Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2016; 7(15):2280-2289. doi:10.7150/jca.15758 This issue Cite
Research Paper
Aberrant methylation of CDH13 can be a diagnostic biomarker for lung adenocarcinoma
Weilin Pu1*, Xin Geng2*, Sidi Chen1, Lixing Tan1, Yulong Tan2, An Wang3, Zhouyi Lu3, Shicheng Guo4 ![]() , Xiaofeng Chen2
, Xiaofeng Chen2 ![]() , Jiucun Wang1
, Jiucun Wang1 ![]()
1. State Key Laboratory of Genetic Engineering and Ministry of Education Key Laboratory of Contemporary Anthropology, School of Life Sciences, Fudan University, Shanghai 200433, China;
2. Department of Cardiothoracic Surgery, Huashan Hospital, Fudan University, Shanghai 200032, China;
3. Department of Chest Surgery, Shanghai Pulmonary Hospital, Shanghai 200433, China.
4. Department of Bioengineering, University of California at San Diego, 9500 Gilman Drive, MC0412, La Jolla, CA 92093-0412.
* co-first author
Received 2016-4-6; Accepted 2016-9-18; Published 2016-11-25
Abstract
Background: Aberrant methylation of CpG islands in tumor cells in promoter regions is a critical event in non-small cell lung carcinoma (NSCLC) tumorigenesis and can be a potential diagnostic biomarker for NSCLC patients. The present study systemically and quantitatively reviewed the diagnostic ability of CDH13 methylation in NSCLC as well as in its subsets. Eligible studies were identified through searching PubMed, Web of Science, Cochrane Library and Embase. The pooled odds of CDH13 promoter methylation in lung cancer tissues versus normal controls were calculated by meta-analysis method. Simultaneously, four independent DNA methylation datasets of NSCLC from TCGA and GEO database were downloaded and analyzed to validate the results from meta-analysis. Results: Thirteen studies, including 1850 samples were included in this meta-analysis. The pooled odds ratio of CDH13 promoter methylation in cancer tissues was 7.41 (95% CI: 5.34 to 10.29, P < 0.00001) compared with that in controls under fixed-effect model. In validation stage, 126 paired samples from TCGA were analyzed and 5 out of the 6 CpG sites in the CpG island of CDH13 were significantly hypermethylated in lung adenocarcinoma tissues but none of the 6 CpG sites was hypermethylated in squamous cell carcinoma tissues. Concordantly, the results from other three datasets, which were subsequently obtained from GEO database consisting of 568 tumors and 256 normal tissues, also consisted with those from TCGA dataset. Conclusion: The pooled data showed that the methylation status of the CDH13 promoter is strongly associated with lung adenocarcinoma. The CDH13 methylation status could be a promising diagnostic biomarker for diagnosis of lung adenocarcinoma.
Keywords: CDH13, DNA methylation, Non-small cell lung cancer, NSCLC, Diagnosis, Adenocarcinoma, Biomarker.
Background
Lung cancer is a complicated disease involving genetic and epigenetic variation, and is the leading cause of cancer death all over the world [1]. Lung cancer is currently poorly diagnosed in the early stages. Non-small cell lung cancer (NSCLC) comprises the majority of lung cancer and has an increasing incidence and mortality in the last two decades in China and in the world [2-4]. The overall five-year survival rates for late stage III and IV of NSCLC patients were just 5%-14% and 1% respectively, however, the rate could come up to 63% for the early stage IA if treated with surgery properly [5, 6].
DNA methylation is one of the key epigenetic modifications in eukaryote, regulating genes and microRNAs expression [7], gene alternative splicing [8], playing an important role in carcinogenesis. Moreover, with the advantages of stable chemical property, detection ability in remote patient media, quantitative signal, relatively low cost in detection, DNA methylation has been regarded as a promising non-invasive biomarker for the early detection of lung cancer [9].
The CDH13 (cadherin 13) gene was isolated recently and has been mapped to 16q24[8]. CDH13 gene is a unique member of the cadherin superfamily due to the devoid of a transmembrane domain. Instead, it uses the glycosylphosphatidylinositol (GPI) anchor to attach to the exterior surface of the plasma membrane [10]. It was shown that the expression of CDH13 could be down-regulated through hypermethylation of gene promoter region [11]. Alterations, like promoter hypermethylation and loss of function of CDH13 gene have been detected in breast cancer [12] and lung cancer[13-15], in pituitary adenoma[16], diffuse large B cell lymphoma[17], and nasopharyngeal carcinoma[18]. Moreover, CDH13 gene has been suggested as an promising early detecting marker for lung cancer [19].
