Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(1):85-96. doi:10.7150/jca.16792 This issue Cite
Review
Human-derived normal mesenchymal stem/stromal cells in anticancer therapies
Cheng Zhang1, 2, 3, Shi-Jie Yang1, 2, 3, Qin Wen1, 2, 3, Jiang F Zhong2, 3, Xue-Lian Chen2, 3, Andres Stucky2, 3, Michael F Press4, Xi Zhang1, 2, 3 ![]()
1. Department of Hematology, Xinqiao Hospital, Third Military Medical University, Chongqing, 400037, People's Republic of China.
2. Division of Periodontology, Diagnostic Sciences & Dental Hygiene, and Division of Biomedical Sciences, Herman Ostrow School of Dentistry, University of Southern California, Los Angeles, CA, 90033, USA.
3. Norris Cancer Center, University of Southern California, Los Angeles, CA, 90033, USA.
4. Department of Pathology, Keck School of Medicine, University of Southern California, Los Angeles, CA, 90033, USA.
Received 2016-7-10; Accepted 2016-9-18; Published 2017-1-1
Abstract
The tumor microenvironment (TME) not only plays a pivotal role during cancer progression and metastasis, but also has profound effects on therapeutic efficacy. Stromal cells of the TME are increasingly becoming a key consideration in the development of active anticancer therapeutics. However, dispute concerning the role of stromal cells to fight cancer continues because the use of mesenchymal stem/stromal cells (MSCs) as an anticancer agent is dependent on the specific MSCs subtype, in vitro or in vivo conditions, factors secreted by MSCs, types of cancer cell lines and interactions between MSCs, cancer cells and host immune cells. In this review, we mainly focus on the role of human-derived normal MSCs in anticancer therapies. We first discuss the use of different MSCs in the therapies for various cancers. We then focus on their anticancer mechanism and clinical application.
Keywords: Mesenchymal stem/stromal cells, Cancer, Tumor microenvironment.
Introduction
The influence of mesenchymal stem/stromal cells (MSCs) and the tumor microenvironment (TME) is now widely appreciated in cancer [1, 2]. However, distinct MSCs in the TME can come from different origins and exhibit a diversity of anticancer properties. Recently, increasing clinical trials have been performed to study the anticancer properties of MSCs. Currently, only MSCs originating from human tissue can be used in the clinic. In this review, we therefore mainly focus on the role of human-derived normal MSCs in anticancer therapies. We first discuss the use of different MSCs in the therapies for various cancers. We then focus on the anticancer mechanism and clinical application of these MSCs.
MSCs in anticancer therapies
Many studies have reported pro- or anti-cancer effects of MSCs on the progression of primary and metastatic tumors. These contradictory results could be associated with differences in the MSCs source used, the type of tumor model and the method of administration or other unknown factors. Growing evidence showed that MSCs can migrate not only to primary tumor sites but also to pre-metastatic sites [3-5].
1. Blood cancers
MSCs of the bone marrow (BM), which we refer to here as BM-MSCs, support primary acute lymphoblastic leukemia (ALL) viability and also protect ALL cells (Reh cell line) from drug-induced apoptosis through an effect on cell cycle arrest via p21 down-regulation [6]. Another report also showed that BM-MSCs can rescue human B-ALL cells from apoptosis. The MSCs of umbilical cord/cord blood (UC-MSCs) inhibit the apoptosis of T-ALL cell line (Jurkat cells) [7]. Another study showed that UC-MSCs inhibited the proliferation of Jurkat cells, but did not induce apoptosis [8]. In contrast MSCs of adipose tissue (AT-MSCs) do not inhibit the proliferation of Jurkat cells [9]. Our study showed that UC-MSCs suppressed proliferation and promoted apoptosis, differentiation, and drug sensitivity of Jurkat cells [10]. We also observed that UC-MSCs can inhibit the growth and induce the apoptosis of tyrosine kinase inhibitor-resistant Philadelphia-positive ALL (unpublished data).
BM-MSCs from unrelated donors have been shown to support the phenotype of a leukemia stem cell derived from AML in a largely quiescent state in a long-term in vitro culture system. AT-MSCs protect acute promyelocytic leukemia (APL) cells from apoptosis induced by serum starvation or by doxorubicin [11]. The viability and activity of neutrophils differentiated from APL cell line (HL-60) were improved when HL-60 cells were cultured with AT-MSCs. However, UC-MSCs can inhibit the proliferation of K562 cell line and increase the cytotoxic effect of doxorubicin without induction of drug resistance [12]. UC-MSCs can induce the apoptotic and proliferative effects of Jurkat cells, HL-60 and K562 cells [13]. Another study showed that UC-MSCs protect HL-60 from Ara-c-induced apoptosis.
BM-MSCs derived from healthy donors protect chronic lymphocytic leukemia (CLL) cell survival with similar capacities to MSCs derived from CLL [14]. Adipocytes have been reported to cause CLL cells to become resistant to dexamethasone by providing lipid factors [15].
BM-MSCs have been seen to exhibit modest effects on cocultured lymphoma cell survival. In vivo, intra-peritoneal injection of BM-MSCs significantly increases the overall survival of xenograft models in nude-SCID mice [16]. AT-MSCs inhibit the growth of EL4 murine T-cell lymphoma by altering cell-cycle progression and inducing apoptosis in vitro, and can migrate to tumor sites and inhibit tumor growth in tumor-bearing nude mice and prolong their survival time. UC-MSCs significantly inhibit the growth of Burkitt's lymphoma cell lines (Ramos, CRL 1596) and increase cell death [17].
BM-MSCs support the growth of multiple myeloma (MM) cells. Both UC-MSCs and AT-MSCs inhibit MM cell growth and clonogenicity in vitro compare to BM-MSCs [18]. When RPMI-8226 MM cells were injected with UC-MSCs into mice, smaller subcutaneous tumormasses were formed, but delayed tumor burden growing was observed as the peritumoral injections of the same MSC subtype. Finally, both microarrays and ELISA revealed different expression of several genes and soluble factors in UC-MSCs as compared to other MSCs. The study also found that the soluble factors involved in MM biology, such as IL-6, IGF-1, and VEGF, were downregulated in UC-MSCs compared to BM-MSCs. The UC-MSCs significantly overexpressed genes involved in cell-to-cell communication, such as RALA, MPG, SMAD-4, OGT, GRB-2, and MAP3K7, while others that were functionally implicated in cell responses to inflammatory stimuli, such as ELK-4, MCL-1, BCL-2, and MSH-2, were significantly downregulated. Furthermore, other genes related to cell motility and chemotactic functions, such as HGS, BCAR-1, PLOD-3, and PAFAH1B1, were also overexpressed in UC-MSCs as compared to BM-MSCs. The intracellular formation of exosomes and microvesicles, as well as a variable modulation of genes involved in the secretion of dense granules and exocytosis of soluble molecules, such as DNAJB-1, DNAJC-5, HPRT-1, HSPA1A, and BCL-2, were also upregulated. Our recent study also showed that UC-MSCs can suppress proliferation and induce apoptosis in KM3 cells by downregulating IL-6R expression and inhibiting NF-κB activity [19].
2. Breast cancers
BM-MSCs promote the proliferation of mouse breast cancer cell line 4T1 and human breast cancer cell line SUM1315 [20]. BM-MSCs promote the production of lysyl oxidase from breast carcinoma cells, which can promote the metastasis of breast cancer (MDAMB-231 and MCF7/Ras). A series of experiments demonstrated that adipocytes could induce both replicative potential and evasion of apoptosis in breast cancer cells [21]. UC-MSCs significantly inhibit the proliferation of breast cancer cell line MDA-MB-231 [22]. They also reduce lung metastasis and inhibit the growth of breast cancer cell line MDA-MB-231 by inducing apoptosis in vitro and in vivo. However, other reports showed that UC-MSCs promote proliferation and migration in breast cancer cell lines MCF-7, MDA-MB-231 and MCF-7 through activation of the ERK pathway and autocrine IL-8 and IL-6 signaling [23].
