Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(10):1744-1749. doi:10.7150/jca.17972 This issue Cite
Short Research Communication
Metformin Inhibits Gemcitabine Induced Apoptosis in Pancreatic Cancer Cell Lines
Dietmar Zechner1* ![]() , Ann-Christin Albert1*, Florian Bürtin1, Brigitte Vollmar1
, Ann-Christin Albert1*, Florian Bürtin1, Brigitte Vollmar1
1. Institute for Experimental Surgery, Rostock University Medical Center, Schillingallee 69a, 18057 Rostock, Germany
*These authors contributed equally to this work
Received 2016-10-18; Accepted 2017-2-24; Published 2017-7-1
Citation:
Zechner D, Albert AC, Bürtin F, Vollmar B. Metformin Inhibits Gemcitabine Induced Apoptosis in Pancreatic Cancer Cell Lines. J Cancer 2017; 8(10):1744-1749. doi:10.7150/jca.17972. https://www.jcancer.org/v08p1744.htm
Other stylesAbstract
Many preclinical and clinical studies are currently evaluating metformin in combination with classical therapeutic agents as anti-cancer therapy. In this study we used three distinct pancreatic cancer cell lines and evaluated cell death by trypan blue assay and Western Blots using antibodies directed against cleaved caspase 3 and PARP. Surprisingly, we observed that 20mM metformin did not enhance, but rather inhibited gemcitabine induced cell death in murine 7265PDA, 6606PDA and 6606l cells. Microenvironmental aspects such as oxygen supply or the pH value did not influence the inhibition of cancer cell apoptosis by metformin. Glucose concentration in the medium, however, had a major effect on the impact of metformin. Medium with 0.5g/L glucose strongly increased metformin induced apoptosis and also prevented the inhibitory effect of metformin on gemcitabine induced cell apoptosis, when compared with medium containing 4.5g/L glucose. We conclude that the combination of metformin with gemcitabine has inappropriate effects for a successful treatment of pancreatic cancer. Thus, it might be more promising to use metformin in combination with other drugs that reduce the uptake or the metabolism of glucose.
Keywords: pancreatic adenocarcinoma, chemotherapy, apoptosis, microenvironment.
Introduction
In spite of the development of novel chemotherapies, the 5-year relative survival rate of pancreatic cancer patients is still only 8 % [1]. Multiple chemotherapies are, therefore, currently evaluated [2, 3]. Lately, the benefit of metformin, a traditional diabetes type II medication, has been assessed in clinical studies for treating pancreatic cancer. Some retrospective studies suggest that metformin improves survival in patients with pancreatic ductal adenocarcinoma [4, 5]. Prospective studies, however, demonstrate that metformin at a low dose typical for glycemic control is unlikely to benefit patients with pancreatic cancer [6, 7, 8]. However, a subgroup of patients with high metformin concentration (>1 mg/L) in the blood seemed to have an improved survival [7]. Currently five clinical studies are still enrolling patients for assessing the benefit of metformin in treating pancreatic cancer [9].
In order to evaluate the feasibility of using metformin in combination with gemcitabine as anti-cancer therapy, we assessed the effect of metformin on gemcitabine induced cell death in three distinct pancreatic cancer cell lines. Surprisingly, we observed that metformin strongly inhibited gemcitabine induced cell death in all cell lines. This suggests, that metformin might not be an ideal agent in combination with gemcitabine for the treatment of pancreatic cancer.
Materials and Methods
Cell culture and evaluation of cell death by trypan blue assay
The murine cell lines, 6606PDA, 7265PDA and 6606l were a gift of Prof. Tuveson at the University of Cambridge, UK. The cells were always seeded on 24 well plates in DMEM high glucose medium (Biochrom GmbH, Berlin, Germany) supplemented with 10 % fetal calf serum.
