from AML patient; M, methylated products; U, unmethylated products; Univ meth: positive control of methylation.
Apoptosis-promoting effect of SET7/9 in AML cells
To investigate the apoptotic effect of SET7/9 in AML cells, SET7/9 overexpressed AML cell line, KG-1a+pSET7/9, and SET7/9 silenced AML cell line, KG-1+pshSET7/9 were established using the pCMV-Tag5B vector. AML cells KG-1a and KG-1 transfected with empty vectors or scrambled siRNA vectors were used as negative controls, respectively. Changes in the expression levels of SET7/9 mRNA and protein were confirmed in SET7/9 overexpressed and silenced cells after transfection (48 h) (Figure 3A). Our results showed significantly decreased viabilities of KG-1a+pSET7/9 cells compared with the control cells. However, the viabilities of KG-1+pshSET7/9 cells and the control cells did now show significant difference (Figure 3B). The apoptotic rates of KG-1a+pSET7/9 cells showed significantly increase [(25.81±0.80)%] compared with those of control cells [(18.25±0.83)%], while the apoptotic rates of KG-1+pshSET7/9 cells showed significant decrease [(4.12±0.95)%] compared with those of control cells [(6.01±0.01)%] (Figure 3C). This suggests a role of SET7/9 in promoting cell apoptosis in AML.
Anti-apoptosis effect of SET7/9 in NSCLC cells independent of p53
To investigate the apoptotic effect of SET7/9 on the p53-deficient NSCLC cells, SET7/9 overexpressed cell line, H1299-pSET7/9 and SET7/9 silenced cell line, H1299-pshSET7/9 were established using the pCMV-Tag5B vector. H1299 cells transfected with empty pCMV-Tag5B vectors were used as negative control. After 48 h of transfection, the efficiencies were confirmed by western blotting and the apoptosis rates were detected by AnnexinV-FITC/PI double-staining followed by flow cytometry (Figure 4). The apoptosis rate of H1299-pSET7/9 cells was significantly decreased [(4.36±0.63)%] compared with that of control cells [(12.11±4.41)%], while the apoptosis rate of H1299-pshSET7/9 was significantly increased [(16.64±1.49)%] (Figure 4B).
To further investigate the apoptotic effect of SET7/9 expression under different genetic backgrounds. The H1299-pSET7/9 and H1299-pshSET7/9 cells as well as the control cells were transfected with p53 overexpression vector, p53/pIRES2-Zs1. Cells transfected with empty pIRES2-Zs1 vectors were used as negative control. Using western blotting, constant expression of p53 protein was detected in the H1299-pSET7/9, H1299-pshSET7/9, and control cells after transfection (Figure 4A). Flow cytometry analyses revealed remarkably higher apoptosis rates in the H1299-pSET7/9, H1299-pshSET7/9, and control cells co-transfected with p53 overexpression vector with [(24.84±2.11)%] or without Adr treatment [(21.70±3.80)%] than those transfected with empty pIRES2-Zs1 vectors [(12.11±4.41)%] (Figure 4B). Meanwhile, overexpression of SET7/9 significantly decreased apoptosis rates in all the groups (p < 0.05), including cells transfected with empty pIRES2-Zs1 vectors [(4.36±0.63)% vs (12.11±4.41)%], cells transfected with p53/pIRES2-Zs1 vectors [(7.80±0.46)% vs (21.70±3.80)%], and cells transfected with p53/pIRES2-Zs1 vectors under Adr treatment [(10.38±1.46)% vs (24.84±2.11)%] (Figure 4B). On the contrary, down-regulation of SET7/9 led to increased apoptosis rates [(16.64±1.49)% vs (12.11±4.41)% for pIRES2-Zs1 transfected cells, (24.32±3.00)% vs (21.70±3.80)% for p53/pIRES2-Zs1 transfected cells, (29.42±2.83)% vs (24.84±2.11)% for p53/pIRES2-Zs1 transfected cells under Adr treatment] (Figure 4B). The same transfection experiments were conducted in the p53-deficient NSCLC cell line, A549, and similar results were observed in all the transfected groups (Figure 4C).