3. Cancers of the digestive system
BM-MSCs can significantly promote the proliferation of hepatocellular carcinoma cell lines with high metastatic potential (MHCC97-H) and low-metastatic potential (MHCC97-L) [24]. They also promote the metastasis of human hepatic cancer cell lines (SMMC7721, SK-Hep1 and Hep-3B). Another report showed that BM-MSCs promote the proliferation of metastatic hepatic cancer cell line MHCC97-H in vivo and vitro, but inhibit its metastasis [25]. AT-MSCs efficiently inhibit human hepatic cancer cell line (HepG2, Huh7, SMMC7721, and Bel7402) proliferation and division, and induce cell death through the downregulation of Akt signaling [26]. UC-MSCs exert a suppressive effect on the growth of a hepatic cancer cell line (Huh7-DsRed2 cells), and have a tumor-specific, antiproliferative effect that is not observed with normal human hepatocyte cells or patient-derived matched normal tissues [27]. UC-MSCs also inhibit the malignant phenotype of human cholangiocarcinoma cell line HCCC-9810.
BM-MSCs promote the progression of colorectal cancer cell lines (HCT-8/E11, HCT 116, SW480, HT-29, LoVo, T84 CRC and ANC-1) [28]. MSCs derived from fetal and adult BM alone or together with human colon cancer cell line SW480 were transplanted subcutaneously into BALB/c-nu/nu mice to observe the outgrowth of tumors. The results showed that both types of MSCs promoted tumor growth in vivo. However, AT-MSCs significantly suppressed the proliferation of colorectal adenocarcinoma (HT-29) in vitro and in mice xenografts. Other reports showed that AT-MSCs enhance the metastatic capacity of colon cancer cell lines (HCT116, LoVo cells, SW480, LS174T and CCD-18Co) in a colon cancer cell co-culture model [29]. AT-MSCs also promote the expression of the growth marker, proliferation cell nuclear antigen, but inhibit apoptosis markers and single-stranded DNA in the well- and poorly differentiated types of gastric adenocarcinoma cell lines (MKN28 and MKN45). UC-MSCs promote the proliferation of gastric epithelial cell lines (GES-1 and the SGC-7901) in vitro and in vivo [30]. They can also promote carcinoma growth and lymph node metastasis when co-injected with esophageal cancer cell lines (Eca109 and TE-1) in nude mice. However, artificial fusion of MSCs with esophageal carcinoma cell line EC9706 resulted in hybrids with declined cell growth, increased apoptosis and suppressed tumorigenicity [31]. UC-MSCs promote the proliferation of mouse pancreatic cancer cell line Pan02 in vivo, however, MSCs producing IL-15 eradicate established pancreatic tumors in syngeneic mice [32].
4. Cancers of the respiratory system
AT-MSCs stimulate adhesion and proliferation of human lung adenocarcinoma cell line A549 in vivo and in vitro [33]. They also promote the proliferation of large cell lung cancer cell line H460, and co-injection of MSCs with H460 promotes tumor growth in vivo along with a reduction in apoptotic cell death. However, another study showed that AT-MSCs do not affect the proliferation of lung adenocarcinoma cell line (A549) in vitro or promote tumor growth in vivo. UC-MSCs can inhibit the proliferation of human bronchioloalveolar carcinoma cell lines (H358 or SW1573) [34].
5. Cancers of the urinary system
BM-MSCs promote growth and angiogenesis of prostate cancer cell line DU145. AT-MSCs can inhibit the proliferation and induce the apoptosis of both androgen-responsive (LNCaP) and androgen-nonresponsive (PC3) human prostate cancer cells [35]. Another study showed that AT-MSCs increase cell viability of prostate tumor cell lines LNCaP and LNCaP-Bic, but no effect was observed for Du145 or PC3. UC-MSCs induce apoptosis in prostate cancer cell line PC-3 and attenuate bladder tumor cell growth in vitro and in vivo [36]. However, BM-MSCs do not inhibit the proliferation of renal cell carcinoma cell lines (RCC42 and RCC52), but increase their migratory ability and invasiveness in vitro [37].
6. Gynecological cancers
AT-MSCs have no effect on the proliferation of the cervical cancer HeLa cell line. They promote cell proliferation and invasion of human epithelial ovarian cancer cell lines (SKOV3, CAOV3, A2780 and ES2) [38]. Both AT-MSCs and BM-MSCs promote vascularization and growth of endometrial tumors [39]. UC-MSCs inhibit the growth of ovarian carcinoma cell line TOV-112D in vitro [40].
7. Cancers of the nervous system
BM-MSCs suppress the growth of human primary glioma cells through inhibition of angiogenesis. AT-MSCs promote the proliferation of human glioma cell line U87MG. Co-injection of MSCs with U87MG promotes tumor growth in vivo and also induces a reduction in apoptotic cell death. AT-MSCs and UC-MSCs can efficiently induce apoptosis and differentiation in human glioma cell line U251 in vitro [41]. UC-MSCs have a significant cytotoxicity against malignant glioma cell line U87MG, and mixed-source MSCs have significantly higher cytotoxicity than UC-MSCs alone for glioma cell line C6. UC-MSCs also inhibit the growth of primary glioblastoma multiforme (GBM) cells and cause apoptosis both in vitro and in vivo; however, AT-MSCs promote GBM growth.
8. Other cancers
AT-MSCs promote migration and early metastasis of head/neck cancer cell lines Cal-27 and SCC-4 in vitro and in xenografts [42]. AT-MSCs inhibit the growth of human melanoma cell lines (A375SM and A375P) by inducing apoptosis and altering cell-cycle distribution in vitro. AT-MSCs also suppress tumor growth and migrate to tumor tissues in tumor-bearing athymic mice [43]. AT-MSCs have no growth-inhibitory effect on human medulloblastoma cell line D283-MED, however, MSCs transfected with neurotrophin-3 significantly inhibit the growth of D283-MED [44].
Altogether, the MSCs derived from different tissues show great diversity in their anticancer properties. It seems that the UC-MSCs are ideal cells for anticancer therapies (Table 1). While complex interactions between these different cell types reshape the surrounding extracellular matrix as cancer progresses, neoplastic and stromal cells also undergo constant changes.
Comparative analysis of the anticancer effects of human tissue-derived normal MSCs in vitro and in vivo.
| Type of MSCs | Tumor propagating effect | Tumor inhibiting effect |
|---|---|---|
| Bone marrow | Acute lymphoblastic leukemia | Hepatocellular carcinoma |
| Acute myeoblastic leukemia | Pancreatic cancer | |
| Chronic lymphocytic leukemia | Glioma | |
| Lymphoma | ||
| Multiple myeloma | ||
| Breast cancer | ||
| Hepatocellular carcinoma | ||
| Colorectal cancer | ||
| Colon cancer | ||
| Prostate cancer | ||
| Renal cell carcinoma | ||
| Endometrial tumors | ||
| Glioma | ||
| Osteosarcoma | ||
| Prostate cancer | ||
| Ovarian cancer | ||
| Gastric cancer | ||
| Pancreatic cancer | ||
| Lung cancer | ||
| Oral tongue tumor | ||
| Adipose tissues | Acute lymphoblastic leukemia | Murine T-cell lymphoma |
| Acute promyelocytic leukemia | Multiple myeloma | |
| Chronic lymphocytic leukemia | Hepatic cancer | |
| Breast cancer | Colorectal adenocarcinoma | |
| Lung cancer | Prostate cancer | |
| Ovarian cancer | Glioma | |
| Prostate cancer | Melanoma | |
| Kaposi sarcoma | ||
| Melanoma tumor | ||
| Colon cancer | ||
| Prostate cancer | ||
| Endometrial tumors | ||
| Glioma | ||
| Head/neck cancer | ||
| Umbilical cord/cord Blood | Acute lymphoblastic leukemia | Acute lymphoblastic leukemia |
| Acute myeoblastic leukemia | Chronic myeloid leukemia in blast crisis | |
| Breast cancer | Acute myeoblastic leukemia | |
| Gastric epithelial cell | Burkitt's lymphoma | |
| Esophageal cancer | Multiple myeloma | |
| Mouse pancreatic cancer | Breast cancer | |
| Ovarian cancer | Hepatic cancer | |
| Cholangiocarcinoma | ||
| Bronchioloalveolar carcinoma | ||
| Prostate cancer | ||
| Bladder tumor | ||
| Ovarian carcinoma | ||
| Glioma | ||
| Esophageal cancer | ||
| Lung cancer |
Note: Distinct MSCs come from different origins and exhibit a diverse range of anticancer properties. The MSCs from bone marrow and adipose tissues mainly show tumor-propagating effects; however, MSCs from umbilical cord/cord blood mainly show tumor-inhibiting effects.