In the past decades, a large number of DNA methylation-based biomarkers of NSCLC have been identified. The diagnostic abilities or risk associations for several of them have been quantitatively evaluated such as SHOX2 [20], APC [21], RASSF1A [22], FHIT [23], MGMT [24], RUNX3 [25], RARbeta [26], E-cadherin [27] and P16 [28]. Zhong and colleagues have conducted a meta-analysis to evaluate the association between CDH13 promoter methylation and NSCLC [29], while only case-control studies were included. We noticed that, recently, the Cancer Genome Atlas project (TCGA) and Gene Expression Omnibus (GEO) databases have collected several independent whole genome DNA methylation microarray datasets of NSCLC with comprehensive clinical and demographic information, providing additional resources that may be without publication bias [30]. Therefore, in order to give a more robust and unbiased conclusion of the association between CDH13 promoter methylation and NSCLC, we innovatively integrated these microarray datasets with the data from published articles to evaluate the diagnostic ability of the CDH13 methylation test in NSCLC comprehensively.
Results
Study characteristics
The electronic search strategy identified 365 potentially relevant articles (PubMed, 73; Web of science, 177; Embase, 115; Cochrane Library, 0), which were further screened for inclusion on the basis of their titles, abstracts, full texts, or a combination of these terms. The electronic search was supplemented from reference lists of relevant articles including reviews. Finally, 13 studies with data on the relationship between CDH13 gene promoter methylation and NSCLC were pooled for analysis (Table 1) [15, 19, 31-41]. All these articles were written in English. Given the diagnostic feature of our research, the quality of the selected papers were critically examined using the QUADAS tool (Table S5) [42]. In total, 1206 lung cancer tissues/serum and 644 normal counterpart tissues/serum were collected (Some articles studied with serum and some studies with plasma, to simplify, we use serum instead of serum/plasma). The age of the subjects in the 13 studies ranged from 26 to 87 years, with mean or median ranging from 59 to 70 years. As for the study aim, 5 articles were especially aiming at diagnosis, while the others were for prognosis, survival research, and so on. Among 13 studies, the proportions of stage I samples differed from 9.52 to 68.57%, and the percentage of male individuals in the NSCLC samples ranged from 52 to 80%. In terms of the methylation detection methods, 8 of 13 inclusions used methylation-specific polymerase chain reaction (MSP), while others used quantitative MSP (qMSP, such as Methylight, Pyrosequencing, and so on). Four kinds of methylation detection primers or probes were found to be utilized for most of the 13 studies (Table S1).
Characteristics of eligible studies considered in the report.
