Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(7):1129-1136. doi:10.7150/jca.16201 This issue Cite
Research Paper
Knockdown of COPS3 Inhibits Lung Cancer Tumor Growth in Nude Mice by Blocking Cell Cycle Progression
Jianan Pang1, Xu Yan2, He Cao1, Lei Qian1, Hua He1, Huimin Tian1, Fujun Han1, Guanjun Wang1, Xiao Chen1, Yuguang Zhao1, Ji-Fan Hu1, 3 ![]() , Jiuwei Cui1
, Jiuwei Cui1 ![]()
1. Cancer Center, the First Hospital of Jilin University, No. 71. Xinmin Street, Changchun, 130021, China;
2. Pathological diagnostic Center, the First Hospital of Jilin University, No.71.Xinmin Street, Changchun, 130021, China;
3. Stanford University Medical School, VA Palo Alto Health Care System, Palo Alto, CA 94304, USA.
Received 2016-5-17; Accepted 2016-9-18; Published 2017-4-9
Citation:
Pang J, Yan X, Cao H, Qian L, He H, Tian H, Han F, Wang G, Chen X, Zhao Y, Hu JF, Cui J. Knockdown of COPS3 Inhibits Lung Cancer Tumor Growth in Nude Mice by Blocking Cell Cycle Progression. J Cancer 2017; 8(7):1129-1136. doi:10.7150/jca.16201. https://www.jcancer.org/v08p1129.htm
Other stylesAbstract
COPS3 encodes the third subunit of the COP9 signalosome and its aberrant expression is associated with many RITE (“Region of Increased Tumor Expression”) genes in lung cancer tissues. To elucidate the specific role of COPS3 in lung cancer, we examined its expression in lung cancer tissues by immunohistochemical staining. We found that COPS3 was overexpressed in most of the lung cancer samples examined, particularly in small cell carcinoma and squamous cell carcinoma. The expression of COPS3 protein was positively correlated with the level of Ki-67 cell proliferation index (p=0.001) and negatively related to the degree of tumor differentiation (p=0.012). In a xenograft tumor model in nude mice, shRNA-knockdown of COPS3 significantly reduced tumor growth. In lung adenocarcinoma A549 cells, shRNA-knockdown of COPS3 induced cell cycle arrest at G0/G1 phase by upregulating the cell cycle regulator protein P21 and downregulating cyclin B1 and CDK4. These data suggest that COPS3 may promote tumor growth by regulating cell-cycle associated proteins.
Keywords: Lung cancer, COPS3, gene expression, xenograft tumor, cell cycle.
Introduction
Lung cancer is the most common cause of cancer-related mortality worldwide [1]. The overall 5-year survival rate of lung cancer is dismal as cancer cases are usually diagnosed at later stages [2]. Lung cancer exhibits the following two predominant pathological types: non-small cell lung cancer (NSCLC) and small cell lung cancer (SCLC). Non-small cell lung cancer accounts for approximately 85% of all lung cancer cases, and adenocarcinoma is the most common subtype of lung cancer. Despite advances in therapeutic methods, the survival rates of lung cancer remain low. Therefore, identification of novel molecular abnormalities that contribute to lung cancer pathogenesis, patient outcome and therapeutic targets, is urgently needed to improve individualized treatment.
The COPS3 (constitutive photomorphogenic homolog subunit 3 of COP9) gene is located on chromosome 17p11.2-12 in humans and encodes the third subunit of the eight-subunit COP9 signalosome (CSN) complex, which was initially identified as a negative regulator of constitutive photomorphogenesis in Arabidopsis thaliana [3]. The COP9 signalosome is an evolutionarily conserved protein complex that exhibits protein kinase and deneddylase activities. The protein encoded by COPS3 acts as a docking site for the signalosome, thereby mediating the phosphorylation of a wide variety of important regulators involved in signal transduction, including I kappa-B alpha, p105, ITPK1, IRF8/ICSBP, c-Jun, p53, and p27 [4].
COPS3 plays an important role in both the tumorigenesis and progression of osteosarcoma and hepatocellular carcinoma [5-7]. The overexpression of COPS3 has been found to be correlated with proliferation, metastasis and apoptosis [7-9]. In addition, clinical data indicate that the overexpression of COPS3 significantly correlates with poor outcome in osteosarcoma patients [10]. Through constructing a transcriptome map of human lung tissue, Zhou and colleagues have discovered the amplification of 17p11 in lung cancer [11], suggesting that COPS3 might contribute to the development of lung cancer.
In a previous cell study, we have demonstrated that COPS3 is highly expressed in lung adenocarcinoma cell lines [11]. Knockdown of COPS3 significantly inhibits tumor cell proliferation in this in vitro model. However, the specific role of COPS3 in vivo remains to be defined. In this extended study, we focused on the role of COPS3 in an in vivo tumor xenograft model. After RNA interference-mediated knockdown of COPS3, we examined the development of lung cancer in nude mice. We further evaluated the correlation between COPS3 expression and clinicopathological features in a cohort of cancer patients.