The mechanism of MSCs in anticancer therapies
In recent years, growing literature has focused on the potential role of MSCs in malignancies. Because many oncological therapies employ MSCs as cellular vehicles, it is necessary to shed light on the mechanism by which MSCs act on the different cells which constitute the tumor microenvironment in anticancer therapies (Fig 1).
Mechanism of mesenchymal stem cell action in anticancer therapies. The primary role of MSCs in anticancer therapies lies in their abilities to change normal fibroblasts into tumor-associated fibroblasts, transition into CAFs, control the EMT, replace host cells, remodel the ECM, regulate the immune system, act in a paracrine fashion and establish cell-cell contacts.
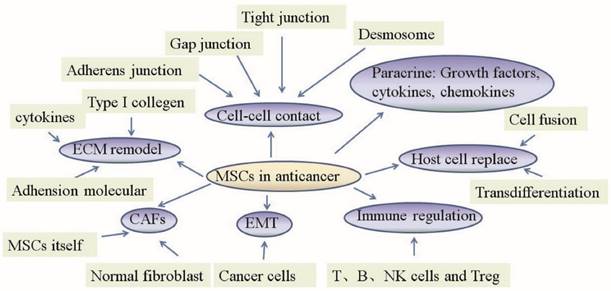
1. MSCs change normal fibroblasts to tumor-associated fibroblasts
The tumor stroma that includes many different types of extracellular matrix (ECM) and non-cancerous cells acts as an essential microenvironment for cancer cells. The major cellular constituents of the tumor stroma are fibroblasts that called as cancer-associated fibroblasts (CAFs). CAFs express α-smooth muscle actin (α-SMA), which is proposed to have strong contractility, and contribute to the stiffening and remodeling of the stromal ECM and also offering a right field for tumor cell invasion.
The surrounding normal tissues were invaded by cancer cells that is necessary for distant metathesis, and is started by the detachment of tumor cells from the primary tumor. Breakdown of the basement membrane around the tumor tissues, the loss of cell-cell adhesion and cell migration into the tumor stroma were required during this process. The EMT of the neighboring cancer cells through paracrine signaling by CAFs secrete soluble factors including THGF and TGF-β. CAFs can also secrete matrix-degrading enzymes, which indicates that control by CAFs is vitally important in anticancer therapies.
BM-MSCs promote the conversion of normal fibroblasts to CAF, which causes increased growth and invasion of prostate cancer cell lines (LNCap and C4-2) through TGFβ-1 in vitro and in vivo [45]. However, interruption with TGFβ-1 inhibitor SB431542 led to alteration of the MSC-induced CAF conversion and the prostate cancer cell growth and invasion was inhibited.
2. MSC transition into CAFs
CAFs as a major constituent of the tumor stroma support malignant cell growth and spreading. Recent study in mouse models of inflammation-induced gastric cancer suggested that at least 20% of CAFs originate from BM-MSCs.
When in direct contact with colon cancer cells, BM-MSCs differentiate into CAFs. BM-MSCs exposed to tumor-conditioned medium (MDAMB231, PANC-1 and U87) over a prolonged period of time assume a CAF-like myofibroblastic phenotype and promote tumor cell growth both in vitro and in an in vivo co-implantation model. However, BM-MSCs induced to differentiate into a myofibroblast-like phenotype using 5-azacytidine do not promote tumor cell growth [46]. Under long-term tumor conditioning in vitro, BM-MSCs can transition into CAFs and stimulate tumor growth through the paracrine production of secreted IL-6 in vivo. BM-MSCs transform into CAFs when cultured with certain cancers (human ovarian cancer TOV-112D; breast cancer MDA-MB-231), but no changes were observed when UC-MSCs were co-cultured in the same conditions [47]. The exosomes released by primary CLL cells and cell line MEC-1 can also induce BM-MSCs to transition into CAFs.
AT-MSCs and UC-MSCs are susceptible to CAF transition under certain conditions as well. The exosomes from breast cancer cells (MCF-7 and MDA-MB231) can convert AT-MSCs into myofibroblast-like cells [48]. The lysophosphatidic acid derived from human ovarian adenocarcinoma cell line OVCAR-3 stimulates differentiation of AT-MSCs into CAFs. The exosomes of gastric cancer (SGC7901, HGC27 and GES-1) also trigger UC-MSCs to CAFs through the TGF-β/Smad pathway.
Although CAFs derived from MSCs block the transition of MSCs into CAFs, they can also abolish their protumor effects. A recent study with customized cDNA microarrays to compare the gene expression profiles of stromal cells from primary tumor tissue, lymph node metastasis and BM obtained from breast cancer patients showed that only small differences were observed in the transcriptional profiles of fibroblasts from the primary tumor and lymph node, whereas greater differences were observed between BM stromal cells and the other two sites [49]. Another study showed that only normal, non-malignant mesenchymal progenitor cells and normal osteosarcoma-derived MSCs but not tumor cells could be cultured from primary human osteosarcoma samples when applying standard MSC culture techniques. An animal study showed that human cancer cells grafted to nu/nu mice can induce the formation of tumor stroma that has the typical smooth muscle actin-containing CAFs that seem to be of the host origin because they are not recognized by an antibody specific for human vimentin, as was also verified in vitro. These showed that the CAFs are not formed from cancer cells [50]. The depletion of CAFs leads to increased tumor invasion associated with decreased survival in transgenic mice with the ability to delete αSMA (+) myofibroblasts in pancreatic ductal adenocarcinoma; fewer myofibroblasts in these tumors also correlated with reduced survival in patients with invasive pancreatic adenocarcinoma [51]. CAFs are derived from both MSCs and local fibroblasts, which suggests that the primitive undifferentiated MSCs can be presented even in advanced cancers. Even within the same type of tumor, the heterogeneous populations of MSCs and CAFs may have different phenotypic characteristics. Taken together, these studies suggest that not all CAFs are malignant, and we should carefully identify and use them.
3. MSCs control the EMT
BM-MSCs have been shown to induce gastric cancer cell EMT and promote transwell and trans-endothelial migration in vitro, as well as remarkably increase tumor mass and liver metastasis in vivo. They also stimulate EMT of breast cancer cell lines (MDA-MB-231, T47D and SK-Br-3) [52]. BM-MSCs also induce EMT in human colorectal cancer cell lines (HCT116, LS180, COLO205, HT29 and SW480) through the expression of surface-bound TGF-β. AT-MSCs can promote the EMT of human breast cancer cell line MCF7 through paracrine and induced autocrine TGF-β [53]. Adipose MSCs can also induce EMT of lung cancer cells (H358). However, glioma stem cells from patients treated with the UC-MSCs revert the EMT by down regulating transcriptional activation of Sox2 and Twist1, which results in a decrease in the invasive ability of tumor cells. In the intracranial glioma stem cell-induced tumors of mice, treatment with UC-MSCs significantly reduces glioma stem cell-induced tumor volumes [54].
MSC-induced EMT of cancer cells contributes to the malignant characteristics of tumors, such as local invasion and distant metastasis. However, normal fibroblasts can also induce the EMT of tumor cells. EMT also regulates the stem cell-like characteristics of both malignant and normal cells [55]. The EMT program seems to be linked to the residence of cells in stem cell-like states in some epithelial tissues on the one hand, but on the other hand, they are also coopted by cancer cells which enable them to acquire cellular traits associated with high-grade malignancy. Therefore, two aspects of the EMT program are worthy of detailed examination.
