Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(9):1586-1597. doi:10.7150/jca.18735 This issue Cite
Review
Redox Imbalance in the Development of Colorectal Cancer
Hao Liu1, Xin Liu1, Chundong Zhang1, Huifang Zhu1, Qian Xu2, Youquan Bu1 ![]() , Yunlong Lei1
, Yunlong Lei1 ![]()
1. Department of Biochemistry and Molecular Biology, and Molecular Medicine and Cancer Research Center, Chongqing Medical University, Chongqing 400016, P. R. China;
2. Department of Anesthesiology, North Sichuan Medical College, Nanchong, Sichuan 637000, China.
Received 2016-12-13; Accepted 2017-2-27; Published 2017-6-3
Abstract
Redox imbalance is resulted from the destruction of balance between oxidants and antioxidants. The dominant oxidants are reactive oxygen species (ROS), which are involved in multiple cellular processes by physiologically transporting signal as a second messenger or pathologically oxidizing DNA, lipids, and proteins. Generally speaking, low concentration of ROS is indispensable for cell survival and proliferation. However, high concentration of ROS is cytotoxic. Additionally, ROS are now known to induce the oxidative modification of macromolecules especially proteins. The redox modification of proteins is involved in numerous biological processes related to diseases including CRC. Herein, we attempt to afford an overview that highlights the crosstalk between redox imbalance and CRC.
Keywords: CRC, oxidative stress, redox modification, cysteine residues.
Introduction
CRC is a major health problem all over the world. Every year, more than 1.2 million patients are diagnosed with CRC, and almost 0.6 million died, making CRC the third most common cancer and the fourth most common cause of cancer-related mortality at present [1, 2].CRC initials a growth called polyp, which begins from the inner surface of the colon or rectum. There are two types of polyps commonly found in the colon or rectum: hyperplastic or inflammatory polyps, and adenomas or adenomatous polyps, which are prone to turn into cancers [3]. In addition, the dysplasia cells in the lining of the colon or rectum may also develop CRC, and is more commonly seen in people with certain IBD like Crohn's disease or ulcerative colitis, in fact, the IBD is the top three high risk factors for CRC [4]. CRC holds other risk factors as well, including age, sex (the risk is higher in women than in men at young ages), smoking, family history of CRC, over drink of alcohol, red meat diet, obesity, diabetes and so on [1]. The CRC therapy strategies are rare at present and mainly dependent on surgery combined with chemotherapy and/or radiotherapy, which is effective against early stage of CRC but poorly effective against advanced stage of CRC especially cancer with metastasis or postoperative recurrence [5]. As a consequence, it is in urgent need of the markers in early diagnosis of CRC and intervention targets in cancer therapy.
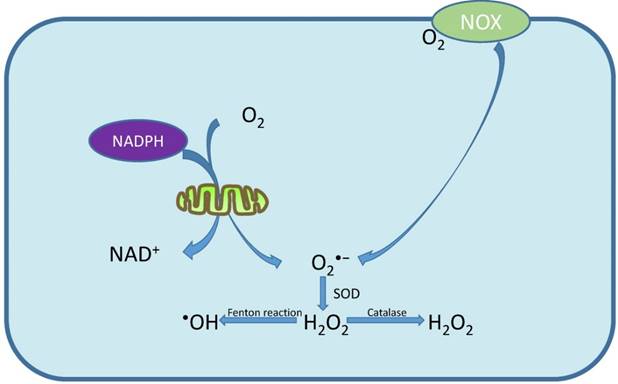
Essentially, oxidative stress is an imbalance between ROS production and the counteractive ability of antioxidants [6]. Free radical involving oxygen can be referred as ROS [7]. Generally, there are two kinds of ROS, the free radicals such as superoxide anion (O2•-), hydroxyl radical (•OH) and the non-radical molecules such as hydrogen peroxide (H2O2), singlet oxygen (1O2) [8]. Mitochondrial respiration is the primary source of ROS and almost 90% of ROS are produced by mitochondria [9, 10]. The electrons are transferred to molecular oxygen by mitochondrial respiratory chain, producing H2O or O2•-, and SOD can catalyze O2•- to H2O2 which is subsequently converted into H2O by catalase or to the highly •OH through Fenton reaction [11] (Fig. 1). Apart from mitochondrial, the NADPH oxidase (NOX), which is mainly response to stress, is another source of intracellular ROS [12] (Fig. 1). As for the elimination of ROS, there are endogenous and exogenous small molecular antioxidants, or enzymatic and nonenzymatic antioxidants [13] (Table 1). The metabolism of these antioxidants regulates the cellular concentration of ROS to prevent cellular damage. For example, O2•- is converted into H2O2 by SOD, and then, H2O2 is disintegrated into water and oxygen by CAT [14].
Mechanism of ROS production. O2 is mainly transformed to O2•- by mitochondrial respiratory chain and NOX. O2•- will be catalyzed to H2O2 by SOD, and H2O2 is subsequently converted into H2O by catalase or to •OH through Fenton reaction.
Antioxidants category
| Type | Name | Refs |
|---|---|---|
| Endogenous | GSH; alpha-lipoic acid; coenzyme Q; ferritin bilirubin; uric acid; metallothionein; melatonin and L-carnitine; | [13, 157] |
| Exogenous | NAC; butylated hydroxytoluene; propyl gallate; tiron; pyruvate; butylated hydroxyanisole; selenium; | [13, 157] |
| Enzymatic | SOD; CAT; GPX;APX | [158] |
On account of the high activity, ROS can react with most of the intracellular substances especially with cysteine residue of proteins, which is called redox modification [15]. In fact, redox modification of proteins are involved in physiology and pathology processes like metabolisms, neurodegenerative diseases and cancers [16, 17]. Considering this, we mainly discuss the function of ROS in the development of CRC and summarize related advances.
The relation between oxidative stress and CRC
In physiological condition, the ROS production and the ROS scavenging ability of antioxidants keep a rough balance, and ROS can assist cell proliferation, migration and differentiation, regulate intermediate products, control the homeostasis of cell and tissue, and activate the survival pathway upon stress response [18, 19]. However, under the pathological condition, excessive level of ROS accumulation due to altered equilibrium between ROS and antioxidants may lead to different kinds of diseases such as atherosclerosis, diabetes, neurodegeneration, and cancer including CRC [20, 21]. Accumulating evidences found that CRC risk factors like smoking and alcohol consumption were involved in ROS production [22, 23]. What's more, studies also revealed that more ROS will be generated in chronic disease of the gastrointestinal tract [24]. For example, oxidative stress is a characteristic of chronic IBD and may increase colon cancer risk [7]. Moreover, through monitoring serum markers such as MPO and oxLDL, researchers observed that the oxidation process begun development in the polyp stage of CRC as well [25]. Thus, it is possible that these risk factors contribute to colorectal carcinogenesis in a ROS dependent way. However, how do these diseases turn into cancer are still not fully understand.
Chronic oxidative stress is a risk factor for CRC [26]. ROS exhibit a high biological activity that could react with substances especially DNA, lipids, and proteins. Thus, excessive level of ROS can affect cancer cell growth, metabolism, invasion and metastasis through gene mutation, DNA damaging, protein conformation transition and so on [22, 27]. For example, through reacting with pyrimidines, purines and chromatin proteins, •OH can induce base modification, genomic instability as well as genetic alteration, all of which contribute to carcinogenesis [28].
ROS-related genetic alteration in colorectal carcinogenesis
It is widely known that ROS-induced DNA damages and genetic mutations are critical causes of cancers including CRC [29]. The main intracellular DNA lesions caused by ROS are single and double strand DNA breaks, and the common genetic mutations include p53, KRAS, APC, and BRAF mutations [30]. For example, a direct relation among oxidative stress, DNA damage and elevated frequency of p53 mutation in CRC has been observed [23]. Most extensively studied endogenous DNA damage by ROS is the formation of 8-oxo-7,8-dihydro-2'-deoxyguanosine (8-oxodG) [31]. As the biomarker of oxidative stress, 8-oxodG level is higher in colorectal tumors than in normal mucosa [31]. Moreover, increased 8-oxodG is also found in leukocytes and urine of CRC patients [32]. In fact, researchers had concluded that levels of 8-oxodG could be applied in clinical practice as an additional and helpful marker to diagnose cancer [33, 34]. Otherwise, 8-oxodG could also induce mismatched pairing and result in switches of cytosine (C) to adenine (A) and/or guanine (G) to thymine (T) [35]. Thus, 8-oxodG is an important cancerogenic factor as well [36]. Fortunately, there are numerous kinds of DNA repair enzymes which could repair the damages induced by 8-oxodG [37]. For example, 8-oxoguanine DNA glycosylase 1(OGG1) and MYH enzyme could repair DNA by detecting and removing the 8-OHdG or mismatched A [38]. However, the activity of OGG1 and MYH enzyme were regulated by ROS, for example, investigators observed that ROS could inhibit the activity of OGG1 through oxidizing the cys326 of OGG1 [39]. In addition, compared with nuclear DNA, mitochondrial DNA is particularly prone to be oxidatively damaged and is more meaningful in colorectal carcinogenesis [40]. Interestingly, apart from that ROS could generate DNA damage, on the contrary, DNA damage could generate ROS as well[41]. For example, study reported that H2AX could regulate Nox1-mediated ROS generation after DNA damage [42]. Thus, a circulated pathway formed, in which ROS and DNA damage promote each other to strengthen the genetic alteration.