For obtaining all data presented in figure 1, the cells were incubated on the following day for another 48 hours with control medium or medium containing therapeutic agents. For evaluating the influence of hypoxia or normoxia on cell lines, the cells (covering about 15% of the well surface) were incubated with DMEM high glucose medium or DMEM high glucose medium containing therapeutic agents for 72 hours either in an Innova CO-48 incubator (New Brunswick Scientific Co, Edison Edison, USA) under 1 % oxygen supply or in an incubator with normoxic conditions. For evaluating the influence of pH-value on cell lines, the cells were incubated for another 48 hours with DMEM high glucose medium adjusted to pH 6.8 or pH 7.8 with 1M HCl or 1M NaOH as previously described [10]. The medium with pH 6.8 was also supplemented with 10 mM lactate (Sigma-Aldrich St Louis, MO, USA) in order to mimic lactate production in the carcinoma. For evaluating the influence of the glucose concentration on cell lines, the cells were incubated for another 24 hours with either DMEM high glucose medium (Biochrom GmbH) or low glucose medium (Dulbeccos MEM from Biochrom after adding 0,5g/L Glucose).
In all experiments the cell culture medium was supplemented with the following therapeutic agents: Gemcitabine (Sigma-Aldrich, G6423, 100 nM), metformin (Sigma-Aldrich, D150959, 20 mM) or gemcitabine (100 nM) plus metformin (20 mM). When treating the cells with gemcitabine plus metformin, the cells were pretreated with 20mM metformin for one hour. The percentage of dead cells was determined after mixing trypsinized cells 1:1 with a trypan blue staining solution using a Neubauer chamber (Life Technologies, CA, USA).
Western Blots
The cells were seeded on 6 well plates in DMEM high glucose medium (Biochrom GmbH) supplemented with 10% fetal calf serum. On the following day the cells covered about 30% of the well surface and were incubated for another 24 or 48 hours with control medium or medium containing therapeutic agents (as described above). After separating the cell lysates on SDS polyacryl gels the proteins were transferred to a polyvinyldifluoride membrane (Immobilon-P; Millipore, Eschborn, Germany) as described previously [10]. The membranes were blocked with 2.5 % (wt/vol.) BSA and incubated overnight at 4 °C with rabbit-anti-cleaved caspase 3 (Cell Signaling, Danvers, MA, USA, code 9661, dilution: 1000x) or rb-anti PARP (Cell Signaling, Danvers, MA, USA, code 9542, dilution: 1000x) followed by incubation with a secondary peroxidase linked anti-rabbit (Cell Signaling, code 7074, dilution: 2000x for cleaved caspase 3 and 20000x for PARP Western Blot). For analysis of β-actin production, membranes were stripped, blocked by 2.5 % (wt/vol.) BSA and incubated with mouse anti-β-actin antibody (Sigma-Aldrich, code A5441, dilution: 20000x) followed by peroxidase-linked anti-mouse antibody (Sigma-Aldrich, USA; code A9044, dilution: 60000x).
Data presentation and statistics
Data presentations and statistics using a Mann-Whitney rank-sum test followed by the Bonferroni correction were described previously [10]. Differences with P ≤ 0.05 or P ≤ 0.08, divided by the number of meaningful comparisons, were considered to be significant or to indicate a tendency.
Results
Metformin inhibits gemcitabine induced apoptosis
In order to evaluate the effect of metformin on gemcitabine induced cell death, we treated the murine cancer cell lines 7265PDA, 6606PDA and 6606l cells with gemcitabine, metformin or both agents. Gemcitabine significantly increased the percentage of trypan blue positive 7265PDA cells when compared to Sham treated control cells (Fig. 1A). Combinatorial treatment of metformin plus gemcitabine did not increase, but rather decreased the percentage of dead cells when compared to gemcitabine treated 7265PDA cells (Fig. 1A). Very similar results were observed with 6606PDA and 6606l cells (Fig. 1B and 1C).