4. Immune regulation
The immunosuppressive property of MSCs is useful for anticancer therapies because they are capable of inhibiting the proliferation of all effector cells of the peripheral blood, such as NK cells, T cells, and B cells. The affected cells are inhibited not only in their proliferative capacity, but also in their cytolysis and antibody production in vitro and in vivo. This immunosuppression at a cellular level is widely considered to be the functional basis for the systemic effects of MSCs and involves several soluble factors, such as galectin-3, galectin-9, IL-10, indoleamine 2, 3-dioxgynease and HLA-G.
In an in vitro study of human gliomas, MSC-like pericytes similar to BM-MSCs displayed inhibitory functions on CD4+ T cells through prostaglandin-E2 and HG. Recently, glioma pericytes were shown to switch from a tumor-suppressive phenotype to a tumor-promoting one in a xenograft model. The antiproliferative effect of MSCs also affects microglia cells by reducing the level of tumour necrosis factor-α [56]. MSCs isolated from tumors showed immunosuppressive properties and can downregulate the activating NK cell receptors and impair tumor lysis by NK cells in vitro [57]. Tumor-associated MSCs producing the immunosuppressive interleukin-10 were markedly increased in cervical cancer compared to normal BM-MSCs, which underpins the role of tumor-associated MSCs in establishing an immunosilenced, quiescent niche. Another report also showed an inhibitory effect of MSCs on various immune effector cells. These data suggest that the tumor-promoting effect of MSCs is due to suppression of immune effector cells. TAFs could also induce CD3/CD28-dependent activation of T cells and do not appear to inhibit these effector cells [58].
One caveat should be noted, however, among these publications, very few experimental designs aimed at a side-by-side comparison of tumor-derived MSCs with fibroblasts or MSCs from adjacent healthy tissue, umbilical cord/cord blood or normal adipose tissues. Without an adequate control population, whether the sole measurement of effector cell proliferation or receptor expression status can faithfully reproduce the conditions in the tumor is questionable.
5. MSCs replace host cells
Transdifferentiation and cell fusion are two explanations for cell replacement. Transdifferentiation means that MSCs can differentiate across lineage barriers into a diverse array of cell types. Cell fusion refers to fusion between MSCs and host cells, which can result in reprogramming of the host nucleus to express MSC-specific genes, which can then regulate the growth, apoptosis and metastasis of cancer cells.
Cell replacement was originally thought to be the major mechanism by which MSCs exert anticancer effects. BM-MSCs acquire epithelial characteristics through fusion with gastrointestinal epithelial cells in vitro and in vivo [59]. BM-MSCs co-cultured with glioma cell lines (U87 and U373) inhibit the proliferation of glioma cells but improve their movement. Another study observed that MSCs can fuse with glioma cells to form a functional and structural syncytium. BM-MSCs spontaneously form tumorigenic hybrids with non-small cell lung cancer cell lines (A549, H460, or SK-MES-1). Hybrids acquire increased motility and invasiveness and form more tumor spheres than the single cancer cells. In NOD/SCID mice, heterotypic hybrids exhibit enhanced tumorigenicity and reproduce the human tumor [60]. Fusion of BM-MSCs with SU3 glioma cells contributes to glioma neovascularization. AT-MSCs fuse with breast carcinoma cell lines (T47D) and the cancer cells express higher transcription levels of typical malignancy markers in co-cultures compared with single cultures [61].
It was found during co-culture that MDA-MB231 and UC-MSCs may adhere upon contact at a variable ratio from 1:1 to 9:1, and all the MDA-MB231 cells in this study shrank following contact with MSCs [61]. More specifically, two different interactions were observed. First, the MDA-MB231 cells shrank immediately upon adhesion and infusion of some substance from MSCs. Secondly, the MSCs internalized into MDA-MB231 to form a cell-in-cell structure, though the two cells separated after a short period. The separated MDA-MB231 cells shrank and died, while the separated MSCs went on to contact other MDA-MB231 cells and shrink them as well. The artificial fusion of UC-MSCs with esophageal cancer cell line EC9706 resulted in hybrids with decreased cell growth, increased apoptosis and suppressed tumorigenicity [31]. UC-MSCs can spontaneously fuse with lung cancer cell line H441 to form synkaryonic hybrids and reprogram the transcriptome of the lung cancer cells through FOXF1, which reduces their growth rate and tumorigenicity [62]. Another study showed that UC-MSCs can exchange membrane proteins and fuse with several tumor cell lines (breast carcinoma cell line MCF-7; Ovarian cancer cell lines NIH OVCAR-3 and SK-OV-3) and enhance the cancers' cell growth. However, some researchers have raised doubts about these findings. In one relevant study, the authors performed double immunostaining of ataxic mouse cerebellum with MAB1287 (UC-MSC marker) and anti-GFAP (astrocyte marker), anti-NeuN (neuron marker) or calbindin (Purkinje cell marker) to display the differentiation of implanted UC-MSCs. The results demonstrated that MAB1287-positive cells rarely co-localized with GFAP, NeuN or calbindin, which implies little relation between the efficiency of UC-MSCs and cell replacement [63].
It should be noted that after transplantation, a large proportion of MSCs can be observed in capillaries within the liver, spleen, lung and other off-target tissues. The number of cells that enter the cancer site is far from being enough for tissue reengineering.
6. MSCs remodel the ECM
Remodeling of the ECM by stimulating epithelial cell transformation induces the migration, invasion, and metastasis of cancer cells. The production of proteolytic ECM affects focal adhesions at cell membranes, cellular cytoskeletal organization, integrin-mediated anchorage of cells, and the signal transduction regulating these structures.
ECM is composed by a complex mixture of components, including proteins, glycoproteins, proteoglycans and polysaccharides. MSCs contribute to formatting or remodeling the tumor stroma, which promotes or inhibits the progression of cancer through focal proteolysis that causes the release of bioactive fragments from ECM molecules, breaks cell-matrix interactions with integrins and receptors, and disrupts the ECM architecture. ECM derived from BM-MSCs increases the survival of prostate cancer cell lines (LNCaP and MDA-PCa-2b) in androgen-depleted conditions, and induces morphological changes and chemoresistance in LNCaP cells through the MEK1/2 or PI3K pathway. BM-MSCs promote the growth of colorectal carcinoma cell line HCT8 with different mutational conditions in vitro and in vivo. MSCs interacting with colorectal carcinoma cells facilitate three-dimensional spheroid formation through a dysfunctional E-cadherin system and secrecte type I collagen [64]. BM-MSCs inhibit the proliferation but increase the invasion of oral tongue tumor cell lines (HSC-3, SAS and DOK) through the secrection of type I collagen. UC-MSCs secrete dickkopf (DKK1) to arrest the growth of breast cancer cell line MDA-MB-231 by upregulating levels of PTEN [65].
7. Paracrine action
Recently, researchers have focused increasing attention on the paracrine actions of MSCs. MSCs can secrete an extensive variety of growth factors, cytokines, and chemokines, including quantities of hepatocyte growth factor, monocyte chemotactic protein-1and stromal-derived factor-1, insulin-like growth factor-1, brain-derived neurotrophic factor, glial cell-derived neurotrophic factor vascular endothelial growth factor and interleukin-8. These released factors may promote or inhibit the growth and metastasis of cancer cells in vitro and in vivo. For example, BM-MSCs and UC-MSCs can inhibit cell cycle progression and induce senescence and differentiation of glioblastoma multiforme stem-like cell lines (NCH421k, NCH644, NIB26, and NIB50). By secrecting soluble NRG1, BM-MSCs promote the progression of colorectal cancer [28]. They also secrete insulin growth factor binding proteins to inhibit the growth of liver cancer cell line (Huh7-DsRed2) through reduced activation of IGF-1R/PI3K/Akt signaling [66]. BM-MSCs secrecte IL-6, which promotes growth of osteosarcoma cell line Saos-2 in vitro, and the proximal tibia of nude mice injected with MSCs combined with Saos-2 could also promote tumor growth and progression. No effect on cell proliferation was observed for breast cancer cell line MCF7 cultured with AT-MSCs, which secrecte TGF-β, but increased invasive and migratory capacities were observed [53]. UC-MSCs inhibit the proliferation of human erythromyeloblastoid leukaemic cell line K562 through the secretion of IL-6 and IL-8 [12]. UC-MSCs also secrete IL-2 and interferon-gamma, which inhibit the proliferation of malignant glioma cell line C6.