ROS induces lipid peroxidation in CRC
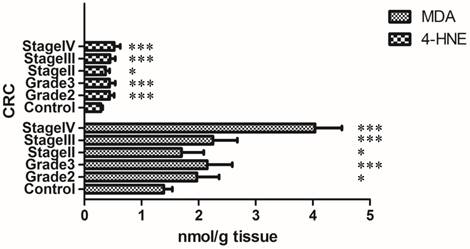
Lipid peroxidation comes from the free radical oxidation of polyunsaturated fatty acids in biological systems [43]. The commonest lipid peroxidation products are MDA and HNE, the levels of which in the CRC tissue are significantly increased with clinical staging [44] (Fig. 2). Although the role of MDA and HNE are still not fully understand, researchers have observed that the HNE could promote the expression of COX-2 which directly induces APC loss and subsequently reduces the degradation of β-catenin, then, the β-catenin translocates to the nucleus and acts as a transcription factor in concert with the T-cell factor-4 (TCF-4) to induce colorectal carcinogenesis [45, 46] (Fig. 3). On the other hand, COX-2 produced prostaglandin can regulate tumor associated angiogenesis, promote cell migration, and inhibit apoptosis, all of those three processes are causes for carcinogenesis [47].
Lipid peroxidation levels with clinical staging of CRC. Lipid peroxidation is significantly increased with clinical staging of CRC. Grade2, 3: histological grade of CRC; Stage II, III, IV: clinical stage of CRC; Control: normal colon mucosa. Collation of data from Elzbieta Skrzydlewska et al [44] (Original data do not include Gade1 and stage I CRC).
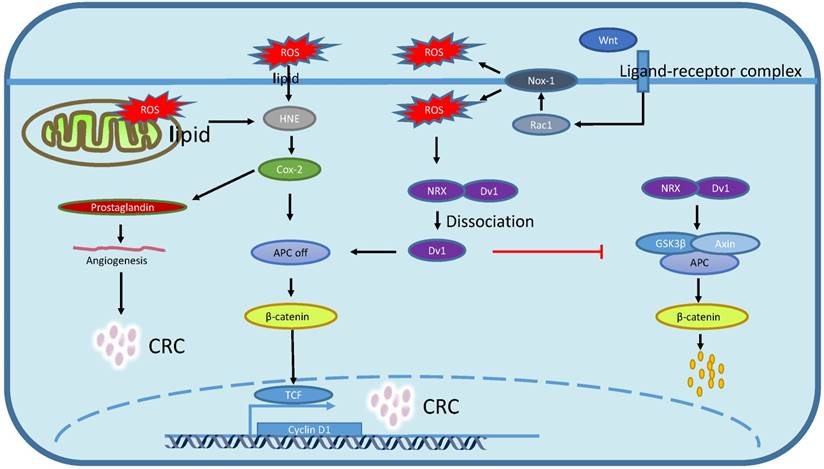
Redox regulation of Wnt/β-catenin signaling pathway and lipid peroxidation. Lipid peroxidation-generated HNE promotes cox-2 expression which induce APC mutation or loss, and the APC loss subsequently inhibit β-catenin degradation that contribute to β-catenin nucleus translocation and targeted genes transcription. Wnt/β-catenin activation aggrandizes Nox1-produced ROS which in turn triggers the dissociation between NRX and Dv1, then dissociated Dv1 suppresses APC expression which results in β-catenin nucleus translocation and targeted genes transcription.
The COX-2 inhibitor such as NSAIDs application is an effective way to prevent CRC. For example, aspirin is used to prevent and treat CRC [48]. NSAIDs, especially acetylsalicylic acid could bring about 50% decrease in CRC incidence and mortality [49]. Moreover, numerous phase III randomized controlled trials that evaluate the role of aspirin in the treatment of CRC are ongoing [50]. In spite of this, NSAIDs application will result in numerous kinds of adverse effects such as ulcers, internal bleeding, kidney failure, heart attack and stroke [51]. Interestingly, latest study demonstrated that some adverse effects of NSAIDs were results from ROS produced by NSAIDs [52]. In addition, MDA could induce DNA damage by directly reacting with DNA, and the product is a DNA adduct called M1G which may contribute to cancer [53].
ROS induces protein oxidation in CRC
Redox modification of thiols on cysteine residues
The protein oxidation in the condition of oxidative stress includes a series of reactions, which are divided into two kinds, the irreversible reaction and the reversible reaction [54]. The irreversible reaction results in protein aggregation and degradation [15]. And the reversible reaction includes methionine side chains oxidation and cysteine side chains oxidation [55]. Most intracellular proteins contain cysteine residues which are usually located in the activity center of proteins. Thus, oxidative cysteine modification is involved in numerous biological events [56]. ROS can reversibly oxidize the active thiol group of cysteine residues into sulfenic acid (R-SOH), inter/intramolecular disulfide bridge (R-S-S-R/R-S-S-R') or protein-S- glutathione (GSH) disulfide, all of which could be reduced to thiol again [57].The most abundant intracellular ROS are hydrogen peroxides (H2O2), thus, the intracellular cysteine residues are usually oxidized by H2O2. In the redox reaction, H2O2 transfers hydroxide radical (OH-) to cysteine thiolate (RS-). According to the concentration of H2O2, the reaction products could be cysteine sulfenic acid (R-SOH), cysteine sulfinic acid (R-SO2H) or cysteine sulfonic acid (R-SO3H) [58]. However, the cysteine sulfenic acid(R-SOH) group and cysteine sulfinic acid(R-SO2H) are not stable, and they could be deoxidized back to cysteine thiolate (RS-) by reducing agents such as thioredoxin, glutaredoxin, peroredoxin and dithiothreitol [59, 60]. Because of the properties, redox modification on cysteine residues could influence function of proteins reversibly and consume ROS by generating cysteine sulfenic acide (R-SOH) and cysteine sulfinic acid (R-SO2H) [61]. Otherwise, cysteine residues modification could cause allosteric interaction in proteins and alter or eliminate protein function permanently as well [62]. Redox modification on protein cysteine thiol has been shown to regulate protein activity involved in transcription, translation and function performing [63]. For example, the redox regulation of Keap1 by oxidation of thiols cause activation of Nrf2 [64]. In addition, a large number of proteins have been identified as redox sensitive proteins in recent years, most of which are involved in the initiation, progress and prognosis of CRC [65]. Thus, it becomes necessary to assess the status of cysteine residues redox in CRC. In this part, we highlight the relation between redox modification in protein cysteine residues and CRC development.
Multiple roles of redox modification in CRC
Accumulating evidences have shown that moderate level of ROS functions as signaling messengers promoting proliferation and invasion of cancer cells, whereas, redox proteins could scavenge basal ROS and function as “tumor suppressors”, or prevent excessive ROS to act as “tumor promoter” [66]. Redox modifications of proteins involved in CRC oncogenesis are through signaling pathways and transcriptional factors modulation [65]. Thus, targeting redox-sensitive signaling pathways, proteins and transcriptional factors as an anticancer strategy offers great promise to prevent and treat CRC.
Redox sensitive/regulate pathways
Cell signaling transduction usually includes 3 steps: upstream transmembrane signal transduction, midstream cytoplasm signaling pathways and intranuclear signal transduction. Extracellular stimulation could often induce alteration of intracellular redox status, which will affect conformation and function of signal molecules to regulate the signal transduction pathways [67]. It is well accepted that intracellular redox imbalance is involved in abnormal activation of some pathways that are closely related with CRC initiation and development, such as the Wnt/β-catenin signaling pathway, PI3K/AKT signaling pathway and the JAK/STAT signaling pathway [68-70].
For example, in the absence of Wnt signaling, NRX binds to Dv1, which will stabilize the Axin, Apc and GSK3β destruction complex, then sequentially phosphorylates β-catenin which subsequently mediates the degradation of β-catenin (Fig. 3). When Wnt signal is activated, ligand-receptor complex triggers Rac1 activation to induce the production of Nox-1-derived ROS which subsequently oxidizes NRX to dissociate Dv1 from NRX and results in suppressing the degradation of β-catenin, which will contribute to CRC carcinogenesis by activating targeted genes like c-Myc [71] (Fig. 3).
PI3K/AKT signaling pathway is closely associated with colon cancer as well, and inhibition of this pathway provides a therapy strategy that may result in curable colon cancer [72]. Studies revealed that ROS could trigger the activation of PI3K and subsequently induce the colorectal carcinogenesis [72-78]. For example, functional studies of oxidative stress observed that the expression of STMN1 and PI3K-AKT-mTOR signaling pathway were involved in ROS-induced and ITGB3-mediated migration and invasion of CRC cells [73]. Furthermore, excessive level of ROS could activate PI3K signaling pathway by oxidizing PTEN cys124, and that will result in CRC [74, 75]. And in vitro study found out that selenite-induced CRC apoptosis was through inhibition of ROS dependent PI3K/AKT pathway [76]. In addition, since EGFR can deregulate the PI3K signaling pathway, the redox modification of EGFR may involve in PI3K pathway activation as well [77]. For example, EGFR can be activated through redox modification of EGFR cys797 [78].