| Author | Sample Type | Agea | Stage I% | Stage (I+II)% | Male% | Patients (M/T) | Control (M/T) | Methods | Aim | Multiple target | Control design | Ad% | Primer set |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Dong et al | tissue | 63 | 0.580 | 0.761 | 0.795 | 26/88 | 7/88 | MSP | Non-Diagnosis | Multi | hom | 0.398 | 1 |
| Feng et al | tissue | 64.3 | 0.429 | 0.776 | 0.531 | 11/49 | 1/49 | qMSP | Non-Diagnosis | Multi | hom | 0.588 | 1 |
| Hanabata et al | tissue | NA | 0.686 | 0.843 | 0.629 | 26/70 | 2/30 | MSP | Non-Diagnosis | Multi | hom | 0.688 | 1 |
| Hsu et alb | tissue | 66.81 | NA | 0.651 | NA | 28/63 | 10/63 | MSP | Diagnosis | Multi | hom | 0.759 | 1 |
| Jin et al | tissue | 66.7 | NA | NA | 0.708 | 25/72 | 2/63 | qMSP | Non-Diagnosis | Multi | hom | 0.652 | 3 |
| Nikolaidis et al | tissue | 65.58 | NA | 0.771 | 0.521 | 11/48 | 0/48 | qMSP | Diagnosis | Multi | hom | 0.500 | 4 |
| Toyooka et al | tissue | NA | NA | NA | NA | 18/42 | 2/25 | MSP | Non-Diagnosis | Single | hom | 0.738 | 1 |
| Toyooka et al | tissue | 63 | NA | NA | 0.691 | 180/514 | 5/84 | MSP | Non-Diagnosis | Multi | hom | 0.606 | 1 |
| Tsou et al | tissue | NA | NA | NA | NA | 39/51 | 24/49 | qMSP | Diagnosis | Multi | both | 1 | 2 |
| Ulivi et al | serum | 70 | 0.557 | 0.721 | 0.803 | 14/61 | 0/15 | qMSP | Diagnosis | Multi | heter | 0.692 | 1 |
| Wang et al | tissue | NA | NA | NA | 0.607 | 15/28 | 3/12 | MSP | Diagnosis | Multi | hom | 0.682 | NA |
| Zhai et al | serum | 62.39 | 0.095 | 0.143 | 0.762 | 23/42 | 0/40 | MSP | Non-Diagnosis | Multi | heter | 0.762 | 1 |
| Zhang et alb | tissue | 59 | 0.321 | 0.744 | 0.744 | 38/78 | 8/78 | MSP | Non-Diagnosis | Multi | hom | 0.455 | 1 |
amean or median age from articles. bwith two records since there are tissue and serum data simultaneously in this article. M and T means number of methylation positive and total samples, respectively. qMSP is short for the quantitative methylation-specific polymerase chain reaction method. MSP is short for the methylation-specific polymerase chain reaction method.
Meta-analysis
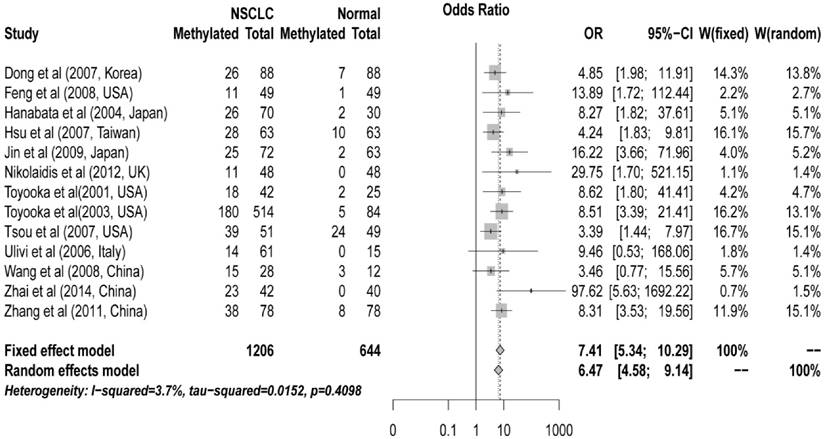
The pooled ORs for CDH13 methylation in cancer samples compared with that in normal controls were 6.47 (95% CI: 4.58 to 9.14, z = 10.59, P < 0.00001) in random effects model using DerSimonian and Laird method, and 7.41 (95% CI: 5.34 to 10.29, z = 11.96, P < 0.0001) in fixed effects model using Mantel-Haenszel method, demonstrating a statistically significant increase in likelihood of methylation in lung cancer tissues comparing to controls. A homogeneity analysis revealed that the variation among them was not significant (I2 = 3.7%, tau2 = 0.015) (Figure 1).
Forest plot of Meta-analysis for association between CDH13 promoter hyper-methylation and non-small cell lung cancer (NSCLC). Author, year, country of the studies and methylated (M) and total number of the sample (T) in case and control, combined odds ratio (OR) with 95% confidence region were labeled in the left column of the figure. The DerSimonian-Laird estimator and Mantel-Haenszel method were selected to conduct combination estimation for the random effects model and fixed effect model, respectively.