Figure 3
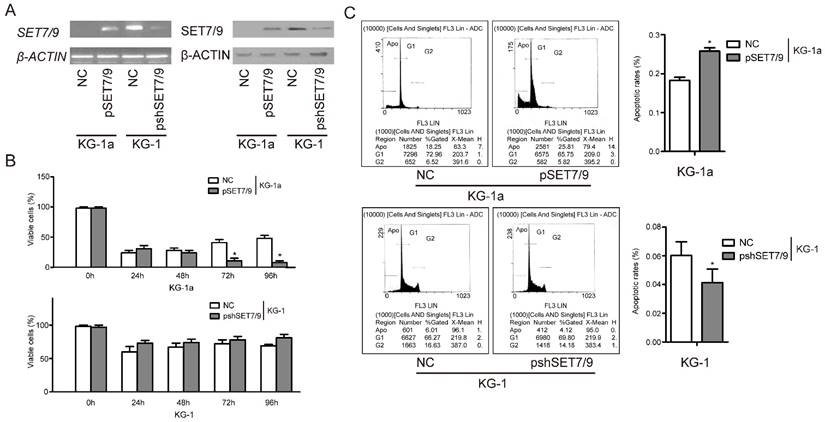
SET7/9 promotes cell apoptosis and affects cell viability in AML cells. (A) Expression SET7/9 at the mRNA level (left) and protein level (right) in SET7/9 overexpressed AML cell line, KG-1a-pSET7/9, SET7/9 silenced AML cell line, KG-1-pshSET7/9, and the control cell lines. (B) Effects of altered SET7/9 expression on cell viabilities of KG-1a-pSET7/9 and KG-1-pshSET7/9 cells. The percentages of viable cells were counted in five randomly selected fields using a light microscope after 0, 24, 48, 72, and 96 h of transfection. (C) Effects of altered SET7/9 expression on the apoptosis rates of KG-1a-pSET7/9 and KG-1-pshSET7/9 cells as determined by flow cytometry analyses after 48 h of transfection. Each error bar represents the mean ± S.D. of three replicates. * p < 0.05.
We also examined cell apoptosis in all the transfected H1299 cells using Hoechst33342 staining. In cells transfected with empty pIRES2-Zs1 vectors, the nuclei were stained less bright. Few apoptotic cells were found in the p53 negative H1299-pshSET7/9 cells. With transfection of the p53/pIRES2-Zs1 vectors, the H1299-pSET7/9, H1299-pshSET7/9, and the control cell lines all exhibited apoptotic cells with condensed or fragmented nuclei. Treatment of Adr combined with p53 expression led to much brighter staining of the nuclei (Figure 4D). On the other hand, overexpression of SET7/9 increased the number of apoptotic cells in both empty-pIRES2-Zs1 transfected cells and p53/pIRES2-Zs1 transfected cells with or without Adr treatment, while silencing of SET7/9 resulted in the opposite effect. The percentages of apoptotic cells were calculated in three randomly selected fields. The minimum apoptosis percentage [(4.63±0.18)%] was found in pIRES2-Zs1 transfected H1299-pSET7/9 cells with negative p53 expression and the maximum apoptosis percentage [(25.09±1.93)%] was found in p53/pIRES2-Zs1 transfected H1299-pshSET7/9 cells with positive p53 expression under Adr treatment. Together with the finding that altered SET7/9 expression did not lead to changes in the expression level of p53 (Figure 4A), our results suggested that SET7/9 inhibited cell apoptosis in NSCLC independent of p53 function and activation of the p53 pathway.
SET7/9 inhibits apoptosis of NSCLC cells through E2F1-mediated pathway
To explore the molecular mechanisms underlying changes in cell apoptosis induced by altered SET7/9 expression, the expression levels of apoptosis-related proteins p21, p65, and E2F1 in the H1299-pSET7/9 and H1299-pshSET7/9 cells lines as well as the control cell lines were examined. The expression levels of p21 and p65 did not change with the overexpression or down-regulation of SET7/9 (Figure 5A). However, the expression level of E2F1 showed opposite changes to that of SET7/9. Decreased E2F1 expression was detected in SET7/9 overexpressed cells, while increased E2F1 expression was detected in SET7/9 silenced cells (Figure 5A). Real-time RT-PCR analysis revealed similar expression changes of E2F1 at the mRNA level (Figure 5B). Our results suggest that SET7/9 may regulate cell apoptosis via p53-independent pathway involving E2F1 function in p53-deficient NSCLC cells.