Since apoptotic cell death is often caused by proteolytic activation of caspase 3 we evaluated cleaved caspase 3 by Western Blots. Indeed we observed proteolytic activation of this proenzyme both 24 as well as 48 hours after gemcitabine administration in 7265PDA (Fig. 2A) 6606PDA (Fig. 2B) and 6606l (Fig. 2C) cells. Proteolytic activation of caspase 3 was substantially reduced after treating the cells with metformin plus gemcitabine at both time points (Fig. 2A-C).
During apoptosis activated caspase 3 cleaves PARP, a protein involved in DNA repair. Indeed we observed the proteolytic inactivation of PARP within 24 to 48 hours after gemcitabine administration in 7265PDA (Fig. 2D) 6606PDA (Fig. 2E) and 6606l (Fig. 2F) cells. Proteolytic inactivation of PARP was reduced after treating the cells with metformin plus gemcitabine (Fig. 2D-F). Thus, metformin inhibits gemcitabine induced apoptosis in these cancer cell lines.
Figure 1
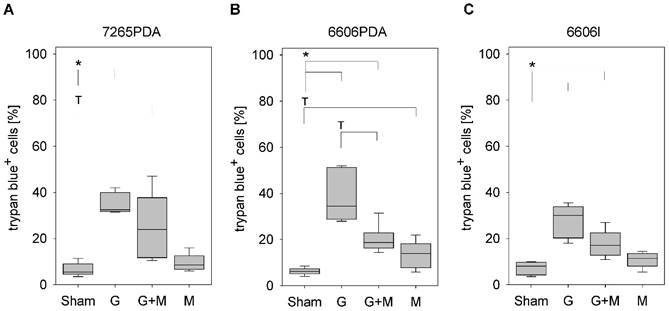
Analysis of cell death. Quantification of trypan blue positive 7265PDA (A), 6606PDA (B), or 6606l (C) cells after incubation with control medium (Sham) or medium containing gemcitabine (G), metformin (M) or gemcitabine plus metformin (G+M). Significant differences: *p≤0.008, tendentious difference: Tp≤0.016.
Figure 2
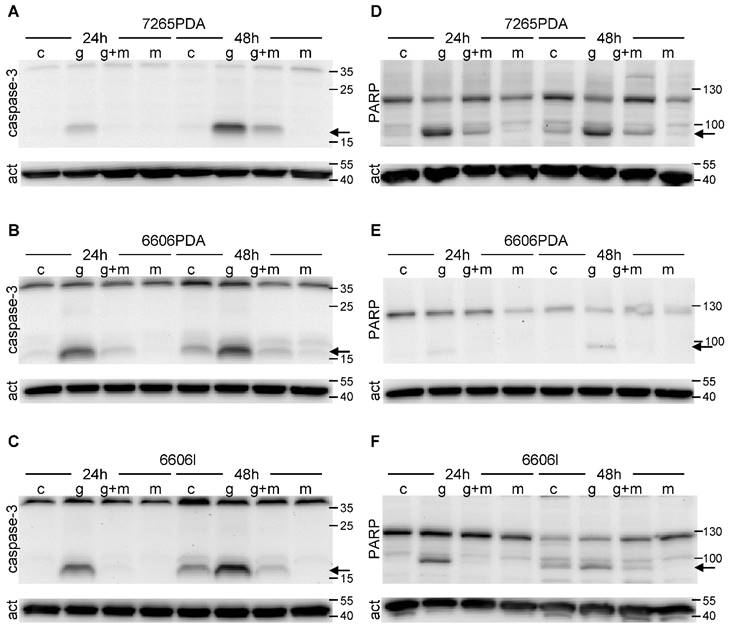
Proteolytic cleavage of caspase 3 and PARP. The 17kDa peptide of proteolytically cleaved caspase 3 (arrow) is visualized by Western Blot in 7265PDA (A), 6606PDA (B), 6606l (C) cell lysate after treating the cells with control medium (Sham) or medium containing gemcitabine (G), metformin (M) or gemcitabine plus metformin (G+M). The 89kDa product of proteolytically cleaved PARP (arrow) is visualized by Western Blot in 7265PDA (D), 6606PDA (E), 6606l (F) cell lysate.