Subgroup analyses were conducted for different subtypes, including sample types (tissue or serum), age, counterpart categories, proportion of early stage, aim of the study (for diagnosis or non-diagnosis), proportion of adenocarcinoma samples (Ad%) and other possible confounding factors (Table 2). Significant differences were found only between the ORs of the diagnosis (4.70, 95% CI: 2.77 to 7.95) and non-diagnosis (9.33, 95% CI: 6.09 to 14.28) subgroups (P = 0.047) (Figure 2A). Both tissue and serum subgroups showed significant association between CDH13 methylation and NSCLC (OR = 6.75 and 9.07, respectively; P = 0.48) (Figure 2B) which suggested that CDH13 methylation can be taken as a potential biomarker for NSCLC diagnosis using either tissue or serum samples. No significant difference was found between subgroups of MSP and qMSP (OR = 7.26 and 7.85, respectively; P = 0.84), suggesting the two methods were equivalent in methylation status detection (Figure 2C). In addition, there were no significant differences between the subgroups of proportion of male samples, proportion of early stage, proportion of adenocarcinoma samples, the primer sets as well as other factors (Table 2).
Subgroup analysis for the main potential confounding factors with fixed effect model.
| Subgroup | Number of study | OR | Lower | Upper | I2 | P-value |
|---|---|---|---|---|---|---|
| Overall | 13 | 7.41 | 5.34 | 10.29 | 0.00% | |
| Age<64.3 | 4 | 8.64 | 5.23 | 14.26 | 31.30% | |
| Age>=64.3 | 5 | 8.50 | 4.50 | 16.04 | 7.30% | 0.97 |
| Stage I<49.3% | 3 | 13.22 | 6.33 | 27.60 | 33.70% | |
| Stage I>=49.3% | 3 | 6.06 | 2.88 | 12.78 | 0.00% | 0.14 |
| Stage I+II<75.2% | 4 | 8.20 | 4.72 | 14.25 | 43.20% | |
| Stage I+II>=75.2% | 4 | 7.73 | 3.88 | 15.38 | 0.00% | 0.90 |
| Male %<70% | 5 | 8.71 | 4.60 | 16.49 | 0.00% | |
| Male %>=70% | 5 | 9.68 | 5.67 | 16.53 | 25.60% | 0.80 |
| MSP | 8 | 7.26 | 4.95 | 10.65 | 0.00% | |
| qMSP | 5 | 7.85 | 4.17 | 14.78 | 30.50% | 0.84 |
| Diagnose | 5 | 4.70 | 2.77 | 7.95 | 0.00% | |
| Non-diagnose | 8 | 9.33 | 6.09 | 14.28 | 0.00% | 0.047 |
| Heterogeneous | 2 | 33.79 | 4.65 | 245.37 | 22.10% | |
| Autogenous | 10 | 7.45 | 5.15 | 10.77 | 0.00% | 0.14 |
| Plasma/Serum | 4 | 9.07 | 4.29 | 19.17 | 58.60% | |
| Tissue | 11 | 6.75 | 4.82 | 9.45 | 0.00% | 0.48 |
| Ad%<65% | 5 | 8.10 | 4.94 | 13.28 | 0.00% | |
| Ad%>=65% | 8 | 6.83 | 4.41 | 10.58 | 25.90% | 0.61 |
| Asian | 7 | 7.44 | 4.92 | 11.24 | 23.60% | |
| Caucasian | 6 | 7.38 | 4.33 | 12.58 | 0.00% | 0.98 |
qMSP is short for the quantitative methylation-specific polymerase chain reaction method. MSP is short for the methylation-specific polymerase chain reaction method. Ad% represents the percent of lung adenocarcinoma samples.
Subgroup meta-analysis and SROC Curve for the relationship between CDH13 promoter hypermethylation and non-small cell lung cancer (NSCLC). A) Subgroup meta-analysis based on aim (Diagnosis vs. Non-diagnosis). B) Subgroup meta-analysis based on sample type (Tissue vs. Non-tissue). C) Subgroup meta-analysis based on method (MSP vs. qMSP), qMSP is short for the quantitative methylation-specific polymerase chain reaction method, and MSP is short for the methylation-specific polymerase chain reaction method. D) Subgroup meta-analysis based on AD% in non-diagnosis subgroup (AD%<65 vs. AD% >=65%), Ad% represents the percent of lung adenocarcinoma samples. E) Diagnostic SROC (bivariate model) for CHD13 in NSCLC.