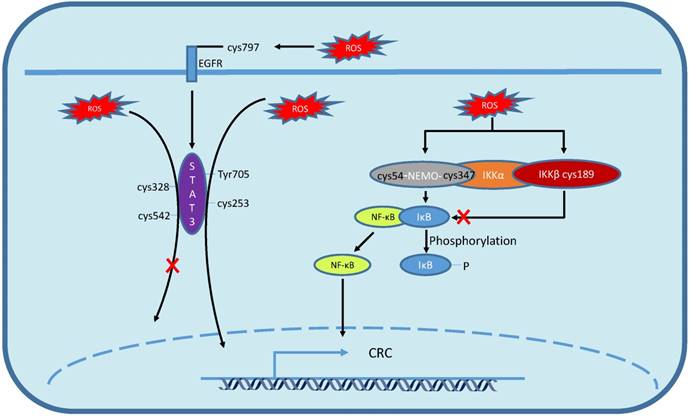
Multiple growth factors and cytokines work as activator in JAK/STAT signaling pathway [79]. Recent studies revealed a prominent role for JAK/STAT pathway in promoting CRC cell growth and survival [68]. In addition, ROS can activate the CRC-related JAK/STAT pathway as well. For example, Sang-Kyu Park demonstrated that short time of H2O2 stimulation induced the activation of STAT pathway by phosphorylation of the Tyr705 of STAT3, which induced the overexpression of cyclinD1 and inhibition of CRC cell apoptosis [80-82] (Fig. 4). EGFR is one of the STAT activators [83], and the redox modification on EGFR cys797 could indirectly activate STAT as well [78] (Fig. 4). Moreover, oxidized low density lipoprotein (oxLDL) could strengthen the combining capacity of STAT1 and STAT3 [84]. And dimerization of STAT3 can be generated by oxidative modification of STAT3 cys253, which promotes its translocation to nucleus [85] (Fig. 4). However, in addition to redox modification-induced STAT pathway activation, oxidative stress can reverse the STAT activation as well, for example, the activation of STAT3 could be impaired by S-glutathionylation in cys328 and cys542 [86] (Fig. 4).
Redox modification on cysteine residues of NF-κB and JAK/STAT signaling pathway. Intracellular ROS induce the phosphorylation of STAT3 Try705 and triggers dimerisation of STAT3, then, dimerisation of STAT3 translocates to nucleus. Extracellular ROS induces STAT3 activation by oxidation of EGFR cys797 while intracellular ROS induces STAT dimerization by oxidation of STAT3 cys253. Whereas ROS-induced S-glutathionylation on cys328 and cys542 impairs STAT3 phosphorylation, damages STAT3 dimerisation and inactivates targeted gene transcription. The NEMO cys347 and cys54 disulfide bond formation leads to IκB phosphorylation and release NF-κB, then NF-κB translocates to nucleus. Whereas ROS induced s-gluthionylation on cys189 of IKKβ inactivates NF-κB.
The MAPK cascades are membrane to nucleus signaling modules that respond to oxidative stress which lead to phosphorylation and activation of down-stream genes required for CRC [87, 88]. For example, the EGFR cys797 can be oxidized and activated by ROS, which initiates the MAPK cascade that is responsible for CRC [89]. Kyoungmun Lee et al analyzed the relation between ROS and MAPK, and found out that the inhibition of PTPs by H2O2 resulted in phosphorylation and activation of ERK, p38 and JNK [90]. Except these, ROS can directly oxidize the constituent parts of MAPK as well, for example, in a CRC model, researchers observed that NOX-generated ROS could induce the activation of Ras by S-glutathionylation on cys118 [91]. However, it is controversial that ROS can work as a trigger to turn off the MAPK cascades either, for example, H2O2 is able to reduce the activatiy and phosphorylation level of p38, ERK1/2 and JNK by inhibiting the MEK1/2 activities [92]. And in vivo study showed that H2O2 could suppress p38 activity by oxidizing p38 cysteine residues [93].
Redox-related transcription factors
Transcriptional factors are a group of proteins which could bind to specific DNA sequences to regulate/trigger genes expression, such as NF-κB, p53, HIF-1α and Nrf2. The increasing production of intracellular or extracellular ROS could regulate the activity of transcriptional factors and play a pivotal role in colorectal carcinogenesis [63, 94, 95]. For example, in vitro study showed that ethanol could enhance arsenic-induced CRC via NF-κB in an ROS dependent way [96]. In addition, emodin inhibits the proliferation of CRC cells by inducing ROS-mediated p53 activation [97].
NF-κB is an important transcription factor in the regulation of inflammation, cell cycling, apoptosis, metabolism and carcinogenesis, while growing evidence also support a major role in CRC [98]. In the canonical pathway, NF-κB binds to IκB and is detained in cytoplasm. While stimulus triggers a cascade of events which lead to IκB phosphorylation by inhibitor κB kinase (IKK) complex, NF-κB is released from IκB and translocated to the nucleus to regulate gene expression [99]. The activity of IKK complex and NF-κB is prone to be regulated through redox modification on cysteine. Study revealed that IKK complex subunit, NEMO, was an essential activator of NF-κB, and ROS could activate the NF-κB through modification of NEMO, that is, inducing the formation of disulfide bond between cys347 and cys54 by H2O2 [74] (Fig. 4). ROS can also inhibit the activation of NF-κB by inducing S-glutathionylation on the cys189 of IKKβ (Fig. 4). Moreover, Takeyuki Nishi et al found that p65 subunit and cys62 of p50 were highly oxidized in cytoplasm and strongly reduced in the nucleus, meanwhile, the reduced form of cys62 was essential for the DNA binding activity of NF-κB [73]. Recent studies demonstrated that the role of ROS on NF-κB activation was cell type dependent [74] [100]. For example, in limphycyte cell, ROS induce the phosphorylation of IΚΚ and subsequently activate NF-κB [75], while in pulmonary epithelial cells, H2O2 oxidizes cysteine residues to inhibit the activation of IKKβ which could reduce the activation of NF-κB [76].
P53 is an important tumor suppressor that induces the apoptosis of malignant tumor cells [101]. Redox-related deletion or mutation of p53 could contribute to colorectal carcinogenesis [102]. For example, study showed that the zinc finger protein 148 (Zfp148) was a potent suppressor of p53 under oxidative stress, which contributed to CRC development [103]. Jenna Scotcher showed that p53 was a multiple cysteine-containing protein [104], thus, it is possible that the redox modification of p53 cysteine residues acts as a switcher that trigger on/off p53 activation. For example, previous study reported that cys182 and cys277 were implicated in p53 redox-regulation, while, cys176, cys182, cys238 and cys242 were found to be oxidized residues in p53 under the treatment of H2O2 [82]. Oxidization suppressed p53 transcriptional activity which subsequently inactivated targeted genes like Bax and bcl-2 expression and resulted in apoptosis inhibition that may induce CRC [105-107]. On the other hand, GSH-induced p53 inactivation may be dependent on S-glutathionylation of p53 on cys141 and lead to colon carcinogenesis [108].
The rapid proliferation of CRC cells can generate a hypoxic microenvironment that activates the transcription factor HIF1-α by redox modification. For example, Klaus Jürgen Schmitz et al reported that under hypoxic condition, the oxidative modification of HIF1-α was downregulated which stabilized the HIF1-α and drove its translocation into the nucleus, where expression of its target genes was upregulated and substantially strengthened the CRC development [109]. While, in basal oxygen condition, ROS can activate and stabilize HIF-1α, for example, John J. Haddad et al reported that cytokine-mediated regulation of HIF-1α stabilization, translocation and activation required a non-hypoxic, ROS-sensitive mechanism [110]. The ROS-sensitive mechanism is basically based on redox modification of cysteine residues. For example, recent studies found that HIF-1α was a potential target for S-nitrosation, and the S-nitrosation of cys800 induced the recruitment of p300 co-activator protein to the HIF-1α C-terminal domain which increased its transcriptional activity [111]. And S-nitrosation of cys162 in pVHL could decrease HIF-1α ubiquitination that benefit the angiogenesis and induce colorectal tumorigenesis [109, 112, 113].
In addition to serving as a ROS-sensitive transcription factor, Nrf2 is the most important intracellular regulator of antioxidants [114]. For example, Nrf2 can directly eliminate ROS via regulating GSH metabolism [115]. Recent study reported that Nrf2 can protect against oxidative stress-derived cancer in stomach, skin, bladder and colon [116, 117]. Under physiological condition, Nrf2 binds to keap1 in cytoplasm where it keeps inactivated and is easy to be degraded [118]. While, in the condition of oxidative stress, redox modification of keap1 cys151, cys273 and cys288 could detach the Nrf2-keap1 complex and lead to the dissociation of Nrf2 from keap1 [64, 119, 120]. Furthermore, oxidants and electrophiles-induced phosphorylation of PKC and ERK can subsequently promote phosphorylation of Nrf2 at serine40 which is necessary for the dissociation of Nrf2 from keap1 [121]. Released Nrf2 will be translocated to nucleus where it binds to genes containing antioxidant responsive element (ARE) or electrophile responsive element (EpRE) that initiate antioxidant responses [64, 119, 120]. And the Nrf2-induced adaptive response to ROS inhibits HIF1-α-VEGF signaling, resulting in diminishing angiogenesis and CRC growth [122]. Furthermore, recent in vivo and in vitro study showed that evolutionarily conserved cysteine residues of Nrf2 like cys119, cys235 and cys506 were necessarily needed in modulation of oxidative stress as well as keap1-induced ubiquitination [123]. Paradoxically, although a large number of evidences indicate that the activation of Nrf2 protect against a variety of cancers, the prolonged activation of Nrf2 has been shown to favor the progression of several types of cancers [124]. For example, researchers observed a continuously elevated Nrf2 expression in lung, breast, ovarian and endometrial cancer [57, 125-128]. And the elevated Nrf2 may be related to cancer proliferation by maintaining redox homeostasis, for example, in A549 cells, researchers found out that Nrf2 could accelerated cancer cell proliferation by promoting GSH synthesis [129]. However, whether a similar effect exists or not in CRC is still uncovered.