Discussion
Located in chromosome 4q28, SET7/9 was initially found to be responsible for mono-methylation of histone 3 lysine 4 (H3K4) [10, 11]. However, recent studies have shown that SET7/9 has only weak activity on nucleosomes [10], while the main targets of the enzyme are non-histone proteins. SET7/9-mediated non-histone lysine methylation can lead to a variety of cellular effects including changes in protein stability and gene expression, maintenance of chromosome structure, and regulation of cell cycle and apoptosis [12]. In agreement with this hypothesis, numerous non-histone proteins such as p53, pRB, TAF10, ERα, DNMT1, pRb, E2F1, AR, and the p65/RelA subunit of NFκB are found to be methylated by SET7/9 [19, 20, 23, 24, 25, 26, 27, 28]. Previous studies have shown that SET7/9 expresses differentially in normal and cancer tissues [13, 18, 29]. Diverged expression patterns and functions of SET7/9 in different cancer types have been reported. SET7/9 acts as a tumor suppressor in breast cancer and gastric cancer with down-regulated expression in tumor tissues [1, 15]. However, SET7/9 shows increased expression in prostate cancer and hepatocellular carcinoma and promotes cell proliferation by regulating cell cycle [19, 30]. It is thus suggested that SET7/9 may play distinct roles in tissue- and cancer-specific manners [1].
Figure 4
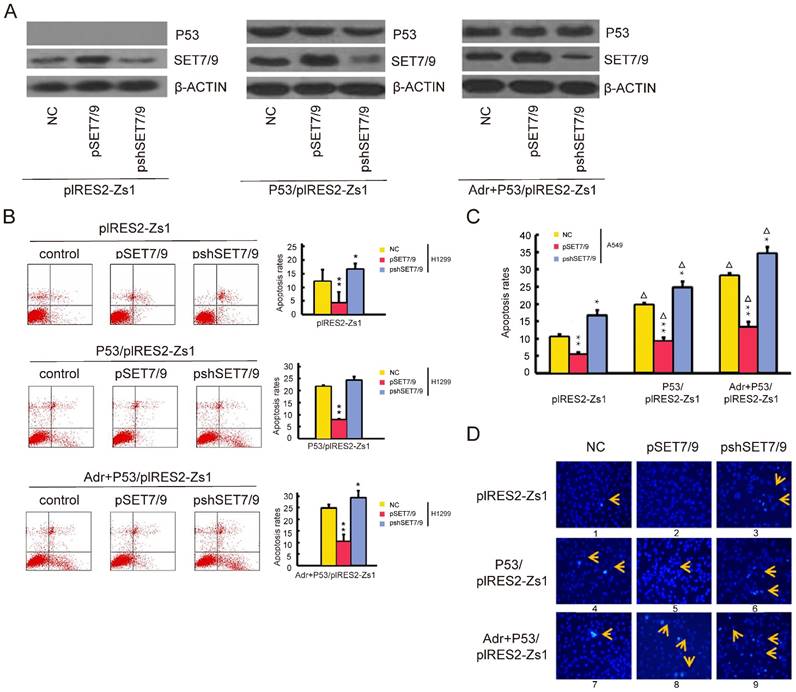
SET7/9 inhibits cell apoptosis in NSCLC cells independent of p53. (A) Expression of SET7/9 and p53 protein in SET7/9 overexpressed cells (H1299-pSET7/9), SET7/9 silenced cells (H1299-pshSET7/9), and the control cells transfected with empty pIRES2-Zs1 vectors (left) or p53/ pIRES2-Zs1 vectors with (right) or without (middle) Adr treatment. (B) Effects of altered SET7/9 and p53 expression and Adr treatment on the apoptosis rates of H1299 cells as determined by flow cytometry analyses. Statistical charts of cell apoptotic rates were shown on the right. (C) Statistical chart showing the effects of altered SET7/9 and p53 expression and Adr treatment on the apoptosis rates of transfected A549 cells, as determined by flow cytometry analyses. (D) Hoechst 33342 staining of transfected H1299 cells and control cells detected by fluorescent microscopy. Highly condensed and bright nuclei represent apoptotic cells. NC, negative control cells transfected with empty pCMV-Tag5B vectors; pSET7/9, cells transfected with SET7/9 overexpression vectors; pshSET7/9, cells transfected with SET7/9 silencing vectors; pIRES2-Zs1, cells transfected with empty pIRES2-Zs1 vectors; p53/pIRES2-Zs1, cells co-transfected with p53 overexpression vectors; Adr+p53/pIRES2-Zs1, cells co-transfected with p53 overexpression vectors and treated with Adr. Student's t-test was used to compare the apoptotic rate of cells between the SET7/9-overexpressed or SET7/9 silenced cells and the control cells (pSET7/9 vs NC, pshSET7/9 vs NC), as well as between the p53-overexpressed cells with or without Adr treatment and the control cells SET7/9 silenced cells and the control cells (p53/pIRES2-Zs1 vs pIRES2-Zs1, Adr+p53/pIRES2-Zs1 vs pIRES2-Zs1). Each error bar represents the mean ± S.D. of three replicates. Compared with the corresponding control groups transfected with empty pCMV-Tag5B vectors, * p < 0.05; ** p < 0.01; Compared with the corresponding control groups transfected with empty pIRES2-Zs1 vectors, ∆ p < 0.05.