Microenvironmental aspects of metformin induced inhibition of cell death
Since microenvironmental aspects within a tumor can have substantial influence on the efficacy of therapeutical agents, we explored if oxygen and glucose supply or the pH value influences the inhibition of cancer cell apoptosis by metformin.
Under normoxic as well as hypoxic conditions gemcitabine significantly increased the percentage of trypan blue positive 6606PDA cells when compared to Sham treated control cells (Fig. 3A and 3B). Under both conditions combinatorial treatment of metformin plus gemcitabine significantly decreased the percentage of dead cells when compared to gemcitabine treated cells (Fig. 3A and 3B). Consistent with this result no significant difference in the percentage of dead cells was observed under normoxic or hypoxic conditions when treating the cells with gemcitabine (Fig. 3C) or metformin (Fig. 3D).
Gemcitabine also significantly increased cancer cell death, in medium with a pH of 7.8 as well as in medium with a pH of 6.8 (Fig. 3E and 3F). Independent of the pH value, metformin plus gemcitabine significantly decreased the percentage of dead cells when compared to gemcitabine treated cells (Fig. 3E and 3F). Interestingly, significant differences were observed with the monotherapies. A lower pH value decreased the percentage of trypan blue positive cells after gemcitabine treatment (Fig. 3G) but significantly increased cell death after metformin treatment (Fig. 3H).
Gemcitabine also increased cancer cell death, in medium with high glucose concentration as well as in medium with low glucose concentration (Fig. 3I and 3J). Metformin plus gemcitabine significantly decreased the percentage of dead cells when compared to gemcitabine treated cells in high glucose medium (Fig. 3I). However, metformin plus gemcitabine did not decrease but significantly increased the percentage of dead cells in low glucose medium (Fig. 3J). However, the combinatorial therapy did not cause more cell death than metformin as monotherapy (Fig 3J). Consistent with these data are observations made after the administration of monotherapies: low glucose concentration reduced gemcitabine induced cell death, but caused a major increase in the percentage of trypan blue positive cells after metformin treatment (Fig. 3K and 3L).
Thus, metformin inhibits gemcitabine induced apoptosis independent of oxygen supply and the pH value. However, inhibition of gemcitabine induced apoptosis by metformin is not observed when the glucose supply is low.
Discussion
The presented data demonstrate that metformin inhibits gemcitabine induced cell death in three distinct cancer cell lines. This conclusion is supported by data derived by trypan blue staining and by Western Blots analyzing the cleavage of caspase 3 and PARP. However, it is one limitation of this study that we have not used additional methods such as annexin V plus propidium iodide staining.
This study casts doubt on the concept that the combination of metformin with gemcitabine might be an appropriate therapy for pancreatic cancer. Surprisingly, few other publications support this point of view. Many publications describe that metformin plus gemcitabine treatment increases cell death in various pancreatic cancer cell lines when compared to the monotherapies [11, 12, 13] or describe other benefits of combining metformin with gemcitabine [10, 14-17]. Only very few publications reported that metformin inhibits for example cisplatin induced apoptosis of cancer cell lines [18] or apoptosis in primary cells [19]. We assume that a publication bias favors publications with a positive message and prevents timely publication of undesirable results.
In our previous study we demonstrated that metformin as monotherapy inhibits proliferation of pancreatic cancer cells in vitro as well as in vivo [10]. In vitro, almost no synergistic effect of metformin and gemcitabine was observed on the inhibition of proliferation [10]. In vivo a synergistic reduction in tumor weight was observed [10]. However, this was caused by inhibition of proliferation in distinct areas within the carcinoma: Metformin inhibited proliferation close to the desmoplastic reaction, whereas gemcitabine preferentially inhibited proliferation distant to the desmoplastic reaction [10]. Our previous study, therefore, suggests that combinatorial therapy of metformin plus gemcitabine can have desirable anti-cancerous effects [10]. The current study describes for the first time that the combination of metformin with gemcitabine also has effects undesirable for pancreatic cancer therapy such as the inhibition of gemcitabine induced cell death.