Modulation of ROS as anticancer strategy
ROS could mediate the colorectal carcinogenesis through gene mutations, redox related signaling pathways and redox related transcription factors, thus dietary and endogenous antioxidants can prevent cancer by reacting with or eliminating oxidizing free radicals [130]. A large randomized trial investigated the putative preventive role of antioxidants on cancer, for example, the consumption of antioxidants like selenium, vitamin E and β-carotene significantly decreased cancer mortality [131]. A similar prevention effect was also observed in CRC. For example, Melissa Y. Wei et al determined that the high level of vitamin D was associated with a decreased risk of colorectal adenoma, including advanced adenoma and recurrent adenoma [132]. Moreover, antioxidants like GSH could inhibit malignant phenotype of CRC [133] and reduce proliferation of CRC by decreasing the expression of cyclooxygenase-2(COX-2) and the production of prostaglandin [134].
On the other hand, increasing ROS as an anticancer therapy has also been well studied these years [135]. Since overaccmulation of ROS can lead to the preferential killing of cancer cell [136], the utilization of oxidants renders a new way in CRC therapy. The study of Yushuang Ding et al supported that promoting ROS overload might be an important strategy for the development of new anticancer drugs [137]. In fact, numerous anti-cancer drugs used by CRC patients are involved in the ROS production (Table 2). For example, 5-Fu is commonly used in the treatment of CRC, especially in CRC at stage III and high risk stage II, alone or together with other drugs [138]. 5-Fu has been used in CRC treatment for more than 40 years and has several mechanisms of anti-cancer effects [139, 140]. In addition to inhibiting the DNA synthesis, altering RNA processing and inducing DNA damage, 5-Fu-activated anti-cancer response can be based on ROS elevation as well [141]. For example, in vitro study observed that 5-Fu treatment in CRC cells generated O2- that positively regulated p53 proteins and thereby induced cancer cell apoptosis [142, 143]. Tamoxifen is a breast cancer drug, whereas, researchers showed that tamoxifen may play a beneficial role in other malignancies treatment [144]. For example, a murine model study revealed that tamoxifen could inhibit colorectal liver metastases [145]. Moreover, in vivo study found that tamoxifen could reverse multidrug resistance of CRC [146]. In addition, a latest study demonstrated that tamoxifen could induce the CRC senescence via antagonizing with CK2α and then promoting ROS generation [147]. Celecoxib is a selective inhibitor of COX-2 and can significantly reduce the risk of colorectal adenomas [148]. The anti-cancer effect of celecoxib is previously considered to base solely on specific inhibition of COX-2 that inhibits angiogenesis by down-regulating VEGF [149]. However, recent study revealed that the anti-cancer effect of celecoxib might be based on ER stress-derived ROS as well [150]. In addition to those specific CRC drugs, there are numerous types of drugs used in cancer therapy including but not limited to CRC, which are also implicated in elevated ROS production. For example, methotrexate can trigger ROS-associated cell apoptosis in different types of cancer [151]. Moreover, irinotecan is a topoisomerases inhibitor that causes oxidative stress among different types of cancer [152]. Meanwhile, the ionizing radiation therapy can also induce accumulation of ROS. For example, after exposure to ionizing radiation, researchers observed an instantaneous and robust release of •OH that oxidized the ETC complex and resulted in mitochondrial dysfunction and CRC cells elimination [153]. In addition, drugs that degenerate antioxidants are also found to be involved in CRC therapy. For example, 6-anicotinamide (6-AN) is an inhibitor of G6PD that reduces GSH in the treatment of colon cancer [154].
Anticancer drugs in the regulation of ROS levels
| Name | Mechanism of action | Effects on ROS | Cancers | Refs |
|---|---|---|---|---|
| 5-Fu | Inhibits thymidylate synthetase and/or incorporates into RNA and DNA | Induces intracellular increase in O2•- levels | CRC | [137] |
| Tamoxifen | Promotes cancer cell senescence | Promotes ROS generation | CRC | [159] |
| Celecoxib | Inhibits COX2 activity, Induces ER stress by causing leakage of calcium from the ER into the cytosol | Induction of ROS owing to ER stress | CRC | [150] |
| Methotrexate | Triggers ROS related cell apoptosis | Promotes ROS generation | Different types of cancer | [160] |
| Irinotecan | Topoisomerases inhibitor | Promotes ROS generation | Different types of cancer | [161] |
| Mitoxantrone | Trigging cell membrane scrambling | Promotes ROS generation | Different types of cancer | [162] |
| Paclitxel(Taxol) | Inhibitor of cell division | Promotes ROS generation | Different types of cancer | [163] |
| Adriamycin | Reduces cell viability through initiating cell apoptosis and strong G2/M phase cell cycle arrest | Promotes ROS generation | Different types of cancer | [164] |
| Imatinib | Protein tyrosine kinase inhibitor that induce apoptosis | Promotes ROS generation | Different types of cancer | [165] |
| Camptothecin | Quinolone alkaloid that induces cytotoxicity | Promotes ROS generation | Different types of cancer | [166] |
| Carboplatin | Cell cycle arrest | Induction of ROS owing to ER stress | Different types of cancer | [167] |
| Capecitabine | Prodrug that is enzymatically converted to 5-Fu in the body | Promotes ROS generation | CRC | [168] |
| Cisplatin | Inducing nuclear DNA adducts | Induces a mitochondrial dependent ROS generation | Different types of cancer | [169] |
| Manumycin | Increasing the ROS production and blocking PI3K/AKT pathway | Promotes ROS generation | CRC | [170] |
| Cribrostatin 6 | Quinone containing product induces apoptotic cell death | Promotes ROS generation | Different types of cancer | [171] |
Conclusion remarks
CRC is a rather complex, multifactorial and multistage disease. As demonstrated previously, the intracellular redox imbalance is a decisive factor in the CRC development and progression. Malignant carcinomas usually characterize as a hypermetabolic state that leads to a persistent oxidative stress state in cellular microenvironment, thus the utilization of antioxidants that antagonize with ROS seems to be a feasible strategy in cancer therapy. But, systematic reviews found out that the use of antioxidants were invalid in cancer therapy, or even reversibly facilitated the progress of cancer [155, 156]. However, the problem is still considered controversial. And the frustrating consequence may partly due to that a certain decreased level of ROS is benefit for proliferation of cancer cells, especially those with very high ROS accumulation but still under the toxic threshold. On the other hand, using oxidants which preferentially kill cancer cells is another way in CRC therapy that has been well studied these years. However, the slight alternation in redox state can be amplified by modification of macromolecules, thus the redox related anticancer substances should be precisely controlled and targeted in body. On the basis of CRC processes, normal colon or rectum cells, polyps, adenocarcinomas and ultimately metastatic CRC, precisely utilizing oxidants and antioxidants with the help of redox sensitive marker is indispensable. Thus, more redox sensitive markers are needed.
Abbreviations
A: Adenine; ARE: Antioxidant response element; C: Cytosine; COX-2: Cyclooxygenase-2; CRC: Colorectal cancer; CAT: Catalase; EpRE: Electrophile responsive element; ER: Endoplasmic reticulum; GSH: Glutathione; GPXs: Glutathione peroxidants; G: Guanine; G6PD: Glucose -6-phosphate dehydrogenase; H2AX: Histone family member x; H2O2: Hydrogen peroxide; HNE: 4-hydroxynonenal; HIF-1α: Hypoxia inducible factor 1α; IBD: Inflammatory bowel diseases; IKK: Inhibitor κB kinase; INrf2: Inhibitor of Nrf2; IFN-α: Interferon-alpha; ROS: Reactive oxygen species; JAK: Janus Kinase; Keap1: Kelch-like ECH-associated protein 1; MPO: Myeloperoxidase; MDA: Malondialdehyde; MAPK: Mitogen-activted protein kinase; mTOR: Mammalian target of rapamycin; NOX: NADPH oxidase; NSAIDs: Nonsteroidal anti-inflammatory drug; NAC: N-acety1 cysteine; NIK: NF-κB inducing kinase; Nrf2: Tanscription factor NFE2-related factor 2; O2•-: Superoxide anion; •OH: Hydroxyl radical; 1O2: Singlet oxygen; oxLDL: Oxidized low-density lipoprotein; OGG1: 8-oxoguanine DNA glycosylase 1; OH-: Hydroxide radical; PRXs: Peroxiredoxins; PTKs: Protein tyrosine kinases; PTPs: Protein tyrosine phosphatases; PI3K: Phosphatidyl inositol 3-OH kinase; PKB: Protein kinase B; PTEN: Tension homolog; RS-: Cysteine thiolate; R-SOH: Sulfenic acid; R-SO2H: Cysteine sulfinic acid; R-SO3H: Cysteine sulfonic acid; R-S-S-R/R-S-S-R': Inter/intramolecular disulfide bridge; RTKs: Receptor tyrosine kinases; SOD: Superoxide dismutase; TRX: Thioredoxin; TCF-4: T-cell factor-4; T: Thymine; VEGF: Vascular endothelial growth factor; Zfp148: Zinc finger protein 148; 5-Fu: 5-fluorouracil; 6-AN: 6-anicotinamide; 8-oxodG: 8-oxo-7, 8-dihydro-2'-deoxyguanosine.
Acknowledgements
This work was supported by grants from the Chinese NSFC (81401951, 81672301 81171879, 81502131, 81501979 and 81401889), Scientific and Technological Research Program of Chongqing Municipal Education Commission (KJ1400207, KJ1500332) and Research Grant from Chongqing Medical University (201412).