Figure 5
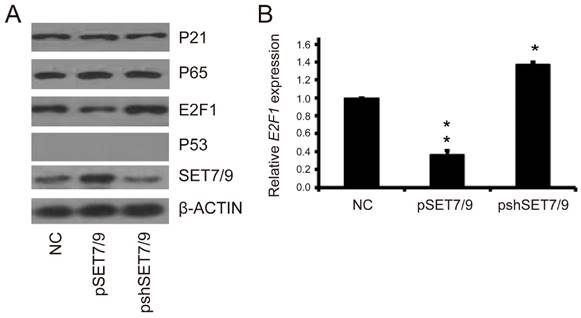
Expression correlations between SET7/9 and apoptosis related proteins. (A) Expression of p21, p65, E2F1, p53, and SET7/9 proteins in SET7/9 overexpressed and silenced cells. (B) Relative expression levels of E2F1 mRNA in SET7/9 overexpressed and silenced cells. The expression levels were quantified using a gray-scale converted from images of semi-quantitative RT-PCR. Each error bar represents the mean ± S.D. of three replicates. NC, negative control of H1299 cells transfected with empty pCMV-Tag5B vectors; pSET7/9, H1299 cells transfected with SET7/9 overexpression vectors; pshSET7/9, H1299 cells transfected with SET7/9 silencing vectors. Student's t-test was used to compare the apoptotic rate of cells between the SET7/9 overexpressed cells and the control cells (pSET7/9 vs NC), as well as between the SET7/9 silenced cells and the control cells (pshSET7/9 vs NC). Each error bar represents the mean ± S.D. of three replicates. * p < 0.05; ** p < 0.01.
In the present study, we examined the expression of SET7/9 in human AML cells and p53-deficient NSCLC cells, as well as clinical samples of AML patients. Evident expression of SET7/9 was detected in all the normal bone marrow samples and 48.6% of the clinical AML samples, but no expression or only weak expression was detected in the other 51.4% clinical AML samples (Figure 1A), indicating down-regulation of SET7/9 expression in AML. Although SET7/9 was proposed to constitute a fine-tuning mechanism for the epigenetic modulation of different gene expression through methylation of promoter regions [31, 32], few studies have focused on the regulatory mechanisms controlling SET7/9 expression itself. Here we found that loss of SET7/9 expression in clinical AML samples and AML cell line KG-1a was related with the methylation of the CpG sites located in SET7/9 promoter (Figure 2). Our results suggested that hypermethylation of SET7/9 promoter may be one of the regulatory mechanisms underlying low expression of SET7/9 in AML. Furthermore, Overexpression of SET7/9 in AML cell line KG-1a led to significantly decreased cell viability and increased apoptosis rate (Figure 3). This coincides with the previously reported tumor suppression function of SET7/9 in breast cancer and gastric cancer [1, 15]. In acute promyelocytic leukemia (APL), SET7/9 was identified as one of the potential regulators of cellular proliferation and apoptosis using GeneChip hybridization. Decreased SET7/9 expression levels in APL patients and involvement of SET7/9 in the modulation of RA response pathway which induces APL remission were observed [33]. Meanwhile, histone H3K4 methylation mediated by SET7/9 also participates in DNA repair through recruitment of ING1 protein, which has been implicated in DNA repair, cellular senescence, apoptosis, and oncogenesis in human diploid fibroblasts, human melanoma, and sporadic breast cancer [34-36]. In this study, we provided the first direct evidence of an apoptosis-promoting effect of SET7/9 on AML cells. The downstream targets of SET7/9 in the apoptotic pathway in AML, however, need to be further investigated.