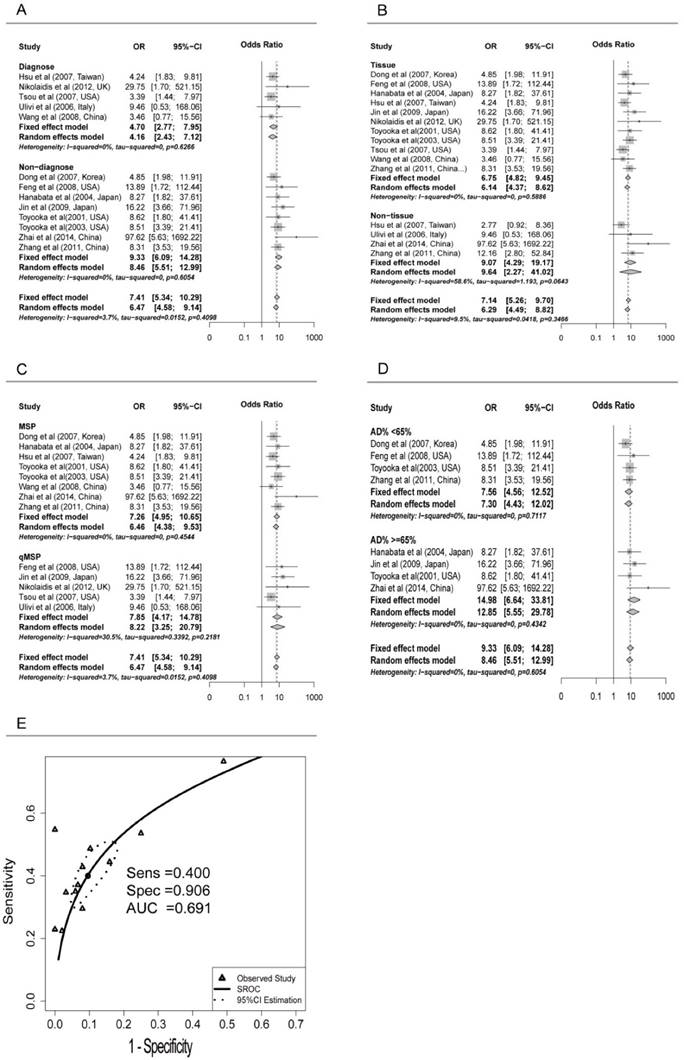
Because of the significant differences between the diagnosis and non-diagnosis subgroup, we conducted further research in the non-diagnosis subgroup. However, when we focused on the studies not aiming at diagnosis, the OR was found to be considerably reduced in Ad% <65% subgroup (7.56, 95% CI: 4.56 to 12.52) than in >=65% subgroup (14.98, 95% CI: 6.64 to 33.81) (P = 0.16) (Figure 2D).
Summary receiver operating characteristic curve for diagnostic capacity of CDH13 methylation
Pooled sensitivity and specificity were 0.400 (95%CI: 0.317 to 0.488) and 0.906 (95%CI: 0.840 to 0.946) for all the studies based on the presupposition of the fixed effects model. The sensitivity of the tissue subgroup was higher than that of the serum subgroup, 0.405 (0.316 to 0.500) versus 0.356 (0.247 to 0.484), while the specificity of the tissue subgroup was lower than that of the serum subgroup, 0.900 (0.823 to 0.945) versus 0.937 (0.803 to 0.982), which suggested that reduced sensitivity but increased specificity could be expected when conducting the methylation test with the serum of patients instead of the cancer tissues in NSCLC.
Although sensitivity and specificity were two of the most important features of a diagnostic test, in some occasions, pooling sensitivity or specificity could be misleading. Therefore, we constructed the summery receiver operating characteristic (SROC) curve to depict the overall stability and accuracy of the methylation test's diagnostic ability. The area under the curve (AUC) of the SROC was 0.691, suggesting a fair ability for NSCLC diagnosis (Figure 2E).
Bias analysis and robust estimation of pooled OR
A funnel plot of methylation status of lung cancer tissues versus normal tissues showed no significant publication bias (Harbord test, t = 1.11, P = 0.29) and no study exceeded the 95% confidence limits (Figure S1).
In order to eliminate the effect of publication bias, trim and fill analysis was performed with the fixed effects model. The adjusted pooled ORs were 5.58 (95% CI: 4.05 to 7.67) in the fixed effects model and 5.64 (95% CI: 3.84 to 8.27) in the random effects model. Both results demonstrated a significantly positive association between CDH13 hypermethylation and NSCLC (Figure S2).