Author contributions
All authors contributed to prepare, review, and write the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Brenner H, Kloor M, Pox CP. Colorectal cancer. The Lancet. 2014;383:1490-502
2. Schreuders EH, Ruco A, Rabeneck L, Schoen RE, Sung JJ, Young GP. et al. Colorectal cancer screening: a global overview of existing programmes. Gut. 2015;64:1637-49
3. Rd BA. Epidemiology, disease progression, and economic burden of colorectal cancer. Journal of Managed Care Pharmacy Jmcp. 2007;13:S5-18
4. Saw CL, Kong AN. Nuclear factor-erythroid 2-related factor 2 as a chemopreventive target in colorectal cancer. Expert Opin Ther Targets. 2011;15:281-95
5. Meyerhardt JA, Mayer RJ. Systemic Therapy for Colorectal Cancer. New England Journal of Medicine. 2005;352:476-87
6. Pruchniak MP, Araźna M, Demkow U. Biochemistry of Oxidative Stress. Advances in Experimental Medicine & Biology. 2015;35:1058-71
7. Aiken CT, Kaake RM, Wang X, Huang L. Oxidative stress-mediated regulation of proteasome complexes. Molecular & Cellular Proteomics Mcp. 2011;10:744-57
8. Sharma P, Jha AB, Dubey RS, Pessarakli M. Reactive Oxygen Species, Oxidative Damage, and Antioxidative Defense Mechanism in Plants under Stressful Conditions. Journal of Botany. 2012. 2012
9. Brookes PS. Mitochondrial H(+) leak and ROS generation: An odd couple. Free Radical Biology & Medicine. 2005;38:12-23
10. Lushchak VI. Free radicals, reactive oxygen species, oxidative stress and its classification. Chemico-Biological Interactions. 2014;224:164-75
11. Kuznetsov AV, Kehrer I, Kozlov AV, Haller M, Redl H, Hermann M. et al. Mitochondrial ROS production under cellular stress: comparison of different detection methods. Analytical and Bioanalytical Chemistry. 2011;400:2383-90
12. Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiological Reviews. 2007;87:245-313
13. Reuter S, Gupta SC, Chaturvedi MM, Aggarwal BB. Oxidative stress, inflammation, and cancer: How are they linked? Free Radical Biology & Medicine. 2010;49:1603-16
14. Vurusaner B, Poli G, Basaga H. Tumor suppressor genes and ROS: complex networks of interactions. Free Radical Biology & Medicine. 2012;52:7-18
15. Yan LJ. Protein redox modification as a cellular defense mechanism against tissue ischemic injury. Oxidative Medicine & Cellular Longevity. 2014;2014:61-78
16. Biswas S, Chida AS, Rahman I. Redox modifications of protein-thiols: Emerging roles in cell signaling. Biochemical Pharmacology. 2006;71:551-64
17. Do KQ, Cabungcal JH, Frank A, Steullet P, Cuenod M. Redox dysregulation, neurodevelopment, and schizophrenia. Current Opinion in Neurobiology. 2009;19:220-30
18. Janssen-Heininger YMW, Mossman BT, Heintz NH, Forman HJ, Kalyanaraman B, Finkel T. et al. Redox-based regulation of signal transduction: Principles, pitfalls, and promises. Free Radical Biology & Medicine. 2008;45:1-17
19. Finkel T. Oxidant signals and oxidative stress. Current Opinion in Cell Biology. 2003;15:247-54
20. Acharya A, Das I, Chandhok D, Saha T. Redox regulation in cancer: a double-edged sword with therapeutic potential. Oxidative Medicine & Cellular Longevity. 2010;3:23-34
21. Ko CH, Yang SLY, Lin CW. Gossypol reduction of tumor growth through ROS-dependent mitochondria pathway in human colorectal carcinoma cells. International Journal of Cancer Journal International Du Cancer. 2007;121:1670-9
22. Ziech D, Franco R, Pappa A, Panayiotidis MI. Reactive oxygen species (ROS)-induced genetic and epigenetic alterations in human carcinogenesis. Mutation Research. 2011;711:167-73
23. Bartsch H, Nair J. Chronic inflammation and oxidative stress in the genesis and perpetuation of cancer: role of lipid peroxidation, DNA damage, and repair. Langenbecks Archives of Surgery. 2006;391:499-510
24. Girgin F, Karaoglu O, Erkuş M, Tüzün S, Ozütemiz O, Dinçer C. et al. Effects of trimetazidine on oxidant/antioxidant status in trinitrobenzenesulfonic acid-induced chronic colitis. Journal of Toxicology & Environmental Health Part A. 2000;59:641-52
25. Crespo-Sanjuán J, Calvo-Nieves MD, Aguirre-Gervás B, Herreros-Rodríguez J, Velayos-Jiménez B, Castro-Alija MJ. et al. Early detection of high oxidative activity in patients with adenomatous intestinal polyps and colorectal adenocarcinoma: myeloperoxidase and oxidized low-density lipoprotein in serum as new markers of oxidative stress in colorectal cancer. Laboratory Medicine. 2015;46:123-35
26. Perše M. Oxidative stress in the pathogenesis of colorectal cancer: cause or consequence? Biomed Research International. 2013;2013:725710 -
27. Forsberg L, Faire UD, Morgenstern R. Oxidative Stress, Human Genetic Variation, and Disease. Archives of Biochemistry & Biophysics. 2001;389:84-93
28. Fruehauf JP Jr MF. Reactive oxygen species: a breath of life or death? Clinical Cancer Research An Official Journal of the American Association for Cancer Research. 2007;13:789-94
29. Kirkali G, Keles D, Canda AE, Terzi C, Jaruga P, Dizdaroglu M. et al. Abstract 1496: Oxidatively induced DNA base damage in colorectal carcinoma. Cancer Research. 2011;70:1496 -
30. Lee JH, Hwang I, Kang YN, Choi IJ, Kim DK. Genetic Characteristics of Mitochondrial DNA Was Associated with Colorectal Carcinogenesis and Its Prognosis. Plos One. 2014:10
31. Matosevic P, Klepac-Pulanic T, Kinda E, Augustin G, Brcic I, Jakic-Razumovic J. Immunohistochemical expression of 8-oxo-7,8-dihydro-2′-deoxyguanosine in cytoplasm of tumour and adjacent normal mucosa cells in patients with colorectal cancer. World Journal of Surgical Oncology. 2015;13:1-9
32. Obtulowicz T, Swoboda ME, Gackowski D, Rozalski R, Siomek A, Janik J. et al. Oxidative stress and 8-oxoguanine repair are enhanced in colon adenoma and carcinoma patients. Mutagenesis. 2010;25:463-71
33. Roszkowski K, Jozwicki W, Blaszczyk P, Mucha-Malecka A, Siomek A. Oxidative damage DNA: 8-oxoGua and 8-oxodG as molecular markers of cancer. Medical Science Monitor International Medical Journal of Experimental & Clinical Research. 2011;17:329-33
34. Thanan R, Murata M, Pinlaor S, Sithithaworn P, Khuntikeo N, Tangkanakul W. et al. Urinary 8-oxo-7,8-dihydro-2'-deoxyguanosine in patients with parasite infection and effect of antiparasitic drug in relation to cholangiocarcinogenesis. Cancer epidemiology, biomarkers & prevention: a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2008;17:518-24
35. Cheng KC, Cahill DS, Kasai H, Nishimura S, Loeb LA. 8-Hydroxyguanine, an abundant form of oxidative DNA damage, causes G--T and A--C substitutions. Journal of Biological Chemistry. 1992;267:166-72
36. Loft S, Svoboda P, Kasai H, Tjønneland A, Vogel U, Møller P. et al. Prospective study of 8-oxo-7,8-dihydro-2'-deoxyguanosine excretion and the risk of lung cancer. Carcinogenesis. 2006;27:1245-50
37. Martinet W, Knaapen MW, De Meyer GR, Herman AG, Kockx MM. Elevated levels of oxidative DNA damage and DNA repair enzymes in human atherosclerotic plaques. Circulation. 2002;106:927-32
38. Goto M, Shinmura K, Yamada H, Tsuneyoshi T, Sugimura H. OGG1, MYH and MTH1 gene variants identified in gastric cancer patients exhibiting both 8-hydroxy-2′-deoxyguanosine accumulation and low inflammatory cell infiltration in their gastric mucosa. Journal of Genetics. 2008;87:181-6
39. Bravard A, Vacher M, Moritz E, Vaslin L, Hall J, Epe B. et al. Oxidation status of human OGG1-S326C polymorphic variant determines cellular DNA repair capacity. Cancer Research. 2009;69:3642-9
40. Liang FQ, Godley BF. Oxidative stress-induced mitochondrial DNA damage in human retinal pigment epithelial cells: a possible mechanism for RPE aging and age-related macular degeneration. Experimental Eye Research. 2003;76:397-403
41. Indo HP, Davidson M, Yen HC, Suenaga S, Tomita K, Nishii T. et al. Evidence of ROS generation by mitochondria in cells with impaired electron transport chain and mitochondrial DNA damage. Mitochondrion. 2007;7:106-18
42. Kang MA, So EY, Simons AL, Spitz DR, Ouchi T. DNA damage induces reactive oxygen species generation through the H2AX-Nox1/Rac1 pathway. Cell Death & Disease. 2012;3:e249
43. Gutteridge JM. Lipid Peroxidation and Antioxidants as Biomarkers of Tissue Damage. Clinical Chemistry. 1995;41:1819-28
44. Skrzydlewska E, Sulkowski S, Koda M, Zalewski B, Kanczuga-Koda L, Sulkowska M. Lipid peroxidation and antioxidant status in colorectal cancer. World Journal of Gastroenterology Wjg. 2005;11:403-6