8. Cell-cell contact
MSCs exist in a special microenvironment in which they can interplay with the host cells via gap junctions, tight junctions, adherens junctions and desmosomes to affect the proliferation, migration and differentiation of host cells.
The expression of connexin 43 (the major component of gap junction-mediated intercellular communication) increases in BM-MSCs co-cultured with multiple myeloma cell line RPMI8226, and the gap junction inhibitor 18a-GA inhibits the migration and adhension of RPMI8226 induced by MSCs [67]. BM-MSCs transfected with TRAIL combined with a gap junction inhibitor can enhance glioma cell death. Direct co-cultures of human glioblastoma cell lines (8-MGBA, 42-MG-BA and U-118 MG) with AT-MSCs transfected with herpes simplex virus-thymidine kinase and the prodrug ganciclovir (TK-MSC/GCV) resulted in a substantial decrease in viability in vitro through the formation of gap junctions between MSCs and glioblastoma cells; however, the inability of breast adenocarcinoma cell line MCF7 and cervical epitheloid carcinoma cell line HeLa to form gap junctions with MSCs renders these cells refractory to TK-MSC/GCV-mediated cytotoxicity [68]. The proliferation and vascular network formation of endothelial cells are decreased through the direct gap junction communication with UC-MSCs, possibly due to the secretion of cytokines and growth factors attributed to cell-to-cell contact [69]. UC-MSCs coculured with breast cancer cell line MDA-MB-231 can spontaneously generate hybrid/chimeric cell populations and induce expression of the GPI-anchored CD90 molecule in breast cancer cells, which can be partially blocked by a gap junction inhibitor in vitro and in vivo, leading to stimulation of breast cancer cell growth [70]. Our previous study showed that increased expression of CX43 on UC-MSCs promotes leukemia apoptosis [71].
Clinical applications of MSCs in anticancer therapies
MSCs are currently under investigation for a variety of clinical therapeutic trials. Despite constant doubt about the antitumor properties of MSCs, application of MSCs is a burgeoning strategy for cancer treatment in clinical settings
Some studies reviewed here have shown that MSCs can promote the growth of tumors. Nevertheless, some researchers have argued that malignant transformation is only a theoretical risk, and there is low probability of tumor formation after MSC transplantation [72]. A systematic review showed that tumorigenicity may occur only in patients suffering from ongoing or previous malignancy [73]. The safety and tolerability of autologous BM-MSCs with herpes simplex transduction has been studied in an ongoing phase I/II study (NCT02008539). The most frequent clinical use of MSCs thus far has been for blood disorders, especially graft-versus-host disease (GVHD) after allogeneic hematopoietic stem cell transplantation. MSC transplantation has been demonstrated to have therapeutic effects in some clinical pilot studies [74]. Twenty patients with hematologic malignancies received MSC transplantation together with allogeneic hematopoietic cell transfusion. The cotransplantation resulted in 80% overall survival and 60% progression-free survival. Similar treatments without MSC cotransplantation resulted in 44% overall survival and 38% progression-free survival. Serial MSC transfusions demonstrated an encouraging 78.1% 100-day survival rate, whereas patients who did not respond to the MSC infusions had 100-day survival of only 31%. The ex vivo culture of MSCs before transfusion decreases the overall survival of the patients, although phase I/II clinical trials have demonstrated the safety of the procedure. Another study of a very small number of cases also showed that co-transplantation of cultured MSCs in vitro could not only result in decreased survival, but also result in disease relapse [75]. Therefore, the co-transplatation of granulocyte-colony stimulating factor-mobilized BM could be a better way to treat cancers because of the higher MSC titer in the collection without the need for ex vivo culture [76, 77].
Currently, new clinical trials are recruiting patients to further explore the potential of MSCs in anticancer therapies (Table 2). Based on the observed data and ongoing studies, MSC transfusion is a promising tool to improve the efficiency of cancer treatments.
Registered trials of mesenchymal stem cells (MSCs) in anticancer therapies.
| Disease | Source of MSCs | Phase | Status | Registered ID |
|---|---|---|---|---|
| Advanced ovarian cancer | Allogeneic bone marrow | 1 | Not yet recruiting | NCT02530047 |
| Localized prostate cancer | Allogeneic bone marrow | 1 | Recruiting | NCT01983709 |
| Head and neck cancer | N/A | 1 | Not yet recruiting | NCT02079324 |
| Recurrent ovarian cancer | Adipose tissue | 1/2 | Not yet recruiting | NCT02068794 |
| Metastatic and refractory tumors. | Autologous bone marrow | 1/2 | Not yet recruiting | NCT01844661 |
| Hematologic malignancies | Third-party bone marrow | 1/2 | Recruiting | NCT01092026 |
| Hematologic malignancies | Allogeneic bone marrow | Not provided | Recruiting | NCT02181478 |
Note: The trials are registered at ClinicalTrials.gov. Search performed on October 28th, 2015.
Future clinical consideration
As described here, human-derived normal MSCs play important roles in anticancer therapies; however, UC-MSCs have produced better outcomes. Nevertheless, some challenges that include the available sources and convenience of obtaining MSCs, the quality control of in vitro-cultured MSCs, the appropriate cell passages, the optimum number of infusions, the optimum cell dose and the efficacy and safety of use remain to be discussed before large-scale clinical application of MSCs. Furthermore, and especially important, is the need to evaluate whether the rates of cancer relapse and infections increase when MSCs are used to treat or prevent diseases such as GVHD.
Acquiring MSCs from BM or adipose tissue involves an invasive and painful procedure. Human umbilical cord tissue seems to be the optimal source of adult multipotent stem cells for clinical application because MSCs from umbilical cord/cord blood are characterized by higher proliferation, faster self-renewal ability, more stable doubling time, lower immunogenicity, more feasible harvest and more effective anticancer properties than MSCs from BM or adipose tissues.
Many investigations have demonstrated that infusion of MSCs is safe and does not carry the risk of ectopic tissue formation or other severe adverse events. However, some preliminary studies also showed that MSC cultures can be infected with both cytomegalovirus and herpes simplex virus-1 in vitro. Even though MSCs of umbilical cord/cord blood are more primitive than MSCs of BM and adipose tissues, the safety of their use remains uncertain with the limited data thus far reported. Immunodeficient patients with ataxia telangiectasia developed a donor-derived brain tumor after transplantation of a mixture of fetally derived neural cells [78]. We are looking forward to obtaining more evidence of the safety and efficacy of MSCs as more and more clinical trials report their results.
One of the characteristics for MSCs is that possess a great propensity for expansion in vitro. Thus, passaged cells are extensively used in experimental and clinical practices, which may potentially lead with increasing transforming events to the establishment of a novel cell. MSCs within six passages are suggested for clinical applications because cells of increasing passages may also gradually lose the properties of early progenitors, such as the abilities of proliferation and differentiation. In addition, it is difficult to guarantee the quality of MSCs for physicians because malignant transformation as well as genomic mutations cannot be completely avoided in the complicated microenvironments in the body after implantation and long-term culture in vitro. Therefore, genetic testing of the donor, strict control of cell-handling procedures and long-term supervision is needed. Banks of cryopreserved/manufactured MSCs, such as cord blood banks, may serve to improve several issues regarding the application of MSCs to clinical practice, including the variability of donors, the time needed to obtain sufficient cells and the risk of contamination, and could provide a ready supply of MSCs for clinical use.
According to recent reports, the majority of patients in clinical trials thus far have received MSCs at dosages of 1-9×106/kg. However, the optimum therapeutic cell dose has not been well established. The immunosuppressive effects of MSCs have been seen to increase in a dose-dependent manner in vitro. However, increased therapeutic effects are not guaranteed with increased MSC infusion. On the other hand, the donor MSCs are gradually eliminated from the recipient after infusion. Accordingly, several MSC infusions may be needed to develop immune tolerance in patients with severe immune disorders. MSCs administered at different ratios to cancer cells show different results. Therefore, further studies are warranted to define the optimum dose of MSCs, as well as the optimum number of infusions.