Sensitivity analysis was performed by omitting one study at a time and calculating the pooled ORs for the remaining studies. The overall ORs after omitting one study were between 7.04 (95% CI: 5.03 to 9.87) and 8.22 (95% CI: 5.74 to 11.77) using fixed effects model, which suggested that the pooled OR was consistent and reliable (Figure S3).
Validation by independent TCGA and GEO lung cancer dataset
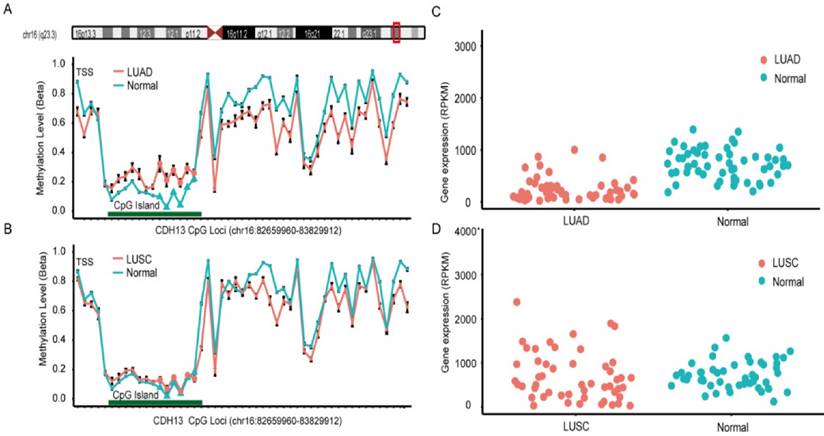
Data from The Cancer Genome Atlas (TCGA) and Gene Expression Omnibus (GEO) was used to validate the findings from the meta-analysis. In TCGA dataset, lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC) methylation datasets were obtained for further analysis [43]. There were 6 CpG sites sharing the same CGI (CpG Island) with the primers used in the included studies of the meta-analysis, and the overall methylation status of the six CpG sites could be used to represent the methylation status of CDH13 gene promoter. Surprisingly, the result from LUAD dataset and LUSC dataset differed and differential methylation profiles were shown between these two subtypes (Figure 3). In LUAD methylation dataset, 5 out of the 6 CpG sites showed significantly differential methylation level between cancer tissues and paired adjacent normal tissues according to the criteria (See Method). While in LUSC dataset, though 5 of the 6 CpG sites had p-values less than 0.05 after multiple correction, the absolute mean differences were less than 0.1 for all and thus couldn't be considered as significantly differential methylation sites as well (Table 3).
CpG sites on the Illumina Infinium HumanMethylation450 Beadchip across CDH13 gene region and Gene expression scatterplot with paired data from TCGA dataset. Methylation and gene expression status for CDH13 gene (TCGA lung cancer dataset). LUAD is short for lung adenocarcinoma, and LUSC represents lung squamous cell carcinoma. A-B each represents the different methylation status of lung cancer subtypes versus normal lung tissues in different datasets. For A-B, the x-axis shows the different CpG sites in CDH13 genes and the y-axis shows the beta value of each CpG site to represent the methylation level of each CpG site. C-D each represents the gene expression status of paired samples. The x-axis of the two figures shows the different types and y-axis shows the gene expression level using RPKM as measurement.
Differential CDH13 methylation, odds ratio between adenocarcinoma, squamous cell carcinoma and their counterparts from TCGA dataset
| Type | CpG site | Position | McaM | McoM | ∆β | P-value |
|---|---|---|---|---|---|---|
| LUAD | cg08747377 | chr16:82660670 | 0.32 | 0.1 | 0.22 | 1.95×10-5 |
| cg05374412 | chr16:82660727 | 0.21 | 0.03 | 0.18 | 2.27×10-5 | |
| cg00806490 | chr16:82660873 | 0.28 | 0.13 | 0.15 | 1.05×10-4 | |
| cg08856946 | chr16:82661421 | 0.19 | 0.04 | 0.15 | 2.27×10-5 | |
| cg09189772 | chr16:82661638 | 0.29 | 0.16 | 0.13 | 8.32×10-4 | |
| cg19369556 | chr16:82661725 | 0.26 | 0.22 | 0.04 | 2.92×10-1 | |
| LUSC | cg08747377 | chr16:82660670 | 0.13 | 0.08 | 0.05 | 1.50×10-2 |
| cg05374412 | chr16:82660727 | 0.07 | 0.02 | 0.05 | 9.00×10-3 | |
| cg00806490 | chr16:82660873 | 0.15 | 0.11 | 0.04 | 6.00×10-3 | |
| cg08856946 | chr16:82661421 | 0.05 | 0.03 | 0.02 | 3.00×10-2 | |
| cg09189772 | chr16:82661638 | 0.16 | 0.14 | 0.02 | 7.61×10-1 | |
| cg19369556 | chr16:82661725 | 0.14 | 0.18 | 0.04 | 2.26×10-5 |
McaM and McoM represent the mean of case methylation (Beta) and mean of control methylation (Beta). Methylation levels are calculated with formula: Beta = (M/M + U).