45. Kumagai T, Matsukawa N, Kaneko Y, Kusumi Y, Mitsumata M, Uchida K. A lipid peroxidation-derived inflammatory mediator: identification of 4-hydroxy-2-nonenal as a potential inducer of cyclooxygenase-2 in macrophages. Journal of Biological Chemistry. 2004;279:48389-96
46. Brown JR, Dubois RN. COX-2: a molecular target for colorectal cancer prevention. Journal of Clinical Oncology. 2005;23:2840-55
47. Yong IC, Dubois RN. NSAIDs and cancer prevention: targets downstream of COX-2. Annual Review of Medicine. 2007;58:239-52
48. Zubiaurre L, Bujanda F. [Aspirin in the prevention of colorectal cancer]. Gastroenterología Y Hepatología. 2011;34:337-45
49. Luk GD. Prevention of gastrointestinal cancer-the potential role of NSAIDs in colorectal cancer. Schweizerische Medizinische Wochenschrift. 1996;126:801-12
50. Coyle C, Cafferty FH, Langley RE. Aspirin and Colorectal Cancer Prevention and Treatment: Is It for Everyone? Current Colorectal Cancer Reports. 2016;12:27-34
51. Naesdal DJ, Brown K. NSAID-Associated Adverse Effects and Acid Control Aids to Prevent Them. Drug Safety. 2006;29:119-32
52. Ghosh R, Alajbegovic A, Gomes AV. NSAIDs and Cardiovascular Diseases: Role of Reactive Oxygen Species. Oxidative Medicine & Cellular Longevity. 2015. 2015
53. Marnett LJ. Lipid peroxidation—DNA damage by malondialdehyde. Mutation Research/fundamental & Molecular Mechanisms of Mutagenesis. 1999;424:83-95
54. Jung T, Höhn A, Grune T. The proteasome and the degradation of oxidized proteins: Part II - protein oxidation and proteasomal degradation. Redox Biology. 2013;2:99-104
55. Hoshi T, Heinemann SH. Regulation of cell function by methionine oxidation and reduction. Journal of Bacteriology. 2001;531:1-11
56. Barford D. The role of cysteine residues as redox-sensitive regulatory switches. Current Opinion in Structural Biology. 2004;14:679-86
57. Wang XJ, Sun Z, Villeneuve NF, Zhang S, Zhao F, Li Y. et al. Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis. 2008;29:1235-43
58. Thomas JA, Mallis RJ. Aging and oxidation of reactive protein sulfhydryls. Experimental Gerontology. 2001;36:1519-26
59. D'Autréaux B, Toledano MB. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nature Reviews Molecular Cell Biology. 2007;8:813-24
60. Jeong J, Jung Y, Na S, Jeong J, Lee E, Kim MS. et al. Novel oxidative modifications in redox-active cysteine residues. Molecular & cellular proteomics. 2011;10:M110 000513
61. Shetty V, Spellman DS, Neubert TA. Characterization by Tandem Mass Spectrometry of Stable Cysteine Sulfenic Acid in a Cysteine Switch Peptide of Matrix Metalloproteinases. Journal of the American Society for Mass Spectrometry. 2007;18:1544-51
62. Den Hertog J. Protein Tyrosine Phosphatases as Mediators of Redox Signaling: Wiley-VCH Verlag GmbH & Co. KGaA. 2009
63. Schieber M, Chandel N. ROS Function in Redox Signaling and Oxidative Stress. Current Biology Cb. 2014;24:R453-R62
64. Yamamoto T, Suzuki T, Kobayashi A, Wakabayashi J, Maher J, Motohashi H. et al. Physiological significance of reactive cysteine residues of Keap1 in determining Nrf2 activity. Molecular & Cellular Biology. 2008;28:2758-70
65. Yang HY, Chay KO, Kwon J, Kwon SO, Park YK, Lee TH. Comparative proteomic analysis of cysteine oxidation in colorectal cancer patients. Molecules & Cells. 2013;35:533-42
66. Chen Y, Zhang H, Zhou HJ, Ji W, Min W. Mitochondrial Redox Signaling and Tumor Progression. Cancers. 2016:8
67. Cullen S, Ponnappan S, Ponnappan U. Redox-Regulated Pathway of Tyrosine Phosphorylation Underlies NF-κB Induction by an Atypical Pathway Independent of the 26S Proteasome. Biomolecules. 2015;5:95-112
68. Wei X, Wang G, Li W, Hu X, Huang Q, Xu K. et al. Activation of the JAK-STAT3 pathway is associated with the growth of colorectal carcinoma cells. Oncology Reports. 2013;31:335-41
69. Sebio A, Kahn M, Lenz HJ. The potential of targeting Wnt/β-catenin in colon cancer. Expert Opinion on Therapeutic Targets. 2014;18:611-5
70. Pandurangan AK. Potential targets for prevention of colorectal cancer: a focus on PI3K/Akt/mTOR and Wnt pathways. Asian Pacific Journal of Cancer Prevention Apjcp. 2013;14:2201-5
71. Kajla S, Mondol AS, Nagasawa A, Zhang Y, Kato M, Matsuno K. et al. A crucial role for Nox 1 in redox-dependent regulation of Wnt-β-catenin signaling. Faseb Journal Official Publication of the Federation of American Societies for Experimental Biology. 2012;26:2049-59
72. Huang XF, Chen JZ. Obesity, the PI3K/Akt signal pathway and colon cancer. Obesity Reviews An Official Journal of the International Association for the Study of Obesity. 2009;10:610-6
73. Lei Y, Huang K, Gao C, Lau QC, Pan H, Xie K. et al. Proteomics Identification of ITGB3 as a Key Regulator in Reactive Oxygen Species-induced Migration and Invasion of Colorectal Cancer Cells. Molecular & Cellular Proteomics: MCP. 2011;10:M110.005397
74. Lee SR, Yang KS, Kwon J, Lee C, Jeong W, Rhee SG. Reversible inactivation of the tumor suppressor PTEN by H2O2. Journal of Biological Chemistry. 2002;277:20336-42
75. Kitagishi Y, Matsuda S. Redox regulation of tumor suppressor PTEN in cancer and aging (Review). International Journal of Molecular Medicine. 2013;31:511-5
76. Luo H, Yang Y, Duan J, Wu P, Jiang Q, Xu C. PTEN-regulated AKT/FoxO3a/Bim signaling contributes to reactive oxygen species-mediated apoptosis in selenite-treated colorectal cancer cells. Cell Death & Disease. 2013;4:e481
77. Wei L, Ren H, Ren J, Yin T, Bing H, Xie S. et al. The Role of EGFR/PI3K/Akt/cyclinD1 Signaling Pathway in Acquired Middle Ear Cholesteatoma. Mediators of Inflammation. 2013;2013:450 -
78. Truong TH, Carroll KS. Redox Regulation of Epidermal Growth Factor Receptor Signaling through Cysteine Oxidation. Biochemistry. 2012;51:9954-65
79. Sengupta TK, Schmitt EM, Ivashkiv LB. Inhibition of cytokines and JAK-STAT activation by distinct signaling pathways. Proceedings of the National Academy of Sciences. 1996;93:9499-504
80. Erkasap N, Özyurt R, Özkurt M, Yaşar F, Erkasap S, Ihtiyar E. The role of JAK/STAT signaling pathway and TNF-α crosstalk in human colorectal cancer. Gene Reports. 2016;3:1-4
81. Ma S. Constitutive activation of Stat3 signaling pathway in human colorectal carcinoma. World Journal of Gastroenterology Wjg. 2004;10:1569-73
82. Park SK, Dahmer MK, Quasney MW. MAPK and JAK-STAT signaling pathways are involved in the oxidative stress-induced decrease in expression of surfactant protein genes. Cellular Physiology & Biochemistry International Journal of Experimental Cellular Physiology Biochemistry & Pharmacology. 2012;30:334-46
83. Spano JP, Milano G, Rixe C, Fagard R. JAK/STAT signalling pathway in colorectal cancer: A new biological target with therapeutic implications. European Journal of Cancer. 2006;42:2668-70
84. Mazière C, Alimardani G, Dantin F, Dubois F, Conte MA, Mazière JC. Oxidized LDL activates STAT1 and STAT3 transcription factors: possible involvement of reactive oxygen species. Febs Letters. 1999;448:49-52
85. Li L, Shaw PE. A STAT3 dimer formed by inter-chain disulphide bridging during oxidative stress. Biochemical & Biophysical Research Communications. 2004;322:1005-11
86. Butturini E, Darra E, Chiavegato G, Cellini B, Cozzolino F, Monti M. et al. S-Glutathionylation at Cys328 and Cys542 impairs STAT3 phosphorylation. Acs Chemical Biology. 2014;9:1885-93
87. Cheruku HR, Mohamedali A, Cantor DI, Tan SH, Nice EC, Baker MS. Transforming growth factor-β, MAPK and Wnt signaling interactions in colorectal cancer. Eupa Open Proteomics. 2015;136:104-15
88. Torres M. Mitogen-activated protein kinase pathways in redox signaling. Frontiers in Bioscience. 2003;8:d369-91
89. Maillet A, Pervaiz S. Redox regulation of p53, redox effectors regulated by p53: a subtle balance. Antioxidants & Redox Signaling. 2012;16:1285-94
90. Torres M, Forman HJ. Redox signaling and the MAP kinase pathways. BioFactors. 2003;17:287-96
91. Wu RF, Terada LS. Ras and Nox: Linked signaling networks? Free Radical Biology & Medicine. 2009;47:1276-81
92. Lee JS, Kim SY, Kwon CH, Kim YK. EGFR-dependent ERK activation triggers hydrogen peroxide-induced apoptosis in OK renal epithelial cells. Archives of Toxicology. 2006;80:337-46
93. Templeton DJ, Aye MS, Rady J, Xu F, Cross JV. Purification of reversibly oxidized proteins (PROP) reveals a redox switch controlling p38 MAP kinase activity. Plos One. 2010;5:e15012