Materials and Methods
Collection of lung cancer samples
A total of 120 formalin-fixed and paraffin-embedded human lung carcinoma samples were collected for COPS3 expression analysis, including 37 cases of squamous carcinoma, 40 cases of adenocarcinoma, and 43 cases of small cell lung carcinoma. All cases were histopathologically and clinically diagnosed at the First Hospital of Jilin University from 2010 to 2015. All samples were reviewed retrospectively and diagnosed according to the 2015 World Health Organization (WHO) Classification of Tumors of the Lung, Pleura, Thymus and Heart [12]. The study protocol was approved by the Human Medical Ethical Review Committee of the hospital at Jilin University. Informed consent was obtained from each cancer patient at the beginning of the study.
Immunohistochemistry of COPS3 expression
Paraffin-embedded human lung cancer tissues were fixed in freshly prepared 10% neutral buffered formalin, embedded in paraffin wax, and cut into 4-μm sections. Tissues at least 3 cm away from the cancer mass were collected as the adjacent “normal lung tissue”. After being baked at 65°C for 1 hour, the sections were deparaffinized and rehydrated. Endogenous peroxidase activity was blocked by 3% hydrogen peroxide for 10 minutes at room temperature. After blocking with 5% bovine serum albumin (BSA), the sections were incubated with anti-COPS3 antibody (2.5 µg/ml) at 37°C for 2 hours, and this was followed by incubation with a second antibody from the MaxVisionTM kit (Maxim, China) for 15 minutes at room temperature. Sections were counterstained with hematoxylin.
The immunostaining was scored blindly by two independent skilled pathologists, based on the intensity and localization of staining. The proportion of tumor cells was scored as follows: 0 (no positive tumor cells), 1 (≤10% positive tumor cells), 2(10-50% positive tumor cells), and 3 (≥50% positive tumor cells). The staining intensity was classified according to the following criteria: 0 (negative staining), 1 (weak staining: weak yellow), 2 (moderate staining: yellow-brown), and 3 (strong staining: brown). For semi-quantitative analysis, the final score of each case was calculated as a percentage of positive cells multiplied by staining intensity (IHC score = Σ(intensity×% positive cells). An optimal cutoff value was identified as follows: a final score of ≥4 was defined as the group with high COPS3 expression, whereas a score of ≤3 was defined as the group with low COPS3 expression.
Cell culture study
The lung adenocarcinoma cell line (A549) and the human embryonic kidney cell line (293T) were purchased from American Type Culture Collection (ATCC, VA). The cell lines were maintained in DMEM supplemented with 10% FBS at 37°C in a humidified incubator with constant air flow of 5% CO2.
Lentivirus packaging and transfection of A549 cells
The short hairpin RNA (shRNA) nucleotide sequences were inserted into the pFH1UGW-GFP vector (Hollybio, Shanghai, China) and were confirmed by DNA sequencing. A random shRNA sequence (shCTRL) was served as a control (Hollybio, Shanghai, China). Lentiviruses were generated via the transfection of 293T cells at 70-80% confluence. Two hours before transfection, the FBS-containing medium was removed and replaced with 200 µl of Opti-MEM containing 20 µg pFH1UGW-GFP-shCOPS3 or -shCTRL, 15 µg pCMV vector and 10 µg pVSVG plasmid prepared with 15 µl of Lipofectamine 2000. After incubation for 8 h, the medium was replaced with 10 ml DMEM supplemented with 10% FBS. The supernatant was collected after 48 hours of transfection, and lentiviral particles were harvested by centrifugation (4,000 g) at 4°C for 10 min. The collected viral particles were filtered through a 45-μm filter, and the filtrate was centrifuged (4, 000 g at 4°C) for 15 min to collect the concentrated lentiviruses.
A549 cells were seeded on a 6-well plate at a concentration of 5×104 cells/well. After 24 hours of incubation, the culture medium was replaced with Opti-MEM medium containing the appropriate amount of lentiviruses. Cells were transfected with the COPS3 shRNA-expressing lentiviruses in the sh-COPS3 group and the control shRNA-expressing lentiviruses in the sh-CTRL group, respectively. Twenty-four hours after transfection, the medium was replaced with fresh medium, and the cells were further incubated for an additional 72 hours. Successful transfection was assessed by fluorescence microscope to examine the expression of green fluorescent protein.