Figure 3
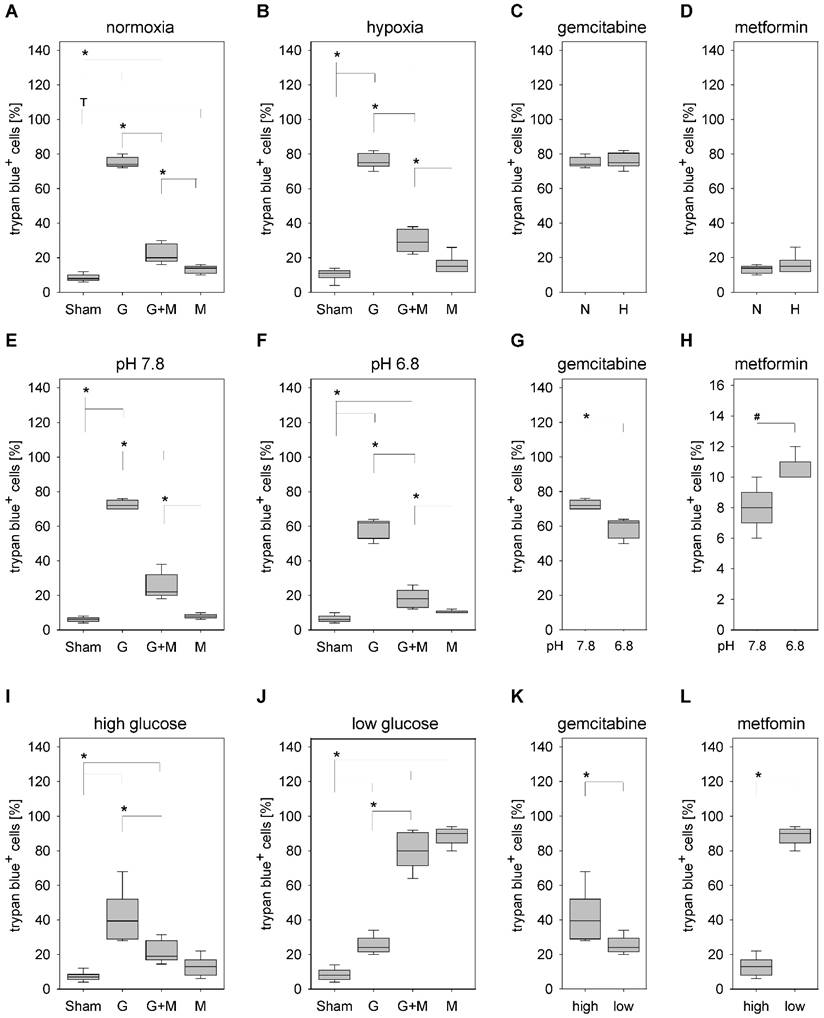
Influence of oxygen supply, pH value and glucose concentration on the death of 6606PDA cells. Quantification of trypan blue-positive cells grown under normoxic (A), or hypoxic (B) conditions after incubation with control medium (Sham) or medium containing gemcitabine (G), metformin (M) or gemcitabine plus metformin (G+M). Quantification of dead cells after treatment with gemcitabine (C) or metformin (D) when grown under normoxia (N) or hypoxia (H). Quantification of trypan blue-positive cells after growth in medium with pH7.8 (E) or pH6.8 (F) containing above mentioned supplements. Comparing death after treating the cells with gemcitabine (G) or metformin (H) between cells grown at pH 7.8 or 6.8. Quantification of death after growing the cells in medium with high glucose (I) or low glucose (J) containing above mentioned supplements. Comparing death after treating the cells with gemcitabine (G) or metformin (H) between cells grown in high or low glucose medium. Significant differences: *p≤0.009, #p= 0.032; tendentious difference: Tp=0.016.