94. Gupta A, Rosenberger SF, Bowden GT. Increased ROS levels contribute to elevated transcription factor and MAP kinase activities in malignantly progressed mouse keratinocyte cell lines. Carcinogenesis. 1999;20:2063-73
95. Pelicci PG, Dalton P, Giorgio M. The Other Face of ROS: a Driver of Stem Cell Expansion in Colorectal Cancer. Cell Stem Cell. 2013;12:635-6
96. Wang L, Hitron JA, Wise JTF, Son YO, Roy RV, Kim D. et al. Ethanol enhances arsenic-induced cyclooxygenase-2 expression via both NFAT and NF-κB signalings in colorectal cancer cells. Toxicology & Applied Pharmacology. 2015;288:232-9
97. Liu B, Yuan B, Zhang L, Mu W, Wang C. ROS/p38/p53/Puma signaling pathway is involved in emodin-induced apoptosis of human colorectal cancer cells. International Journal of Clinical & Experimental Medicine. 2015;8:15413-22
98. Viennois E, Chen F, Merlin D. NF-κB pathway in colitis-associated cancers. Translational Gastrointestinal Cancer. 2013;2:21-9
99. Hayden MS, Ghosh S. Shared Principles in NF-κB Signaling. Cell. 2008;132:344-62
100. Loukili N, Rosenblattvelin N, Rolli J, Levrand S, Feihl F, Waeber B. et al. Oxidants positively or negatively regulate nuclear factor kappaB in a context-dependent manner. Journal of Biological Chemistry. 2010;285:15746-52
101. Vazquez A, Bond EE, Levine AJ, Bond GL. The genetics of the p53 pathway, apoptosis and cancer therapy. Nature Reviews Drug Discovery. 2008;7:979-87
102. Wu HH, Yuan YC, Momand J, Sherman M. Direct redox modulation of p53 protein: potential sources of redox control and potential outcomes. Gene Therapy & Molecular Biology. 1999
103. Sayin V. Pathophysiological impact of targeting the ROS-p53 axis. Gothenburg, Sweden: Ineko AB Publisher. 2014
104. Scotcher Jenna. Study of the molecular details of p53 redox-regulation using Fourier transform ion cyclotron resonance mass spectrometry. Journal of Engineering & Applied Sciences. 2011:6
105. Chipuk JE, Kuwana T, Bouchier-Hayes L, Droin NM, Newmeyer DD, Schuler M. et al. Direct Activation of Bax by p53 Mediates Mitochondrial Membrane Permeabilization and Apoptosis. Science. 2004;303:1010-4
106. Shen Y, White E. p53-Dependent apoptosis pathways. Advances in Cancer Research. 2001;82:55-84
107. Giatromanolaki A, Sivridis E, Stathopoulos G, Fountzilas G, Kalofonos H, Tsamandas A. et al. Bax protein expression in colorectal cancer: association with p53, bcl-2 and patterns of relapse. Anticancer Research. 2001;21:253-9
108. Richie-Jp J, Komninou D. Induction of colon tumorigenesis by glutathione depletion in p53-knock-out mice. International Journal of Oncology. 2007;30:1539-43 (5)
109. Schmitz KJ, Müller CI, Reis H, Alakus H, Winde G, Baba HA. et al. Combined analysis of hypoxia-inducible factor 1 alpha and metallothionein indicates an aggressive subtype of colorectal carcinoma. International Journal of Colorectal Disease. 2009;24:1287-96
110. Haddad JJ, Land SC. A non-hypoxic, ROS-sensitive pathway mediates TNF-α-dependent regulation of HIF-1α. Febs Letters. 2001;505:269-74
111. Yasinska IM, Sumbayev VV. S -nitrosation of Cys-800 of HIF-1α protein activates its interaction with p300 and stimulates its transcriptional activity. Febs Letters. 2003;549:105-9
112. Lisa A. Palmer AD, Preeti Chhabra, Mary Lynn Sheram, Victor E. Laubach, Molly Z. Karlinsey, Michael S. Forbes, Timothy Macdonald, Benjamin Gaston. S-nitrosothiols signal hypoxia-mimetic vascular pathology. Journal of Clinical Investigation. 2007;117:2592-601
113. Zhang J, Zhu L, Fang J, Ge Z, Li X. LRG1 modulates epithelial-mesenchymal transition and angiogenesis in colorectal cancer via HIF-1α activation. Journal of Experimental & Clinical Cancer Research Cr. 2016;35:1-11
114. Sporn MB, Liby KT. NRF2 and cancer: the good, the bad and the importance of context. Nature Reviews Cancer. 2012;12:564-71
115. Thimmulappa RK, Mai KH, Srisuma S, Kensler TW, Yamamoto M, Biswal S. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Research. 2002;62:5196-203
116. Jaramillo MC, Zhang DD. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes & Development. 2013;27:2179-91
117. Chang LC, Fan CW, Tseng WK, Chen JR, Chein HP, Hwang CC. et al. Immunohistochemical study of the Nrf2 pathway in colorectal cancer: Nrf2 expression is closely correlated to Keap1 in the tumor and Bach1 in the normal tissue. Applied Immunohistochemistry & Molecular Morphology Aimm. 2013;21:511-7
118. Leak RK. Heat shock proteins in neurodegenerative disorders and aging. Journal of Cell Communication & Signaling. 2014;8:293-310
119. Buckley B, Li S. Keap1 modification and nuclear accumulation in response to S-nitrosocysteine. Free Radical Biology & Medicine. 2008;44:692-8
120. Kobayashi A, Kang MI, Okawa H, Ohtsuji M, Zenke Y, Chiba T. et al. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Molecular & Cellular Biology. 2004;24:7130-9
121. Bloom DA, Jaiswal AK. Phosphorylation of Nrf2 at Ser40 by protein kinase C in response to antioxidants leads to the release of Nrf2 from INrf2, but is not required for Nrf2 stab. Journal of Biological Chemistry. 2003;278:44675-82
122. Kim TH, Hur EG, Kang SJ, Kim JA, Thapa D, Lee YM. et al. NRF2 blockade suppresses colon tumor angiogenesis by inhibiting hypoxia-induced activation of HIF-1α. Cancer Research. 2011;71:2260-75
123. He X, Ma Q. NRF2 cysteine residues are critical for oxidant/electrophile-sensing, Kelch-like ECH-associated protein-1-dependent ubiquitination-proteasomal degradation, and transcription activation. Molecular Pharmacology. 2009;76:1265-78
124. Leinonen HM, Kansanen E, Pölönen P, Heinäniemi M, Levonen AL. Role of the Keap1-Nrf2 pathway in cancer. Advances in Cancer Research. 2014;122C:281-320
125. Singh A, Boldinadamsky S, Thimmulappa RK, Rath SK, Ashush H, Coulter J. et al. RNAi-mediated silencing of nuclear factor erythroid-2-related factor 2 gene expression in non-small cell lung cancer inhibits tumor growth and increases efficacy of chemotherapy. Cancer Research. 2008;68:7975-84
126. Jiang T, Chen N, Zhao F, Wang XJ, Kong B, Zheng W. et al. High levels of Nrf2 determine chemoresistance in type II endometrial cancer. Cancer Research. 2010;70:5486-96
127. Solis LM, Behrens C, Dong W, Suraokar M, Ozburn NC, Moran CA. et al. Nrf2 and Keap1 abnormalities in non-small cell lung carcinoma and association with clinicopathologic features. Clinical Cancer Research. 2010;16:3743-53
128. Ming Z, Fahl WE. Functional Characterization of Transcription Regulators That Interact with the Electrophile Response Element ☆. Biochemical & Biophysical Research Communications. 2001;289:212-9
129. Mitsuishi Y, Taguchi K, Kawatani Y, Shibata T, Nukiwa T, Aburatani H. et al. Nrf2 Redirects Glucose and Glutamine into Anabolic Pathways in Metabolic Reprogramming. Cancer Cell. 2012;22:66-79
130. Slaga TJ. Inhibition of the Induction of Cancer by Antioxidants. Advances in Experimental Medicine & Biology. 1995;369:167-74
131. Blot WJ, Li JY, Taylor PR, Guo W, Dawsey S, Wang GQ. et al. Nutrition intervention trials in Linxian, China: supplementation with specific vitamin/mineral combinations, cancer incidence, and disease-specific mortality in the general population. Jnci Journal of the National Cancer Institute. 1993;85:340-6
132. Wei MY, Garland CF, Gorham ED, Mohr SB, Giovannucci E. Vitamin D and prevention of colorectal adenoma: a meta-analysis. Cancer Epidemiology Biomarkers & Prevention. 2008;17:2958-69
133. Liu J, Du J, Zhang Y, Sun W, Smith BJ, Oberley LW. et al. Suppression of the malignant phenotype in pancreatic cancer by overexpression of phospholipid hydroperoxide glutathione peroxidase. Human Gene Therapy. 2006;17:105-16
134. Chinery R, Beauchamp R, Shyr Y, Kirkland S, Coffey R, Morrow J. Antioxidants reduce cyclooxygenase-2 expression, prostaglandin production, and proliferation in colorectal cancer cells. Cancer Research. 1998;58:2323-7
135. Yang Y, Karakhanova S, Hartwig W, D'Haese JG, Philippov PP, Werner J. et al. Mitochondria and Mitochondrial ROS in Cancer: Novel Targets for Anticancer Therapy. Journal of Cellular Physiology. 2016
136. Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nature Reviews Drug Discovery. 2013;12:931-47