Enhancing the efficiency of delivering MSCs and cell homing could improve their therapeutic effects. It is necessary to improve approaches to promoting migration and longevity of MSCs in specific tissues. Bioengineering approaches and genetic modifications may provide opportunities to solve these problems in the future. Furthermore, at the present, it is very difficult to mimic human baseline conditions that are associated with the occurrence, development and prognosis of diseases. Therefore, laboratory disease models are in dire need of common standards.
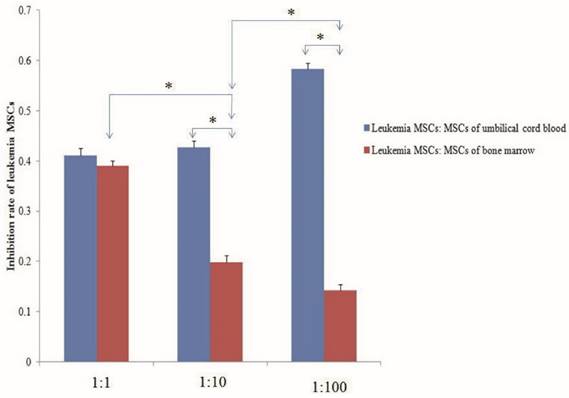
Despite the ability of MSCs to differentiate into multiple cell lineages in vitro, recent evidence suggests that the fate of MSCs may be restricted in BM in vivo, with only the ability to replenish osteogenic lineages, such as the osteoblasts that form part of the endosteal niche. A recent report described how malignant myeloid cells profoundly reprogram the endosteal BM osteoblasts into a pro-inflammatory BM niche that supports LSCs while creating an inhospitable environment for normal HSCs. Indeed, human LSCs have been transplanted in vivo and been shown to home to the epiphyseal osteoblastic surface of the endosteum before later dispersion to perivascular niches in vessels near the endosteum and diaphysis. MSC-derived osteoblasts are more harbinger than host in haematopoiesis. However, the abnormal differentiation and location in the niche is more related to the growth of LSCs than to normal hematopoietic stem cells. Recent research showed that AML induces MSCs to differentiate into osteoblasts, increasing their number above normal levels; however, the number of osteoblasts normalized after removal of AML. Larger amounts of osteoblasts have also been observed in patients with AML, with osteoblast numbers higher during relapse than in remission. Gene expression and in vitro functions of MSCs from leukemia also change. Thus, we should pay attention to whether MSCs in vivo and in vitro can eliminate leukemia MSCs, re-educate the leukemia MSCs to act as normal MSCs, or induce osteoblasts to reduce to normal levels. Preliminary studies from our lab have shown that both the MSCs of umbilical cord blood and of normal BM co-cultured with leukemia MSCs can decrease the numbers of leukemia MSCs. The inhibition rate of leukemia MSCs was significantly higher with the MSCs of umbilical cord blood than with the MSCs of normal BM. The inhibition rate increased with the number of UC-MSCs used, but decreased with increasing numbers of MSCs from normal BM [Fig 2].
Normal mesenchymal stem cells (MSCs) inhibit the proliferation of leukemia MSCs. MSCs of umbilical cord blood and normal bone marrow and leukemia MSCs were isolated and cultured according to our previous report. The second passage leukemia MSCs stained with CMDil (1×104) were cultured for 24 h, then the MSCs of umbilical cord blood and bone marrow were co-cultured with the leukemia MSCs with the ratio of 1:1, 10:1 and 100:1, respectively. After five days, the numbers of leukemia MSCs were detected by flow cytometry. The inhibition rate of leukemia MSCs was calculated according to the following: inhibition rate = (numbers before co-culture - numbers after co-culture) / numbers before co-culture. The inhibition rate of the MSCs from umbilical cord blood was significantly higher than that of the MSCs from normal BM. The inhibition rate increased as the number of umbilical cord MSCs increased, but decreased as the number of BM-MSCs increased. * p<0.01.
In addition to their primary effects, MSCs could be employed as carriers of multiple anticancer agents because they can track microscopic tumors and pathological lesions without incurring the risk of spreading tumor cells. Therefore, they could be used as a vehicle for anticancer drugs or tumor-suppressor genes when drug resistance occurs or systemic treatments fail. Because of their larger loading capacity, better durability and lower risk of spreading tumors, MSCs of the umbilical cord/cord blood can be considered as an ideal delivery vector for multiple anticancer agents.
Recently, two phenotypes of MSCs, called MSC1 and MSC2, have been isolated from a mixed population of MSCs using a completely novel approach, and the researchers found that MSC1s are pro-inflammatory while MSC2s are immunosuppressive in nature [79]. Researchers in the same group reported that MSC1s are anti-tumorigenic while MSC2s are pro-tumorigenic in nature [80]. The appropriate culture, isolation and identification of the MSC1 and MSC2 subpopulations may help resolve some of the surrounding controversies to advance the safe use of MSCs in anticancer therapies.
In summary, although controversy exists and cell-based therapy will not completely take the place of conventional therapeutic tools, researchers still hold the view that cancer therapy based on MSCs has a promising future. However, many problems still remain to be solved before clinical applications of MSCs can be more widely implemented. We believe that the era of the MSCs as a new clinical paradigm of anticancer therapies is upcoming.
Acknowledgements
This study was funded by grants from the National Natural Science Foundation (No. 81170529), the China Scholarship Council (No. 201308505213) and the special foundation for the “1130 project” of Xinqiao Hospital of Third Military Medical University.
Authorship contributions: All authors designed the experiment and collected data; ZC and ZJF wrote the paper; and all authors discussed and commented on the manuscript.
Conflict of interest
None declared.