Real-time PCR analysis
RNA was extracted from virally infected lung cancer cells on day 5. Cells were lysed using TRIzol reagent (Invitrogen, CA), and total RNA was extracted from the lysate following the manufacturer's protocol. cDNA was synthesized from the extracted RNA using a Promega M-MLV cDNA synthesis kit, according to the manufacturer's instruction. Real time PCR analysis was performed to evaluate COPS3 mRNA expression in sh-COPS3 and sh-CTRL infected cells. β-ACTIN was used as the internal reference control for PCR. Relative gene expression levels were calculated using 2-△△CT analysis, as previously described [13, 14] .
Western blotting
The expression of COPS3 protein, cyclin B1, cyclin D1, CDK4 and CDK2 was detected by western blotting. Briefly, cells were infected with sh-COPS3 and sh-CTRL for 5 days and were lysed with lysis buffer (0.15 M NaCl, 5 mM EDTA, pH 8, 10 mM Tris-Cl, PH 7.5, 1% Triton-X100). Protein contents were measured using the Lowry method. The protein concentration of each sample was adjusted to 2 µg/µl, and 20-µg protein samples were electrophoresed on 10% SDS-PAGE and transferred onto polyvinylidenedifluoride membranes. Protein expression was detected with the indicated antibodies. Band densities were quantified with the Image J program[15].
Cell cycle analysis
Cell cycle was analyzed by flow cytometric analysis after popidium iodide (PI) staining [16, 17]. Infected cells were seeded onto 6-well plates at a concentration of 1×106/well and incubated for 24 hours. Cells were collected by trypsinization, washed with ice cold PBS, and then fixed by resuspension in 0.7 ml of 70% ethanol for 30 min at 4°C. The ethanol was removed by centrifugation and a PI solution containing 10 µg/ml of DNase-free RNase A was added to stain the cells for 30 min. Cell suspensions were filtered through a 50-µm nylon mesh and then analyzed with flow cytometry (FAS Calibur, BD Biosciences, CA).
Tumorigenesis in nude mice
The role of COPS3 in lung cancer tumorigenesis was examined in BALB/c nude mice (8-12 weeks old, 22±3 g). Animals were purchased from the Experimental Animal Center of College of Basic Medical Sciences, Jilin University and were randomized into two groups (20 mice in each group). A549 cell suspensions (0.25 ml) infected with sh-COPS3 or sh-CTRL (5×106 cells/ mouse) were subcutaneously injected into the armpits or the backs of the mice. Tumor sizes were recorded on days 10, 17, 24 and 31 as maximum diameter (A, mm) and vertical short diameter (B, mm). The tumor volumes were calculated using the formula V (mm3) = ½(AxB2) as previously described [18]. After 31 days of observation, the mice were sacrificed, and the xenograft tumors were removed and weighed for analysis. The animal protocol was approved by the Animal Care and Use Committee of the Fist Affiliated Hospital in Jilin University.
Statistical analysis
All data were statistically analyzed using the SPSS 16.0 software package. Correlation analysis between COPS3 expression and clinicopathological data was performed with Fisher's exact probability test. After variables were confirmed to be related to COPS3 levels, a Chi-Square test among the three groups was performed. The correlation between Ki-67 indices and COPS3 scores was calculated by Spearman's rank correlation test. P values <0.05 were considered to be statistically significant.
Results
COPS3 expression is correlated with clinicopathological characteristics in lung cancer patients
The clinicopathological characteristics and COPS3 expression levels in patients with lung cancer are summarized in Table 1. We found that the expression level of COPS3 protein was significantly associated with the histological type (P =0.001), with higher COPS3 expression in small cell carcinoma (86%) and squamous cell carcinoma (86.5%) than that in adenocarcinoma (52.5%). We found that COPS3 expression was also significantly higher in the low-differentiation tumors (86%) than that in the high-differentiation tumors (50%) (P =0.012). Interestingly, COPS3 was also positively associated with Ki-67 index in lung cancer patients (P =0.001).
Overall, COPS3 protein was more abundant in squamous carcinoma and small cell carcinoma than that in adenocarcinoma. However, no significant association of COPS3 expression was found with age, tumor size, N classification, status of distant metastases (M classification), or clinical staging in 120 cancer cases.
COPS3 is upregulated in lung cancer tissues
Figure 1 shows the representative images of the overexpressed COPS3 in lung cancer tissues. We found that COPS3 was overexpressed in cancer tissues (red arrow) compared with adjacent normal lung tissues (right panels). In agreement with the data in Table 1, COPS3 expression was significantly higher in small cell lung carcinoma and squamous carcinomas than that in adenocarcinoma.