Loss of functional p53 results in impaired cell cycle control and abrogated cell apoptosis [37]. Specific methylation of p53 at one residue within the carboxyl-terminus regulatory region by SET7/9 methyltransferase has been identified as an important mechanism maintaining the stability of p53 protein [20]. In the human osteosarcoma cell line U2OS, overexpression of wild-type SET7/9 resulted in 'hyper-stabilization' and activation of nuclear p53 and induction of cell-cycle arrest and apoptosis, suggesting that SET7/9 contributes to the control of cell cycle and apoptosis in a p53-dependent manner [20]. However, structural alterations of the p53 gene associated with adverse prognosis are detected in about 45-50 % of tumors of NSCLC patients [38, 39], but the apoptotic effect of SET7/9 on p53-deficient NSCLC has not been investigated previously. In this study, we examined the expression of SET7/9 in two p53-deficient NSCLC cell lines, H1299 and A549. Higher protein abundance of SET7/9 was detected in H1299 and A549 cells compared with that in normal human embryonic cells HEK293 (Figure 1E). Overexpression or silencing of SET7/9 in H1299 and A549 cell lines resulted in significantly decreased or increased apoptosis rates, respectively (Figure 4B, C, D), suggesting that SET7/9 inhibits cell apoptosis in these cell lines. To test whether the apoptosis-suppression effect of SET7/9 relies on specific genetic background of p53 deficiency, the SET7/9 overexpression or silencing cells lines were co-transfected with p53-overexpression plasmid and treated with or without Adr, a chemical that induces DNA damage resulting in the activation of a p53-responsive pathway. Significantly increased cellular apoptosis rates were detected after either ectopic expression of p53 or down-regulation of SET7/9 compared with the corresponding control groups (Figure 4B, C, D). No synergistic or potentiating effect on the apoptosis rates induced by SET7/9 silencing and ectopic p53 expression was observed. Meanwhile, the expression level of p53 protein did not show evident difference between the SET7/9 overexpressed and silenced cell lines (Figure 4A), suggesting independent mechanisms of SET7/9 and p53 in controlling cellular apoptosis in NSCLC cells. Interestingly, SET7/9 was found to methylate the FOXO3 transcription factor with a central role in oxidative stress-induced neuronal cell death at lysine 270 [40]. Lysine methylation of FOXO3 inhibits its DNA-binding activity and reduces oxidative stress-induced and FOXO3-mediated Bim expression, which leads to neuronal apoptosis in neurons [40]. Distinct effects of SET7/9 on the methylation of target proteins, such as p65, have also been reported [41]. Therefore, SET7/9 may have distinct effects on cellular behavior and cancer progression depending on the different recruitment of its numerous substrates as well as the context of other covalent modifications, such as acetylation, phosphorylation and ubiquitination [41, 42].
To investigate the molecular basis underlying the apoptosis-promoting effect of SET7/9 independent of p53 function, we further examined the expression level of p21, p65, and E2F1 in SET7/9 overexpressed and silenced H1299 cells. These proteins are considered pivotal for multiple biological functions including inflammation, immunity, cell proliferation, and apoptosis [40, 41, 43, 44]. SET7/9 regulates the activities of p65 and E2F1 through specific lysine methylation [41, 45] and activates p21 expression through direct binding to its promoter [46]. In this study, the expression levels of p21 and p65 did not show evident changes after overexpression or silencing of SET7/9 in H199 cells, while the expression level of E2F1 decreased in SET7/9 overexpressed cells and increased in SET7/9 silenced cells (Figure 5). Our results suggested that SET7/9 may inhibit cell apoptosis through p21- and p65-independent mechanisms in the p53-deficient NSCLC cells. Instead, E2F1 may be one of the co-regulators that function together with SET7/9 in preventing cell apoptosis. In fact, SET7/9 has long been identified as a critical co-activator of E2F1-dependent transcription in response to DNA damage [47]. Beside direct modification of E2F1 protein, SET7/9 also affects the activity of E2F1 by indirectly modulating histone modifications in the promoters of E2F1-dependent genes [47]. Consistent with our results, SET7/9 was found to regulate DNA damage-induced cell death in a manner opposite to that observed in p53+/+ cells via modulation of E2F1 stabilization under circumstance of p53 deficiency. Methylation of E2F1 at lysine-185 by SET7/9 makes the protein prone to ubiquitination and degradation. This process prevents E2F1 accumulation during DNA damage and negatively influences E2F1-mediated cell death [27]. Furthermore, SET7/9 can differentially affect E2F1 transcription targets, promoting cell proliferation via expression of the CCNE1 gene and represses apoptosis by inhibiting the TP73 gene [47]. These studies together with our findings illustrate that the function of methyltransferases SET7/9 can have opposing biological outcomes depending on the specificity of transcription factor targets.