References
1. Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21:309-22
2. Gao L, Chen XH, Zhang X. et al. Contribution of SDF-1/CXCR4 axis on proliferation of megakaryocyte co-cultured with human umbilical cord blood-derived stromal cells. Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2009;17:412-6
3. Yuan Y, Lu X, Chen X. et al. Jagged1 contributes to the drug resistance of jurkat cells in contact with human umbilical cord derived mesenchymal stem cells. Oncol Lett. 2013;6:1000-6
4. Liu Y, Chen XH, Si YJ. et al. Reconstruction of hematopoietic inductive microenvironment after transplantation of VCAM-1-modified human umbilical cord blood stromal cells. PLoS One. 2012;7:e31741
5. Boutter J, Huang Y, Marovca B. et al. Image-based RNA interference screening reveals an individual dependence of acute lymphoblastic leukemia on stromal cysteine support. Oncotarget. 2014;5:11501-12
6. Zhang Y, Hu K, Hu Y. et al. Bone marrow mesenchymal stromal cells affect the cell cycle arrest effect of genotoxic agents on acute lymphocytic leukemia cells via p21 down-regulation. Ann Hematol. 2014;93:1499-508
7. Yuan Y, Lu X, Chen X. et al. Jagged1 contributes to the drug resistance of jurkat cells in contact with human umbilical cord derived mesenchymal stem cells. Oncol Lett. 2013;6:1000-6
8. Yuan Y, Chen D, Chen X. et al. Human umbilical cord-derived mesenchymal stem cells inhibit proliferation but maintain survival of Jurkat leukemia cells in vitro by activating Notch signaling. Nan Fang Yi Ke Da Xue Xue Bao. 2014;34:441-7
9. Mousavi Niri N, Jaberipour M, Razmkhah M. et al. Mesenchymal stem cells do not suppress lymphoblastic leukemic cell line proliferation. Iran J Immunol. 2009;6:186-94
10. Liang X, Hao L, Chen X. et al. Effects of bone marrow stromal cells and umbilical cord blood-derived stromal cells on daunorubicin-resistant residual Jurkat cells. Transplant Proc. 2010;42:3767-72
11. Tabe Y, Konopleva M, Munsell MF. et al. PML-RARα is associated with leptin-receptor induction: the role of mesenchymal stem cell-derived adipocytes in APL cell survival. Blood. 2004;103:1815-22
12. Fonseka M, Ramasamy R, Tan BC. et al. Human umbilical cord blood-derived mesenchymal stem cells (hUCB-MSC) inhibit the proliferation of K562 (human erythromyeloblastoid leukaemic cell line). Cell Biol Int. 2012;36:793-801
13. Yuan Y, Lu X, Tao CL. et al. Forced expression of indoleamine-2, 3-dioxygenase in human umbilical cord-derived mesenchymal stem cells abolishes their anti-apoptotic effect on leukemia cell lines in vitro. In Vitro Cell Dev Biol Anim. 2013;49:752-8
14. Wang H, Zhou J, Xu JJ. et al. Comparison and analysis between CLL-hBMSC and N-hBMSC. Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2014;22:914-9
15. Tung S, Shi Y, Wong K. et al. PPARα and fatty acid oxidation mediate glucocorticoid resistance in chronic lymphocytic leukemia. Blood. 2013;122:969-80
16. Secchiero P, Zorzet S, Tripodo C. et al. Human bone marrow mesenchymal stem cells display anti-cancer activity in SCID mice bearing disseminated non-Hodgkin's lymphoma xenografts. PLoS One. 2010;5:e11140
17. Lin HD, Fong CY, Biswas A. et al. Human Wharton's jelly stem cells, its conditioned medium and cell-free lysate inhibit the growth of human lymphoma cells. Stem Cell Rev. 2014;10:573-86
18. Ciavarella S, Caselli A, Tamma AV. et al. A peculiar molecular profile of umbilical cord-mesenchymal stromal cells drives their inhibitory effects on multiple myeloma cell growth and tumor progression. Stem Cells Dev. 2015;24:1457-70
19. Gao L, Zhang C, Zhang X. et al. Human umbilical cord blood-derived stromal cells: A new resource for the proliferation and apoptosis of myeloma cells. Hematology. 2014;19:148-57
20. Goldstein RH, Reagan MR, Anderson K. et al. Human bone marrow-derived MSCs can home to orthotopic breast cancer tumors and promote bone metastasis. Cancer Res. 2010;70:10044-50
21. Iyengar P, Combs TP, Shah SJ. et al. Adipocyte-secreted factors synergistically promote mammary tumorigenesis through induction of anti-apoptotic transcriptional programs and protooncogene stabilization. Oncogene. 2003;22:6408-23
22. Leng L, Wang Y, He N. et al. Molecular imaging for assessment of mesenchymal stem cells mediated breast cancer therapy. Biomaterials. 2014;35:5162-70
23. Ma F, Chen D, Chen F. et al. Human umbilical cord mesenchymal stem cells promote breast cancer metastasis by interleukin-8 and interleukin-6 dependent induction of CD44+/CD24- cells. Cell Transplant. 2015;24:2585-99
24. Li T, Zhao S, Song B. et al. Effects of transforming growth factor β-1 infected human bone marrow mesenchymal stem cells on high- and low-metastatic potential hepatocellular carcinoma. Eur J Med Res. 2015;20:56
25. Li T, Song B, Du X. et al. Effect of bone-marrow-derived mesenchymal stem cells on high-potential hepatocellular carcinoma in mouse models: an intervention study. Eur J Med Res. 2013;18:34
26. Zhao W, Ren G, Zhang L. et al. Efficacy of mesenchymal stem cells derived from human adipose tissue in inhibition of hepatocellular carcinoma cells in vitro. Cancer Biother Radiopharm. 2012;27:606-13
27. Yulyana Y, Ho IA, Sia KC. et al. Paracrine factors of human fetal MSCs inhibit liver cancer growth through reduced activation of IGF-1R/PI3K/Akt signaling. Mol Ther. 2015;23:746-56
28. De Boeck A, Pauwels P, Hensen K. et al. Bone marrow-derived mesenchymal stem cells promote colorectal cancer progression through paracrine neuregulin 1/HER3 signalling. Gut. 2013;62:550-60
29. Chen D, Liu S, Ma H. et al. Paracrine factors from adipose-mesenchymal stem cells enhance metastatic capacity through Wnt signaling pathway in a colon cancer cell co-culture model. Cancer Cell Int. 2015;15:42
30. Wang M, Cai J, Huang F. et al. Pre-treatment of human umbilical cord-derived mesenchymal stem cells with interleukin-6 abolishes their growth-promoting effect on gastric cancer cells. Int J Mol Med. 2015;35:367-75
31. Wang Y, Fan H, Zhou B. et al. Fusion of human umbilical cord mesenchymal stem cells with esophageal carcinoma cells inhibits the tumorigenicity of esophageal carcinoma cells. Int J Oncol. 2012;40:370-7
32. Jing W, Chen Y, Lu L. et al. Human umbilical cord blood-derived mesenchymal stem cells producing IL-15 eradicate established pancreatic tumor in syngeneic mice. Mol Cancer Ther. 2014;13:2127-37
33. Shin SH, Kim J, Heo SC. et al. Proteomic identification of betaig-h3 as a lysophosphatidic acid-induced secreted protein of human mesenchymal stem cells: paracrine activation of A549 lung adenocarcinoma cells by betaig-h3. Mol Cell Proteomics. 2012;11:M111.012385
34. Matsuzuka T, Rachakatla RS, Doi C. et al. Human umbilical cord matrix-derived stem cells expressing interferon-β gene significantly attenuate bronchioloalveolar carcinoma xenografts in SCID mice. Lung Cancer. 2010;70:28-36
35. Takahara K, Ii M, Inamoto T. et al. Adipose-derived stromal cells inhibit prostate cancer cell proliferation inducing apoptosis. Biochem Biophys Res Commun. 2014;446:1102-7
36. Han I, Yun M, Kim EO. et al. Umbilical cord tissue-derived mesenchymal stem cells induce apoptosis in PC-3 prostate cancer cells through activation of JNK and downregulation of PI3K/AKT signaling. Stem Cell Res Ther. 2014;5:54
37. Hsiao WC, Sung SY, Liao CH. et al. Vitamin D3-inducible mesenchymal stem cell-based delivery of conditionally replicating adenoviruses effectively targets renal cell carcinoma and inhibits tumor growth. Mol Pharm. 2012;9:1396-408
38. Chu Y, Tang H, Guo Y. et al. Adipose-derived mesenchymal stem cells promote cell proliferation and invasion of epithelial ovarian cancer. Exp Cell Res. 2015;337:16-27
39. Klopp AH, Zhang Y, Solley T. et al. Omental adipose tissue-derived stromal cells promote vascularization and growth of endometrial tumors. Clin Cancer Res. 2012;18:771-82
40. Gauthaman K, Yee FC, Cheyyatraivendran S. et al. Human umbilical cord Wharton's jelly stem cell (hWJS) extracts inhibit cancer cell growth in vitro. J Cell Biochem. 2012;113:2027-39
41. Yang C, Lei D, Ouyang W. et al. Conditioned media from human adipose tissue-derived mesenchymal stem cells and umbilical cord-derived mesenchymal stem cells efficiently induced the apoptosis and differentiation in human glioma cell lines in vitro. Biomed Res Int. 2014;2014:109389