Figure 1
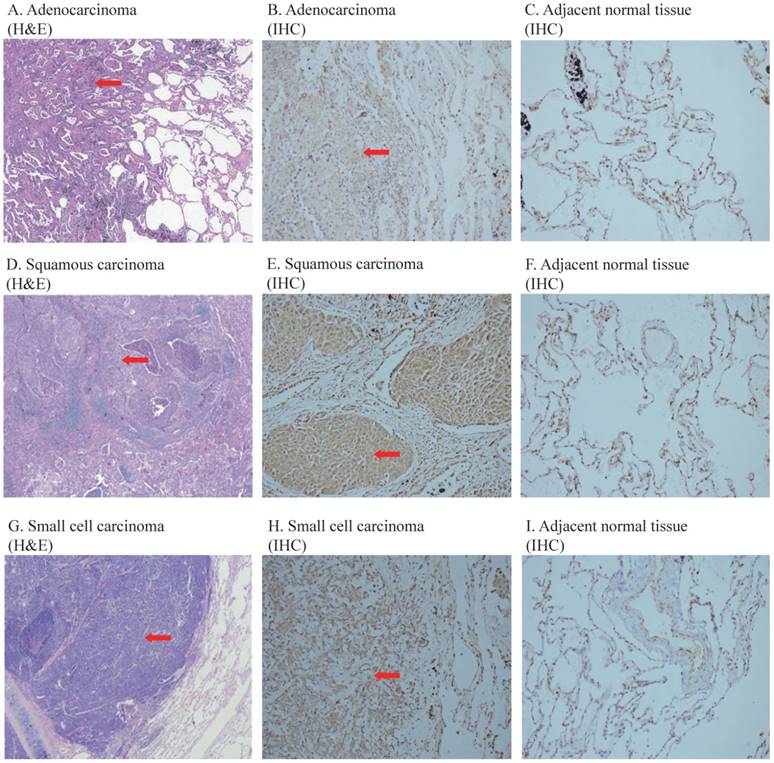
Representative images of COPS3 immunohistochemical staining in lung cancer tissues and adjacent normal lung tissues. A-C. H&E and immunohistochemical (IHC) staining of adenocarcinoma and adjacent normal lung tissues; D-F: H&E and immunohistochemical (IHC) staining of squamous carcinoma and adjacent normal lung tissues; G-I: H&E and immunohistochemical (IHC) staining of SCLC and adjacent normal lung tissues. The images of H&E staining were taken at 40× magnification, and the images of IHC at 200× magnification. Tissues at least 3 cm away from the cancer mass were collected as adjacent “normal lung tissue”. Note that the COPS3 immunostaining was more immunoreactive in cancer tissues than that in adjacent normal lung tissues.
Figure 2
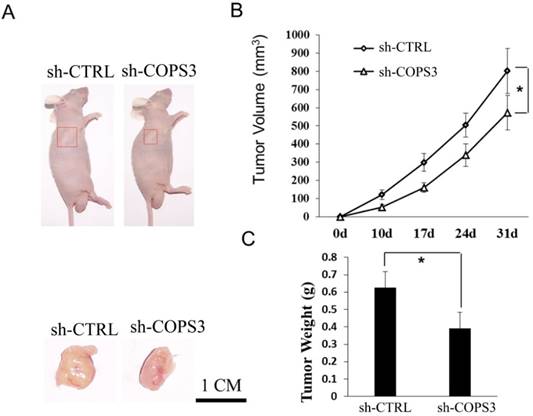
COPS3 shRNA knockdown inhibits tumor growth in nude mice. A. Representative images of xenograft tumors in nude mice treated with sh-COPS3 and sh-CTRL. Red boxes indicate the location and size of the tumors. B. Reduction of tumor growth in nude mice treated with sh-COPS3. *P<0.05 compared with the sh-CRTL control. C. Tumor weights in the nude mice treated with sh-COPS3. *P<0.05 compared with the sh-CRTL control.
COPS3 knockdown inhibits tumor growth in nude mice
In a previous in vitro study, we reported that COPS3 promoted cell proliferation in two lung cancer cell lines, including A549 (adenocarcinoma) and 95D (metastatic lung cancer) [11]. In this extended study, we focused on the role of COPS3 in tumorigenesis in nude mice. For this, adenocarcinoma A549 cells were used to establish the xenograft tumor model.
In this in vivo study, we found that tumor volume was remarkably smaller in the sh-COPS3 group than that in the sh-CTRL control group (Figs.2A and 2B, P<0.05). Similarly, the mice in the sh-COPS3 group showed a significant reduction in tumor weight, as compared with those in the sh-CTRL controls (Fig.2C, P<0.05).
Knockdown of COPS3 expression induces cell-cycle arrest at G0/G1 phase
To examine the mechanism underlying the role of COPS3 in tumorigenesis, we used flow cytometry (FACS) to repeat cell cycle analysis as previously reported in these cells [11]. We found that lung cancer cells treated with sh-COPS3 were predominantly arrested at G0/G1 phase (Fig. 3A). Cell cycle quantification showed that compared with the sh-CTRL control, knockdown of COPS3 with shRNA significantly blocked A549 lung cancer cells at G0/G1 phase (Fig. 3B, P<0.01).