To conclude, in this study we found down-regulated expression of SET7/9 in human AML cells and clinical AML samples and up-regulated expression of SET7/9 in human NSCLC cells. Possible correlation between loss of SET7/9 expression and the methylation of SET7/9 promoter region was detected. We also reported an apoptosis-promoting effect of SET7/9 in AML cells. On the contrary, SET7/9 inhibited cell apoptosis in NSCLC cells independent of p53. Opposite changes in the expression levels of E2F1 and SET7/9 were observed in SET7/9 overexpressed and silenced cells. Therefore, we propose that SET7/9 may regulate cell apoptosis via modulation of E2F1 under circumstance of p53 deficiency in NSCLC cells. Further investigation of SET7/9 in a wide range of cancer and tissue types may provide better understanding of the molecular mechanisms leading to its oncogenic or tumor-suppressor effects and shed new light on the development of new therapeutic strategies in human cancers.
Acknowledgements
We are grateful to Dr. John DeCoteau for providing laboratory space and reagents, to Mr. Mark Boyd and Mrs Susan Bergen for their technical assistance for part of our work. This work was supported by the Saskatchewan Health Research Foundation of Canada and the National Natural Science Foundation of China (81071654).
Competing Interests
None of the authors have financial or other potential conflicts of interest.
References
1. Akiyama Y, Koda Y, Byeon SJ. et al. Reduced expression of SET7/9, a histone mono-methyltransferase, is associated with gastric cancer progression. Oncotarget. 2016;7:3966
2. Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nature Reviews Genetics. 2002;3:415-428
3. Baylin SB, Jones PA. A decade of exploring the cancer epigenome — biological and translational implications. Nature Reviews Cancer. 2011;11:726-734
4. Greer EL, Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nature reviews. Genetics. 2012;13:343-357
5. Visser HP, Gunster MJ, Kluinnelemans HC. et al. The Polycomb group protein EZH2 is upregulated in proliferating, cultured human mantle cell lymphoma. British Journal of Haematology. 2001;112:950-958
6. Varambally S, Dhanasekaran SM, Zhou M. et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. 2002;419:624-629
7. Kleer CG. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proceedings of the National Academy of Sciences. 2003;100:11606-11611
8. Albert M, Helin K. Histone methyltransferases in cancer. Seminars in Cell & Developmental Biology. 2010;21:209-220
9. Chi P, Allis CD, Wang GG. Covalent histone modifications-miswritten, misinterpreted and mis-erased in human cancers. Nature Reviews Cancer. 2010;10:457-469
10. Wang H, Huang ZQ, Xia L. et al. Methylation of histone H4 at arginine 3 facilitating transcriptional activation by nuclear hormone receptor. Science. 2001;293:853-857
11. Nishioka K, Chuikov S, Sarma K. et al. Set9, a novel histone H3 methyltransferase that facilitates transcription by precluding histone tail modifications required for heterochromatin formation. Genes & Development. 2002;16:479-489
12. Pradhan S, Hang GC, Estève PO. et al. SET7/9 mediated methylation of non-histone proteins in mammalian cells. Epigenetics Official Journal of the Dna Methylation Society. 2009;4:383-387
13. S. Munro. Lysine methylation regulates the pRb tumour suppressor protein. Oncogene. 2010;29:2357-2367
14. Liu X, Wang D, Zhao Y. et al. Methyltransferase Set7/9 regulates p53 activity by interacting with Sirtuin 1 (SIRT1). Proceedings of the National Academy of Sciences. 2011;108:1925-1930