137. Ding Y, Wang H, Niu J, Luo M, Gou Y, Miao L. et al. Induction of ROS Overload by Alantolactone Prompts Oxidative DNA Damage and Apoptosis in Colorectal Cancer Cells. International Journal of Molecular Sciences. 2016:17
138. Huang Y, Yu H, Lei H, Xie C, Zhong Y. Matrix metalloproteinase 7 is a useful marker for 5-fluorouracil-based adjuvant chemotherapy in stage II and stage III colorectal cancer patients. Medical Oncology. 2014;31:1-8
139. Jonge RD, Maftouh M, Griffioen P. Polymorphisms correlated with the clinical outcome of locally advanced or metastatic colorectal cancer patients treated with ALIRI vs. FOLFIRI. Pteridines. 2013;24:69-79
140. Iwagaki H, Fuchimoto S, Shiiki S, Miyake M, Orita K. A mechanism for the potentiation of the cytotoxic effects of antimetabolites drugs (FT-207, 5FU) by hyperthermia. Research Communications in Chemical Pathology & Pharmacology. 1988;62:353-60
141. Fan C, Chen J, Wang Y, Wong YS, Zhang Y, Zheng W. et al. Selenocystine potentiates cancer cell apoptosis induced by 5-fluorouracil by triggering ROS-mediated DNA damage and inactivation of ERK pathway. Free Radical Biology & Medicine. 2013;65:305-16
142. Hwang PM, Bunz F, Yu J, Rago C, Chan TA, Murphy MP. et al. Ferredoxin reductase affects p53-dependent, 5-fluorouracil-induced apoptosis in colorectal cancer cells. Nat Med. 2001;7:1111-7
143. Longley DB, Harkin DP, Johnston PG. 5-fluorouracil: mechanisms of action and clinical strategies. Nature Reviews Cancer. 2003;3:330-8
144. Kallio A, Zheng A, Dahllund J, Heiskanen KM, Härkönen P. Role of mitochondria in tamoxifen-induced rapid death of MCF-7 breast cancer cells. Apoptosis An International Journal on Programmed Cell Death. 2005;10:1395-410
145. Kuruppu D, Christophi C, Bertram JF, O'Brien PE. Tamoxifen inhibits colorectal cancer metastases in the liver: A study in a murine model. Journal of Gastroenterology & Hepatology. 1998;13:521-7
146. Li-ZongShen Yi-BingHua, Xue-MingYu QingXu, TaoChen Jian-HuaWang. et al. Tamoxifen can reverse multidrug resistance of colorectal carcinoma in vivo. World Journal of Gastroenterology. 2005;11:1060-4
147. Lee YH, Kang BS, Bae YS. Premature senescence in human breast cancer and colon cancer cells by tamoxifen-mediated reactive oxygen species generation. Life Sciences. 2014;97:116-22
148. Arber N, Eagle CJ, Spicak J, Rácz I, Dite P, Hajer J. et al. Celecoxib for the prevention of colorectal adenomatous polyps. New England Journal of Medicine. 2006;355:885-95
149. Abdelrahim M, Safe S. Cyclooxygenase-2 inhibitors decrease vascular endothelial growth factor expression in colon cancer cells by enhanced degradation of Sp1 and Sp4 proteins. Molecular Pharmacology. 2005;68:317-29
150. Xu B, Wang Y, Yang J, Zhang Z, Zhang Y, Du H. Celecoxib induces apoptosis but up-regulates VEGF via endoplasmic reticulum stress in human colorectal cancer in vitro and in vivo. Cancer Chemotherapy & Pharmacology. 2016:1-10
151. Chen P, Wang H, Duan Z, Zou JX, Chen H, He W. et al. Estrogen-Related Receptor Alpha Confers Methotrexate Resistance via Attenuation of Reactive Oxygen Species Production and P53 Mediated Apoptosis in Osteosarcoma Cells. Biomed Research International. 2014;2014:223-32
152. Arifa RDN, Paula TPD, Madeira MFM, Lima RL, Garcia ZM, Ávila TV. et al. The reduction of oxidative stress by nanocomposite Fullerol decreases mucositis severity and reverts leukopenia induced by Irinotecan. Pharmacological Research. 2016;107:102-10
153. Yoshida T, Goto S, Kawakatsu M, Urata Y, Li TS. Mitochondrial dysfunction, a probable cause of persistent oxidative stress after exposure to ionizing radiation. Free Radical Research. 2012;46:147-53
154. Belfi CA, Chatterjee S, Gosky DM, Berger SJ, Berger NA. Increased sensitivity of human colon cancer cells to DNA cross-linking agents after GRP78 up-regulation. Biochemical & Biophysical Research Communications. 1999;257:361-8
155. Watson J. Oxidants,antioxidants and the current incurability of metastatic cancers. Open Biology. 2013;3:806-11
156. Policastro LL, Ibañez IL, Notcovich C, Duran HA, Podhajcer OL. The tumor microenvironment: characterization, redox considerations, and novel approaches for reactive oxygen species-targeted gene therapy. Antioxidants & Redox Signaling. 2013;19:854-95
157. Sosa V, Moliné T, Somoza R, Paciucci R, Kondoh H, Lleonart ME. Oxidative stress and cancer: an overview. Ageing Research Reviews. 2013;12:376-90
158. Ahmad P, Jaleel CA, Salem MA, Nabi G, Sharma S. Roles of enzymatic and nonenzymatic antioxidants in plants during abiotic stress. Critical Reviews in Biotechnology. 2010;30:161-75
159. Shah VP, Chegini HA, Vishneski SR, Weatherman RV, Blackmore PF, Dobrydneva Y. Tamoxifen promotes superoxide production in platelets by activation of PI3-Kinase and NADPH oxidase pathways. Thrombosis Research. 2012;129:36-42
160. Chibber S, Hassan I, Farhan M, Naseem I. In vitro pro-oxidant action of Methotrexate in presence of white light. Journal of Photochemistry & Photobiology B Biology. 2011;104:387-93
161. Halliwell B. Oxidative stress and cancer: have we moved forward? Biochemical Journal. 2007;401:1-11
162. Arnold M, Bissinger R, Lang F. Mitoxantrone-induced suicidal erythrocyte death. Cellular Physiology & Biochemistry International Journal of Experimental Cellular Physiology Biochemistry & Pharmacology. 2014;34:1756-67
163. † WL, † JG, Qi J, Zeng XN, Ji J, Chen ZZ. et al. Lentinan exerts synergistic apoptotic effects with paclitaxel in A549 cells via activating ROS-TXNIP-NLRP3 inflammasome. Journal of Cellular & Molecular Medicine. 2015;19:1949-55
164. Wang J, Tan X, Yang Q, Zeng X, Zhou Y, Luo W. et al. Inhibition of autophagy promotes apoptosis and enhances anticancer efficacy of adriamycin via augmented ROS generation in prostate cancer cells. International Journal of Biochemistry & Cell Biology. 2016;77:80-90
165. Tibullo D, Barbagallo I, Giallongo C, La CP, Parrinello N, Vanella L. et al. Nuclear translocation of heme oxygenase-1 confers resistance to imatinib in chronic myeloid leukemia cells. Current Pharmaceutical Design. 2013;19:2765-70
166. Jayasooriya RGPT, Choi YH, Jin WH, Kim GY. Camptothecin sensitizes human hepatoma Hep3B cells to TRAIL-mediated apoptosis via ROS-dependent death receptor 5 upregulation with the involvement of MAPKs. Environmental Toxicology & Pharmacology. 2014;38:959-67
167. Brozovic A, Vuković L, Polančac DS, Arany I, Köberle B, Fritz G. et al. Endoplasmic reticulum stress is involved in the response of human laryngeal carcinoma cells to Carboplatin but is absent in Carboplatin-resistant cells. Plos One. 2013;8:e76397-e
168. Ryung-Ah Hyun-Ah, Bo-Young Kang, Kwang-Ho. Hemoglobin induces colon cancer cell proliferation by release of reactive oxygen species. World Journal of Gastroenterology. 2006;12:5644-50
169. Marullo R, Werner E, Degtyareva N, Moore B, Altavilla G, Ramalingam SS. et al. Cisplatin induces a mitochondrial-ROS response that contributes to cytotoxicity depending on mitochondrial redox status and bioenergetic functions. Plos One. 2013;8:e81162
170. Zhang J, Jiang H, Li X, Hu J, Li L, Yang M. et al. Antitumor effect of manumycin on colorectal cancer cells by increasing the reactive oxygen species production and blocking PI3K-AKT pathway. Oncotargets & Therapy. 2016;9:2885-95
171. Hoyt MT, Palchaudhuri R, Hergenrother PJ. Cribrostatin 6 induces death in cancer cells through a reactive oxygen species (ROS)-mediated mechanism. Investigational New Drugs. 2010;29:562-73
Author contact
![]() Corresponding authors: Dr. Yunlong Lei, Department of Biochemistry and Molecular Biology,ChongQing Medical University, 1# Yixueyuan Road, Yuzhong District, ChongQing, 400016, China. Email: leiyunlongcom Phone: +86 13648323625 Prof. Youquan Bu, Department of Biochemistry and Molecular Biology, and Molecular Medicine and Cancer Research Center, Chongqing Medical University, Chongqing 400016, P. R. China. Email: buyqcncom Phone: +86-23-68485991
Corresponding authors: Dr. Yunlong Lei, Department of Biochemistry and Molecular Biology,ChongQing Medical University, 1# Yixueyuan Road, Yuzhong District, ChongQing, 400016, China. Email: leiyunlongcom Phone: +86 13648323625 Prof. Youquan Bu, Department of Biochemistry and Molecular Biology, and Molecular Medicine and Cancer Research Center, Chongqing Medical University, Chongqing 400016, P. R. China. Email: buyqcncom Phone: +86-23-68485991