Table 1
Correlation between the clinicopathologic characteristics and expression of COPS3 protein in lung cancer
| Characteristics | n | COPS3 (%) | P | ||
|---|---|---|---|---|---|
| High expression | Low expression | ||||
| Gender | |||||
| Male | 84 | 61 (72.6%) | 23 (27.4%) | ||
| Female | 36 | 29 (80.6%) | 7 (19.4%) | 0.491 | |
| Age(y) | ≥65 | 27 | 18 (66.7%) | 9 (33.3%) | |
| <65 | 93 | 72 (77.4%) | 21 (22.6%) | 0.313 | |
| Histological classification | |||||
| Small cell carcinoma | 43 | 37 (86.0%) | 6 (14.0%) | ||
| adenocarcinoma | 40 | 21 (52.5%) | 19 (47.5%) | ||
| squamous cell carcinoma | 37 | 32 (86.5%) | 5 (13.5%) | 0.001 | |
| Tumor size (cm) | |||||
| ≤3 | 59 | 44 (74.6%) | 15 (25.4%) | ||
| 4-7 | 57 | 43 (75.4%) | 14 (24.6%) | ||
| >7 | 4 | 3 (75%) | 1 (25%) | 1.000 | |
| Differentiation | |||||
| Low | 57 | 49 (86.0%) | 8 (14.0%) | ||
| Moderate | 54 | 36 (66.7%) | 18 (33.3%) | ||
| High | 8 | 4 (50.0%) | 4 (50.0%) | 0.012 | |
| N classification | |||||
| N0 | 68 | 50 (73.5%) | 18 (26.5%) | ||
| N1 | 32 | 26 (81.2%) | 6 (18.8%) | ||
| N2 | 19 | 14 (73.7%) | 5 (26.3%) | 0.762 | |
| Distant metastasis | |||||
| M0 | 114 | 85 (74.6%) | 29 (25.4%) | ||
| M1 | 2 | 2 (100.0%) | 0 (0.0%) | 1.000 | |
| Ki-67 index | |||||
| <50% | 52 | 31 (59.6%) | 21 (40.4%) | ||
| ≥50% | 68 | 59 (86.8%) | 9 (13.2%) | 0.001 | |
| Clinical stage | |||||
| Ⅰ | 56 | 42 (75.0%) | 14 (25. 0%) | ||
| Ⅱ | 43 | 34 (79.1%) | 9 (20.9%) | ||
| Ⅲ | 18 | 12 (66.7%) | 6 (33.3%) | ||
| Ⅳ | 2 | 2 (100.0%) | 0 (0.0%) | 0.708 | |
Figure 3
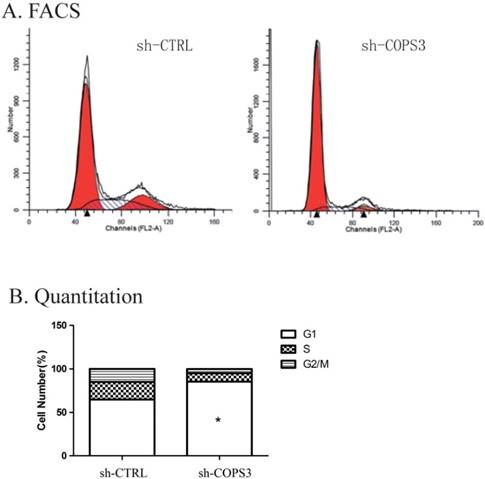
Down-regulation of COPS3 induces cell cycle arrest at G0/G1 phase. A. The cell cycle distribution of A549 cells as assessed by flow cytometry. B. G0/G1 phase blockage in sh-COPS3 A549 cells. Values were presented as the mean values ±standard deviation of three independent experiments. ** P<0.01 compared with the sh-CRTL control group.
COPS3 knockdown alters expression of the cell-cycle pathway proteins
To further investigate the underlying molecular mechanisms, several cell-cycle associated factors were quantified by Western blotting. In the sh-COPS3-infected A549 cells, the expression of P21 was significantly upregulated (Figs.4A-4F, P<0.05). In contrast, the knockdown of COPS3 dramatically reduced the expression of cyclin B1 and CDK4 (P<0.05). However, knockdown of COPS3 did not affect the expression of other two cell-cycle genes cyclin D1 and CDK2 in A549 cells.
Discussion
COPS3 has been reported to participate in the progression of several tumors [5-7, 19]. However, the role of COPS3 in human lung cancer remains to be defined. In this study, we examined the expression of COPS3 in a total of 120 lung cancer patients. By comparison with adjacent lung tissues, we demonstrate that COPS3 is highly expressed in lung cancer tissues, particularly in small cell carcinoma and squamous cell carcinoma. The upregulation of COPS3 was associated with poor differentiation. Therefore, our data suggest that COPS3 might be another molecular marker in lung cancer, particularly small cell carcinoma and squamous cell carcinomas.