15. Song Y, Zhang J, Tian T. et al. SET7/9 inhibits oncogenic activities through regulation of Gli-1 expression in breast cancer. Tumor Biology. 2016;37:1-12
16. Kassner I, Andersson A, Fey M. et al. SET7/9-dependent methylation of ARTD1 at K508 stimulates poly-ADP-ribose formation after oxidative stress. Open biology. 2013;3:120173
17. Kastan MB, Onyekwere O, Sidransky D. et al. Participation of p53 protein in the cellular response to DNA damage. Cancer Research. 1991;51:6304-6311
18. Kurash JK, Lei H, Shen Q. et al. Methylation of p53 by Set7/9 mediates p53 acetylation and activity in vivo. Molecular Cell. 2008;29:392-400
19. Gaughan L, Stockley J, Wang N. et al. Regulation of the androgen receptor by SET9-mediated methylation. Nucleic Acids Research. 2011;39:1266-1279
20. Chuikov S, Kurash JK, Wilson JR. et al. Regulation of p53 activity through lysine methylation. Nature. 2004;432:353-360
21. Rice P, Longden I, Bleasby A. EMBOSS: the European molecular biology open software suite. Trends in genetics. 2000;16:276-277
22. Li LC, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics. 2002;18:1427-1431
23. Carr SM, Shonagh M, Benedikt K. et al. Interplay between lysine methylation and Cdk phosphorylation in growth control by the retinoblastoma protein. Embo Journal. 2011;30:317-327
24. Kouskouti A, Scheer E, Staub A. et al. Gene-Specific Modulation of TAF10 Function by SET9-Mediated Methylation. Molecular Cell. 2004;14:175-182
25. Subramanian K, Jia D, Kapoorvazirani P. et al. Regulation of Estrogen Receptor Alpha by the SET7 lysine methyltransferase. Molecular Cell. 2008;30:336-347
26. Esteve PO, Chin HG, Benner J. et al. Regulation of DNMT1 stability through SET7-mediated lysine methylation in mammalian cells. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:5076-81
27. Kontaki H, Talianidis I. Lysine methylation regulates E2F1-induced cell death. Molecular Cell. 2010;39:152-160
28. Lu T, Jackson MW, Wang B. et al. Regulation of NF κB by NSD1/FBXL11-dependent reversible lysine methylation of p65. Cytokine. 2009;48:19-20
29. Campaner S, Spreafico F, Burgold T. et al. The methyltransferase Set7/9 (Setd7) is dispensable for the p53-mediated DNA damage response in vivo. Molecular Cell. 2011;43:681-688
30. Chen Y, Yang S, Hu J. et al. Increased Expression of SETD7 Promotes Cell Proliferation by Regulating Cell Cycle and Indicates Poor Prognosis in Hepatocellular Carcinoma. PloS one. 2016;11:e0154939
31. He S, Owen DR, Jelinsky SA. et al. Lysine Methyltransferase SETD7 (SET7/9) Regulates ROS Signaling through mitochondria and NFE2L2/ARE pathway. Scientific Reports. 2015;5:14368
32. Montenegro MF, Sánchez-Del-Campo L, González-Guerrero R. et al. Tumor suppressor SET9 guides the epigenetic plasticity of breast cancer cells and serves as an early-stage biomarker for predicting metastasis. Oncogene. 2016;35:6143-6152
33. Meani N, Minardi S, Licciulli S. et al. Molecular signature of retinoic acid treatment in acute promyelocytic leukemia. Oncogene. 2005;24:3358-3368
34. Garkavtsev I, Riabowol K. Extension of the replicative life span of human diploid fibroblasts by inhibition of the p33ING1 candidate tumor suppressor. Molecular & Cellular Biology. 1997;17:2014-2019
35. Toyama T, Iwase H, Watson P. et al. Suppression of ING1 expression in sporadic breast cancer. Oncogene. 1999;18:5187-5193
36. Campos EI, Martinka M, Mitchell DL. et al. Mutations of the ING1 tumor suppressor gene detected in human melanoma abrogate nucleotide excision repair. International Journal of Oncology. 2004;25:73-80