In this study we also explored, if microenvironmental aspects within a tumor, such as oxygen supply, glucose concentration, or distinct pH values, can influence the effect of metformin on cancer cell death. The evaluated concentrations of oxygen (normoxia versus 1% oxygen) were based on the very heterogeneous distribution of oxygen levels, which were observed within tumors [20]. The evaluated concentrations of glucose (0,5g/L versus 4,5g/L) are below or above normal glucose concentrations in the blood and mimic a bad supply of nutrients due to hypovascularization of pancreatic cancer or diabetes, which is often associated with pancreatic cancer [21]. The evaluated pH values (pH 7.8 versus pH 6.8) were based on the heterogeneous distribution of pH values within PDAs analyzed in an orthotopic pancreatic cancer model [10]. Our data (Fig. 3A to E) demonstrate that metformin inhibits gemcitabine induced cell death independent of oxygen supply, or the pH value. Since identical results were observed using distinct experimental settings, the conclusion that metformin does not aggravate, but rather inhibits gemcitabine induced cell death, is very robust.
The observation, that metformin primarily induces cell death of cancer cells when low glucose concentration is present in the medium, might give us a hint, that metformin should be especially useful in combination with other agents that inhibit glucose uptake or glucose metabolism in cancer cells [22]. Thus, combining metformin with drugs inhibiting glucose uptake, lactate dehydrogenase or lactate export might be more successful than combining metformin with classical chemotherapeutic agents such as gemcitabine.
Acknowledgements
We thank Berit Blendow, Dorothea Frenz, Eva Lorbeer-Rehfeldt and Maren Nerowski (Institute for Experimental Surgery, University of Rostock) for excellent technical assistance. We also thank the B. BRAUN-STIFTUNG (project: BBST-D-15-00003) for financing parts of this study.
Competing Interests
The authors declare that there are no competing interests.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7-30
2. Goel G, Sun W. Novel approaches in the management of pancreatic ductal adenocarcinoma: potential promises for the future. J Hematol Oncol. 2015;8:44
3. Cohen R, Neuzillet C, Tijeras-Raballand A. et al. Targeting cancer cell metabolism in pancreatic adenocarcinoma. Oncotarget. 2015;6:16832-47
4. Amin S, Mhango G, Lin J. et al. Metformin Improves Survival in Patients with Pancreatic Ductal Adenocarcinoma and Pre-Existing Diabetes: A Propensity Score Analysis. Am J Gastroenterol. 2016;111:1350-7
5. Cerullo M, Gani F, Chen SY. et al. Metformin Use Is Associated with Improved Survival in Patients Undergoing Resection for Pancreatic Cancer. J Gastrointest Surg. 2016;20:1572-80
6. Reni M, Dugnani E, Cereda S. et al. (Ir)relevance of metformin treatment in patients with metastatic pancreatic cancer: an open-label, randomized phase 2 trial. Clin Cancer Res. 2016;22:1076-85
7. Kordes S, Pollak MN, Zwinderman AH. et al. Metformin in patients with advanced pancreatic cancer: a double-blind, randomised, placebo-controlled phase 2 trial. Lancet Oncol. 2015;16:839-47
8. Braghiroli MI, de Celis Ferrari AC. et al. Phase II trial of metformin and paclitaxel for patients with gemcitabine-refractory advanced adenocarcinoma of the pancreas. Ecancermedicalscience. 2015;9:563
9. U.S. National Library of Medicine at the U.S. National Institutes of Health: Bethesda, Maryland, USA. Information retrieved on 10th October 2016. https://clinicaltrials.gov/