Previously, we have shown that COPS3 plays an important role in stimulating cell proliferation in lung cancer cells [19]. In this cohort study, we also found that the level of COPS3 expression was positively correlated with Ki-67 index in lung cancer patients. The Ki-67 protein is a well-known cellular marker for proliferation [20]. Thus, COPS3 might be an important regulator in cellular proliferation. To further demonstrate the role of COPS3 in proliferation in vivo, we used a nude mouse model that was xenografted with A549 cells in which COPS3 was knocked down by shRNA. Our results show that knockdown of COPS3 expression dramatically reduces both tumor size and weight. Our in vivo tumor study further confirms the hypothesis that COPS3 promotes tumorigenesis by regulating proliferation. Most published reports have focused on COPS3-mediated mechanisms of metastasis and apoptosis [7, 9]. However, we did not find a correlation between COPS3 and tumor metastasis in our model.
The cell cycle is governed by two specific checkpoints: G1-S and G2-M phases. Cell cycle progression is regulated by Cyclins and Cyclin-dependent kinases (CDKs) [21]. Transition from G1 to S phase requires the activation of Cyclin D/CDK4 and Cyclin E/CDK2 complexes [22, 23]. CDK4 is often overexpressed in lung cancer, particularly in adenocarcinoma, and is a significant predictor of poor prognosis for lung cancer patients [24]. In this study, A549 cells treated with sh-COPS3 were predominantly arrested at G0/G1 phase. By examining the CDKs, we found that the sh-COPS3 treatment induced significant downregulation of CDK4 (Fig. 4), with simultaneous G0/G1 arrest in the treated cells. However, CDK2 was not significantly affected by sh-COPS3 knockdown. Our data therefore suggest that the G0/G1 cell cycle arrest induced by COPS3 knockdown was primarily mediated through the down-regulation of CDK4.
Figure 4
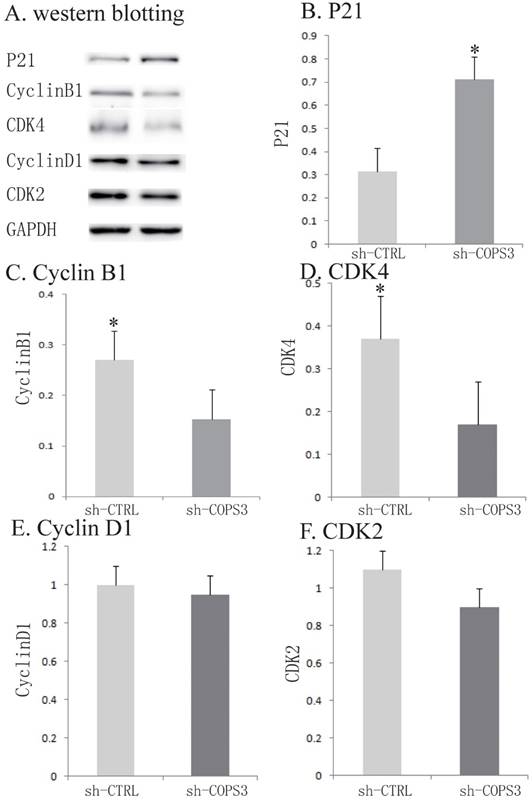
Activation of cell cycle related proteins by COPS3 knockdown. A. Activation of cell cycle related proteins. Expression of Cyclin D1, CDK4, Cyclin B1, CDK2 and P21 was detected by Western blotting. GADPH protein was used as an internal control. B. Quantification of cell cycle related proteins expression. * P<0.05 compared with the sh-CTRL A549 cells.
As a protein that suppresses Cyclin E/A-CDK2 activity, P21 inhibits the activities of the Cyclin E/CDK2 and CyclinA/CDK2 complexes at G1/S and G2/M phase checkpoints [25, 26]. The COP9 signalosome has also been reported to control E3 ubiquitin (Ub) ligases, through which CSN controls the degradation of a wide variety of proteins, such as P53. Consequently, the levels of target proteins of P53, such as P21, are reduced. In this study, we showed that knockdown of COPS3 also significantly upregulates the expression of P21. Thus, the upregulation of P21 may also contribute to cell cycle arrest at G0/G1 phase.
During G2 phase, Cyclin A/CDK2 and Cyclin B1/CDK1 complexes regulate M phase entry [21]. Using flow cytometry, we found that the sh-COPS3-treated A549 cells, compared with the sh-CTRL-treated A549 cells, showed a decreased proportion of cells in G2/M phase. By assessing G2 phase regulators, we found that Cyclin B1 was significantly downregulated in the sh-COPS3-treated A549 cells, thus suggesting that the down-regulation of Cyclin B1 might be related to the reduced proportion of sh-COPS3 A549 cells in G2/M phase.