42. Rowan BG, Lacayo EA, Sheng M. et al. Human adipose tissue-derived stromal/stem cells promote migration and early metastasis of head and neck cancer xenografts. Aesthet Surg J. 2016;36:93-104
43. Ahn JO, Coh YR, Lee HW. et al. Human adipose tissue-derived mesenchymal cells inhibit melanoma growth in vitro and in vivo. Anticancer Res. 2015;35:159-68
44. Kim YH, Cho SH, Lee SJ. et al. Growth-inhibitory effect of neurotrophin-3-secreting adipose tissue-derived mesenchymal stemcells on the D283-MED human medulloblastoma cell line. J Neurooncol. 2012;106:89-98
45. Wen S, Niu Y, Yeh S. et al. BM-MSCs promote prostate cancer progression via the conversion of normal fibroblasts to cancer-associated fibroblasts. Int J Oncol. 2015;47:719-27
46. Mishra PJ, Mishra PJ, Humeniuk R. et al. Carcinoma-associated fibroblast-like differentiation of human mesenchymal stem cells. Cancer Res. 2008;68:4331-9
47. Subramanian A, Shu-Uin G, Kae-Siang N. et al. Human umbilical cord Wharton's jelly mesenchymal stem cells do not transform to tumor-associated fibroblasts in the presence of breast and ovarian cancer cells unlike bone marrow mesenchymal stem cells. J Cell Biochem. 2012;113:1886-95
48. Cho JA, Park H, Lim EH. et al. Exosomes from breast cancer cells can convert adipose tissue-derived mesenchymal stem cells into myofibroblast-like cells. Int J Oncol. 2012;40:130-8
49. Del Valle PR, Milani C, Brentani MM. et al. Transcriptional profile of fibroblasts obtained from the primary site, lymph node and bone marrow of breast cancer patients. Genet Mol Biol. 2014;37:480-9
50. Dvořánková B, Smetana K Jr, Říhová B. et al. Cancer-associated fibroblasts are not formed from cancer cells by epithelial-to-mesenchymal transition in nu/nu mice. Histochem Cell Biol. 2015;43:463-9
51. Özdemir BC, Pentcheva-Hoang T, Carstens JL. et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. 2014;25:719-34
52. Martin FT, Dwyer RM, Kelly J. et al. Potential role of mesenchymal stem cells (MSCs) in the breast tumour microenvironment: stimulation of epithelial to mesenchymal transition (EMT). Breast Cancer Res Treat. 2010;124:317-26
53. Xu Q, Wang L, Li H. et al. Mesenchymal stem cells play a potential role in regulating the establishment and maintenance of epithelial-mesenchymal transition in MCF7 human breast cancer cells by paracrine and induced autocrine TGF-β. Int J Oncol. 2012;41:959-68
54. Velpula KK, Dasari VR, Tsung AJ. et al. Cord blood stem cells revert glioma stem cell EMT by down regulating transcriptional activation of Sox2 and Twist1. Oncotarget. 2011;2:1028-42
55. Ye X, Weinberg RA. Epithelial-mesenchymal plasticity: A central regulator of cancer progression. Trends Cell Biol. Trends Cell Biol. 2015;25:675-86
56. Jose S, Tan SW, Ooi YY. et al. Mesenchymal stem cells exert anti-proliferative effect on lipopolysaccharide-stimulated BV2 microglia by reducing tumour necrosis factor-α levels. J Neuroinflammation. 2014;11:149
57. Johann PD, Vaegler M, Gieseke F. et al. Tumour stromal cells derived from paediatric malignancies display MSC-like properties and impair NK cell cytotoxicity. BMC Cancer. 2010;10:501
58. Barnas JL, Simpson-Abelson MR, Brooks SP. et al. Reciprocal functional modulation of the activation of T lymphocytes and fibroblasts derived from human solid tumors. J Immunol. 2010;185:2681-92
59. Ferrand J, Noël D, Lehours P. et al. Human bone marrow-derived stem cells acquire epithelial characteristics through fusion with gastrointestinal epithelial cells. PLoS One. 2011;6:e19569
60. Xu MH, Gao X, Luo D. et al. EMT and acquisition of stem cell-like properties are involved in spontaneous formation of tumorigenic hybrids between lung cancer and bone marrow-derived mesenchymal stem cells. PLoS One. 2014;9:e87893
61. Kuhbier JW, Bucan V, Reimers K. et al. Observed changes in the morphology and phenotype of breast cancer cells indirect co-culture with adipose-derived stem cells. Plast Reconstr Surg. 2014;134:414-23
62. Wei HJ, Nickoloff JA, Chen WH. et al. FOXF1 mediates mesenchymal stem cell fusion-induced reprogramming of lung cancer cells. Oncotarget. 2014;5:9514-29
63. Zhang MJ, Sun JJ, Qian L. et al. Human umbilical mesenchymal stem cells enhance the expression of neurotrophic factors and protect ataxic mice. Brain Res. 2011;1402:122-31
64. Widder M, Lutzkendorf J, Caysa H. et al. Multipotent mesenchymal stromal cells promote tumor growth in distinct colorectal cancer cells by a β-integrin-dependent mechanism. Int J Cancer. 2015
65. Sun B, Yu KR, Bhandari DR. et al. Human umbilical cord blood mesenchymal stem cell-derived extracellular matrix prohibits metastatic cancer cell MDA-MB-231 proliferation. Cancer Lett. 2010;296:178-85
66. Chan JK, Gnecchi M, Huynh H. et al. Paracrine factors of human fetal MSCs inhibit liver cancer growth through reduced activation of IGF-1R/PI3K/Akt signaling. Mol Ther. 2015;23:746-56
67. Zhang XH, Sun Y, Wang ZY. et al. Alterations of connexin 43 expression and SDF-1α secretion of bone marrow mesenchymal stem cells co-cultured with myeloma cells. Zhonghua Xue Ye Xue Za Zhi. 2013;34:788-93
68. Matuskova M, Hlubinova K, Pastorakova A. et al. HSV-tk expressing mesenchymal stem cells exert bystander effect on human glioblastoma cells. Cancer Lett. 2010;290:58-67
69. Menge T, Gerber M, Wataha K. et al. Human mesenchymal stem cells inhibit endothelial proliferation and angiogenesis via cell-cell contact through modulation of the VE-Cadherin/beta-catenin signaling pathway. Stem Cells Dev. 2013;22:148-57
70. Mandel K, Yang Y, Schambach A. et al. Mesenchymal stem cells directly interact with breast cancer cells and promote tumor cell growth in vitro and in vivo. Stem Cells Dev. 2013;22:3114-27
71. Yang S, Wen Q, Liu Y. et al. Increased expression of CX43 on stromal cells promotes leukemia apoptosis. Oncotarget. 2015;6:44323-31
72. Prockop DJ, Brenner M, Fibbe WE. et al. Defining the risks of mesenchymal stromal cell therapy. Cytotherapy. 2010;12:576-8
73. Lalu MM, McIntyre L, Pugliese C. et al. Safety of cell therapy with mesenchymal stromal cells (SafeCell): a systematic review and meta-analysis of clinical trials. PLoS One. 2012;7:e47559
74. Cammarota F, Laukkanen MO. Mesenchymal stem/stromal cells in stromal evolution and cancer progression. Stem cell Int. 2015;2015:427350
75. Ning H, Yang F, Jiang M. et al. The correlation between cotransplantation of mesenchymal stem cells and higher recurrence rate in hematologic malignancy patients: outcome of a pilot clinical study. Leukemia. 2008;22:593-9
76. Zhang C, Zhang X, Chen XH. Cellular mechanism for granulocyte-colony stimulating factor in the prevention of graft-versus-host disease in combined bone marrow and peripheral blood transplantation for hematological malignancies: The composition in collection. Transfus Apher Sci. 2013;48:3-9
77. Zhang C, Zhang X, Chen XH. Granulocyte-colony stimulating factor-mobilized mesenchymal stem cells: A new resource for rapid engraftment in hematopoietic stem cell transplantation. Medical Hypotheses. 2011;6:241-3
78. Glenn JD, Whartenby KA. Mesenchymal stem cells: Emerging mechanisms of immunomodulation and therapy. World Journal of Stem Cells. 2014;6:526-39
79. Waterman RS, Tomchuck SL, Henkle SL, Betancourt AM. A new mesenchymal stem cell (MSC) paradigm: polarization into a pro-inflammatory MSC1 or an Immunosuppressive MSC2 phenotype. PLoS One. 2010;5:e10088
80. Waterman RS, Henkle SL, Betancourt AM. Mesenchymal stem cell 1 (MSC1)-based therapy attenuates tumor growth whereas MSC2-treatment promotes tumor growth and metastasis. PLoS One. 2012;7:e45590
Author contact
![]() Corresponding author: Department of Hematology, Xinqiao Hospital, Third Military Medical University, Chongqing, 400037, People's Republic of China. E-mail: zhangxxicom (Zhang X). Fax: +86-023-6875-5609.
Corresponding author: Department of Hematology, Xinqiao Hospital, Third Military Medical University, Chongqing, 400037, People's Republic of China. E-mail: zhangxxicom (Zhang X). Fax: +86-023-6875-5609.