10. Zechner D, Bürtin F, Albert AC. et al. Intratumoral heterogeneity of the therapeutical response to gemcitabine and metformin. Oncotarget. 2015;7:56395-407
11. Feng YH, Velazquez-Torres G, Gully C. et al. The impact of type 2 diabetes and antidiabetic drugs on cancer cell growth. J Cell Mol Med. 2011;15:825-36
12. Kawanami T, Takiguchi S, Ikeda N. et al. A humanized anti-IGF-1R monoclonal antibody (R1507) and/or metformin enhance gemcitabine-induced apoptosis in pancreatic cancer cells. Oncol Rep. 2012;27:867-72
13. Shi Y, He Z, Jia Z. et al. Inhibitory effect of metformin combined with gemcitabine on pancreatic cancer cells in vitro and in vivo. Mol Med Rep. 2016;14:2921-8
14. Lonardo E, Cioffi M, Sancho P. et al. Metformin targets the metabolic achilles heel of human pancreatic cancer stem cells. PLoS One. 2013;8:e76518
15. Fasih A, Elbaz HA, Hüttemann M. et al. Radiosensitization of pancreatic cancer cells by metformin through the AMPK pathway. Radiat Res. 2014;182(1):50-9
16. Baron B, Wang Y, Maehara S. et al. Resistance to gemcitabine in the pancreatic cancer cell line KLM1-R reversed by metformin action. Anticancer Res. 2015;35(4):1941-9
17. Chai X, Chu H, Yang X. et al. Metformin Increases Sensitivity of Pancreatic Cancer Cells to Gemcitabine by Reducing CD133+ Cell Populations and Suppressing ERK/P70S6K Signaling. Sci Rep. 2015;5:14404
18. Janjetovic K, Vucicevic L, Misirkic M. et al. Metformin reduces cisplatin-mediated apoptotic death of cancer cells through AMPK-independent activation of Akt. Eur J Pharmacol. 2011;651:41-50
19. Conde de la Rosa L, Vrenken TE, Buist-Homan M. et al. Metformin protects primary rat hepatocytes against oxidative stress-induced apoptosis. Pharmacol Res Perspect. 2015;3:e00125
20. Bratasz A, Pandian RP, Deng Y. et al. In vivo imaging of changes in tumor oxygenation during growth and after treatment. Magn Reson Med. 2007;57:950-9
21. Donghui Li. Diabetes and Pancreatic Cancer. Mol Carcinog. Mol Carcinog. 2012;51:64-74
22. Butler EB, Zhao Y, Muñoz-Pinedo C. et al. Stalling the engine of resistance: targeting cancer metabolism to overcome therapeutic resistance. Cancer Res. 2013;73(9):2709-17
Author contact
![]() Corresponding author: PD Dr. rer. nat. Dietmar Zechner, Institute for Experimental Surgery, Rostock University Medical Center, Schillingallee 69a, 18057 Rostock, Germany. E-mail: dietmar.zechnerde, phone: +49 381 494 2512, fax: +49 381 494 2502.
Corresponding author: PD Dr. rer. nat. Dietmar Zechner, Institute for Experimental Surgery, Rostock University Medical Center, Schillingallee 69a, 18057 Rostock, Germany. E-mail: dietmar.zechnerde, phone: +49 381 494 2512, fax: +49 381 494 2502.
Citation styles
APA
Zechner, D., Albert, A.C., Bürtin, F., Vollmar, B. (2017). Metformin Inhibits Gemcitabine Induced Apoptosis in Pancreatic Cancer Cell Lines. Journal of Cancer, 8(10), 1744-1749. https://doi.org/10.7150/jca.17972.
ACS
Zechner, D.; Albert, A.C.; Bürtin, F.; Vollmar, B. Metformin Inhibits Gemcitabine Induced Apoptosis in Pancreatic Cancer Cell Lines. J. Cancer 2017, 8 (10), 1744-1749. DOI: 10.7150/jca.17972.
NLM
Zechner D, Albert AC, Bürtin F, Vollmar B. Metformin Inhibits Gemcitabine Induced Apoptosis in Pancreatic Cancer Cell Lines. J Cancer 2017; 8(10):1744-1749. doi:10.7150/jca.17972. https://www.jcancer.org/v08p1744.htm
CSE
Zechner D, Albert AC, Bürtin F, Vollmar B. 2017. Metformin Inhibits Gemcitabine Induced Apoptosis in Pancreatic Cancer Cell Lines. J Cancer. 8(10):1744-1749.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.