In summary, this study suggests that COPS3 might be a novel molecular marker in lung cancer, for its high expression in lung cancer. COPS3 promotes the proliferation of lung cancer cells, increasing S-phase cells by regulating cell cycle-associated proteins. These data suggest an important role of COPS3 in regulating cell cycle progression of lung cancer cells. Further studies are required to confirm the molecular regulatory mechanism, clinical significance and prognostic value of COPS3 in lung cancer.
Acknowledgements
This work was supported by grants from the National Science Fund for Young Scholars (30901702) to YX, the California Institute for Regenerative Medicine (CIRM) (RT2-01942), the National Natural Science Foundation of China (81272294, 31430021), the National Basic Research Program of China (973 Program)(2015CB943303) to JFH; the National Natural Science Foundation of China (#81372835) and Jilin Provincial Science and Technology Department (20160101128JC) to WL; and the Key Project of Chinese Ministry of Education (311015), the Bethune Program B (2012202) of the Jilin University, Jilin Provincial Science and Technology Department (20111807, 20140414014GH, and 20150101176), the Platform Construction Project of Development and Reform Commission of Jilin Province (2014N147) to CJ.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel R, Ma JM, Zou ZH, Jemal A. Cancer Statistics, 2014. Ca-Cancer J Clin. 2014;64:9-29
2. National Lung Screening Trial Research T, Aberle DR, Berg CD, Black WC, Church TR, Fagerstrom RM. et al. The National Lung Screening Trial: overview and study design. Radiology. 2011;258:243-53
3. Yan J, Walz K, Nakamura H, Carattini-Rivera S, Zhao Q, Vogel H. et al. COP9 signalosome subunit 3 is essential for maintenance of cell proliferation in the mouse embryonic epiblast. Mol Cell Biol. 2003;23:6798-808
4. Bech-Otschir D, Seeger M, Dubiel W. The COP9 signalosome: at the interface between signal transduction and ubiquitin-dependent proteolysis. J Cell Sci. 2002;115:467-73
5. Henriksen J, Aagesen TH, Maelandsmo GM, Lothe RA, Myklebost O, Forus A. Amplification and overexpression of COPS3 in osteosarcomas potentially target TP53 for proteasome-mediated degradation. Oncogene. 2003;22:5358-61
6. van Dartel M, Redeker S, Bras J, Kool M, Hulsebos TJM. Overexpression through amplification of genes in chromosome region 17p11.2 similar to p12 in high-grade osteosarcoma. Cancer Genet Cytogen. 2004;152:8-14
7. Yu YS, Tang ZH, Pan QC, Chen XH, Liu XN, Zang GQ. Inhibition of Csn3 expression induces growth arrest and apoptosis of hepatocellular carcinoma cells. Cancer Chemoth Pharm. 2012;69:1173-80
8. Bech-Otschir D, Kraft R, Huang XH, Henklein P, Kapelari B, Pollmann C. et al. COP9 signalosome-specific phosphorylation targets p53 to degradation by the ubiquitin system. Embo J. 2001;20:1630-9
9. Yan T, Tang G, Ren T, Shen D, Sun K, Liang W. et al. RNAi-mediated COPS3 gene silencing inhibits metastasis of osteogenic sarcoma cells. Cancer Gene Ther. 2011;18:450-6
10. Yan TQ, Wunder JS, Gokgoz N, Gill M, Eskandarian S, Parkes RK. et al. COPS3 amplification and clinical outcome in osteosarcoma. Cancer. 2007;109:1870-6
11. Zhou Y, Luoh SM, Zhang Y, Watanabe C, Wu TD, Ostland M. et al. Genome-wide identification of chromosomal regions of increased tumor expression by transcriptome analysis. Cancer Res. 2003;63:5781-4
12. Travis WD, Brambilla E, Burke AP, Marx A, Nicholson AG. World Health Organization (WHO) classification of tumours of the lung, pleura, thymus and heart, 4th ed. Lyon: International Agency for Research on Cancer. 2015
13. Sun J, Li W, Sun Y, Yu D, Wen X, Wang H. et al. A novel antisense long noncoding RNA within the IGF1R gene locus is imprinted in hematopoietic malignancies. Nucleic acids research. 2014;42:9588-601
14. Wang H, Li W, Guo R, Sun J, Cui J, Wang G. et al. An intragenic long noncoding RNA interacts epigenetically with the RUNX1 promoter and enhancer chromatin DNA in hematopoietic malignancies. International journal of cancer. 2014;135:2783-94