37. Levine AJ. p53, the Cellular Gatekeeper for Growth and Division. Cell. 1997;88:323-331
38. Takahashi T, Minna JD. p53: a frequent target for genetic abnormalities in lung cancer. Science. 1989;246:491-494
39. Nishio M, Koshikawa T, Kuroishi T. et al. Prognostic significance of abnormal p53 accumulation in primary, resected non-small-cell lung cancers. Journal of Clinical Oncology Official Journal of the American Society of Clinical Oncology. 1996;14:497-502
40. Xie Q, Hao Y, Tao L. et al. Lysine methylation of FOXO3 regulates oxidative stress-induced neuronal cell death. EMBO reports. 2012;13:371-377
41. Ea CK, Baltimore D. Regulation of NF-kappaB activity through lysine monomethylation of p65. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:18972-18977
42. Rizzo PAD, Trievel RC. Substrate and product specificities of SET domain methyltransferases. Epigenetics Official Journal of the Dna Methylation Society. 2011;6:1059-1067
43. Gartel AL, Tyner AL. The role of the cyclin-dependent kinase inhibitor p21 in apoptosis. Molecular Cancer Therapeutics. 2002;1:639-649
44. Stark LA, Dunlop MG. Nucleolar sequestration of RelA (p65) regulates NF-κB-driven transcription and apoptosis. Molecular and cellular biology. 2005;25:5985-6004
45. Lezina L, Aksenova V, Fedorova O. et al. KMT Set7/9 affects genotoxic stress response via the Mdm2 axis. Oncotarget. 2015;6:25843
46. Li X, Li C, Li X. et al. Involvement of Histone Lysine Methylation in p21 Gene Expression in Rat Kidney In Vivo and Rat Mesangial Cells In Vitro under Diabetic Conditions. Journal of Diabetes Research. 2016;10:1-12
47. Lezina L, Aksenova V, Ivanova T. et al. KMTase Set7/9 is a critical regulator of E2F1 activity upon genotoxic stress. Cell Death Differ. 2014;21:1889-1899
Author contact
![]() Corresponding author: Weiping Yu, Department of Pathophysiology, Medical school of Southeast University, 87 Ding Jia Qiao Road, Nanjing 210009, Jiangsu, China. Tel: +86-25-83272508; Fax: +86-25-84454318; Email: wpylgcom. Wei-Feng Dong, Department of Laboratory Medicine, Cross Cancer Institute, University of Alberta, 11560 University Avenue, Edmonton, AB T6G 1Z2, Canada. Tel: +1 780 432-8780; Fax: +1 780 432-8780; Email: wei-feng.dongab.ca
Corresponding author: Weiping Yu, Department of Pathophysiology, Medical school of Southeast University, 87 Ding Jia Qiao Road, Nanjing 210009, Jiangsu, China. Tel: +86-25-83272508; Fax: +86-25-84454318; Email: wpylgcom. Wei-Feng Dong, Department of Laboratory Medicine, Cross Cancer Institute, University of Alberta, 11560 University Avenue, Edmonton, AB T6G 1Z2, Canada. Tel: +1 780 432-8780; Fax: +1 780 432-8780; Email: wei-feng.dongab.ca
Citation styles
APA
Gu, Y., Wang, Y., Wang, X., Gao, L., Yu, W., Dong, W.F. (2017). Opposite Effects of SET7/9 on Apoptosis of Human Acute Myeloid Leukemia Cells and Lung Cancer Cells. Journal of Cancer, 8(11), 2069-2078. https://doi.org/10.7150/jca.19143.
ACS
Gu, Y.; Wang, Y.; Wang, X.; Gao, L.; Yu, W.; Dong, W.F. Opposite Effects of SET7/9 on Apoptosis of Human Acute Myeloid Leukemia Cells and Lung Cancer Cells. J. Cancer 2017, 8 (11), 2069-2078. DOI: 10.7150/jca.19143.
NLM
Gu Y, Wang Y, Wang X, Gao L, Yu W, Dong WF. Opposite Effects of SET7/9 on Apoptosis of Human Acute Myeloid Leukemia Cells and Lung Cancer Cells. J Cancer 2017; 8(11):2069-2078. doi:10.7150/jca.19143. https://www.jcancer.org/v08p2069.htm
CSE
Gu Y, Wang Y, Wang X, Gao L, Yu W, Dong WF. 2017. Opposite Effects of SET7/9 on Apoptosis of Human Acute Myeloid Leukemia Cells and Lung Cancer Cells. J Cancer. 8(11):2069-2078.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.