15. Song W, Li W, Li L, Zhang S, Yan X, Wen X. et al. Friend leukemia virus integration 1 activates the Rho GTPase pathway and is associated with metastasis in breast cancer. Oncotarget. 2015;6:23764-75
16. Zhai Y, Chen X, Yu D, Li T, Cui J, Wang G. et al. Histone deacetylase inhibitor valproic acid promotes the induction of pluripotency in mouse fibroblasts by suppressing reprogramming-induced senescence stress. Experimental cell research. 2015;337:61-7
17. Yin H, Chen N, Guo R, Wang H, Li W, Wang G. et al. Antitumor potential of a synthetic interferon-alpha/PLGF-2 positive charge peptide hybrid molecule in pancreatic cancer cells. Scientific reports. 2015;5:16975
18. Zhang S, Zhong B, Chen M, Yang L, Yang G, Li Y. et al. Epigenetic reprogramming reverses the malignant epigenotype of the MMP/TIMP axis genes in tumor cells. International journal of cancer. 2014;134:1583-94
19. Wang XM, Cui JW, Li W, Cai L, Song W, Wang GJ. Silencing of the COPS3 Gene by siRNA Reduces Proliferation of Lung Cancer Cells Most Likely via Induction of Cell Cycle Arrest and Apoptosis. Asian Pac J Cancer P. 2012;13:1043-8
20. Scholzen T, Gerdes J. The Ki-67 protein: from the known and the unknown. Journal of cellular physiology. 2000;182:311-22
21. Musat M, Morris DG, Korbonits M, Grossman AB. Cyclins and their related proteins in pituitary tumourigenesis. Mol Cell Endocrinol. 2010;326:25-9
22. Lim S, Kaldis P. Cdks, cyclins and CKIs: roles beyond cell cycle regulation. Development. 2013;140:3079-93
23. Wikman H, Kettunen E. Regulation of the G1/S phase of the cell cycle and alterations in the RB pathway in human lung cancer. Expert Rev Anticancer Ther. 2006;6:515-30
24. Wu AB, Wu B, Guo JS, Luo WR, Wu D, Yang HL. et al. Elevated expression of CDK4 in lung cancer. J Transl Med. 2011:9
25. Abbas T, Dutta A. p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer. 2009;9:400-14
26. Romanov VS, Pospelov VA, Pospelova TV. Cyclin-dependent kinase inhibitor p21(Waf1): contemporary view on its role in senescence and oncogenesis. Biochemistry (Mosc). 2012;77:575-84
Author contact
![]() Corresponding authors: Jiuwei Cui, Cancer Center, the First Hospital of Jilin University, No.71, Xinmin Street, Changchun, 130021, China, Tel: 86-43188782178, Fax: 86-43188786134, e-mail: cuijwedu.cn; or Ji-Fan Hu, Stanford University Medical School, Palo Alto Veterans Institute for Research, Palo Alto, CA 94304, USA, Tel: 650-493-5000, x63175, Fax: 650-849-1213, e-mail: jifanedu.
Corresponding authors: Jiuwei Cui, Cancer Center, the First Hospital of Jilin University, No.71, Xinmin Street, Changchun, 130021, China, Tel: 86-43188782178, Fax: 86-43188786134, e-mail: cuijwedu.cn; or Ji-Fan Hu, Stanford University Medical School, Palo Alto Veterans Institute for Research, Palo Alto, CA 94304, USA, Tel: 650-493-5000, x63175, Fax: 650-849-1213, e-mail: jifanedu.
Citation styles
APA
Pang, J., Yan, X., Cao, H., Qian, L., He, H., Tian, H., Han, F., Wang, G., Chen, X., Zhao, Y., Hu, J.F., Cui, J. (2017). Knockdown of COPS3 Inhibits Lung Cancer Tumor Growth in Nude Mice by Blocking Cell Cycle Progression. Journal of Cancer, 8(7), 1129-1136. https://doi.org/10.7150/jca.16201.
ACS
Pang, J.; Yan, X.; Cao, H.; Qian, L.; He, H.; Tian, H.; Han, F.; Wang, G.; Chen, X.; Zhao, Y.; Hu, J.F.; Cui, J. Knockdown of COPS3 Inhibits Lung Cancer Tumor Growth in Nude Mice by Blocking Cell Cycle Progression. J. Cancer 2017, 8 (7), 1129-1136. DOI: 10.7150/jca.16201.
NLM
Pang J, Yan X, Cao H, Qian L, He H, Tian H, Han F, Wang G, Chen X, Zhao Y, Hu JF, Cui J. Knockdown of COPS3 Inhibits Lung Cancer Tumor Growth in Nude Mice by Blocking Cell Cycle Progression. J Cancer 2017; 8(7):1129-1136. doi:10.7150/jca.16201. https://www.jcancer.org/v08p1129.htm
CSE
Pang J, Yan X, Cao H, Qian L, He H, Tian H, Han F, Wang G, Chen X, Zhao Y, Hu JF, Cui J. 2017. Knockdown of COPS3 Inhibits Lung Cancer Tumor Growth in Nude Mice by Blocking Cell Cycle Progression. J Cancer. 8(7):1129-1136.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.