LUAD is short for lung adenocarcinoma, and LUSC is short for lung squamous cell carcinoma
Position represents the chromosome position of each CpG site according to GRCh37/hg19.
P-values are calculated from Wilcoxon signed-rank test after false discovery rate (FDR adjustment).
In order to draw a more robust conclusion, GEO dataset GSE39279 as well as GSE52401 were further downloaded and combined from Gene Expression Omnibus. There were 322 lung adenocarcinomas and 122 lung squamous cell carcinomas and 244 normal tissues in the combined dataset, which is of sufficient sample size to be an independent validation database. We performed the same analysis as before and obtained the result consistent with the TCGA dataset (Figure S4). Due to the large sample size, p-values of all the CpG sites in LUAD and LUSC datasets were less than 0.05 after multiple corrections. However, In LUAD dataset, 5 out of the 6 CpG sites showed absolute mean differences above 0.1 and thus could be regarded as significantly differential methylated sites while none of the CpG sites passed the criteria in LUSC dataset (Table S2). Moreover, another independent GEO dataset GSE56044 with 83 lung adenocarcinomas and 23 lung squamous cell carcinoma tissues and 12 adjacent normal tissues, was also downloaded for further validation (Figure S5). Unsurprisingly, the result was nearly the same with the previous two datasets. According to our criteria, 6 out of 7 CpG sites were significantly hypermethylated in LUAD dataset while none of the 7 CpG sites was differentially methylated in LUSC dataset (Table S3). Further, we then combined the TCGA and GEO datasets for evaluation of the diagnosis ability of CDH13 methylation status. After quality control, four shared CpG sites within the CpG island were selected. The AUCs of logistic regression models based on the CpG sites were 0.83-0.94 for Ads and 0.60-0.75 for SqCs, showing that the diagnostic ability of CDH13 methylation status is much better in Ads than in SqCs (Table S4).
Gene Expression data with TCGA RNA-Seq dataset
DNA methylation is one of the key regulators for gene expression. It is found that DNA methylation change correlates with gene expression level inversely, especially in gene promoter region. As a result, we downloaded the level 3 RNA-Seq dataset from TCGA. Reads per kilo base per million mapped reads (RPKM) was used as the measurement for gene expression quantification. After calculating the fold change and p-value with multiple correction, significantly differential expression of CDH13 was shown in LUAD (P = 5.03×10-8, fold change = 0.437) but not shown in LUSC (P = 0.97, fold change = 1.138) samples when compared with normal tissues. And the expression profiles of CDH13 were in accordance with the methylation profiles drawn from the microarray datasets, further strengthening our conclusions (Figure 3).
Discussion
The CDH13 gene has been reported to be hypermethylated in many types of cancers. In this study, we performed an integrated analysis to quantify the ability for the CDH13 promoter methylation test in NSCLC diagnosis, and a significant association was identified between CDH13 methylation and NSCLC (OR = 7.41, 95% CI: 5.34 to 10.29, P < 0.0001). Four imputed studies were filled when trim and fill test was performed to eliminate the influence of publication bias in the fixed effects model, and the overall OR (5.58, 95% CI: 4.05 to 7.67) was still significant, indicating the existence of a strong association between CDH13 promoter methylation and NSCLC.
In order to validate the result from the meta-analysis, we downloaded four independent datasets from TCGA and GEO database. And unexpectedly, the methylation profile in the two subsets of lung cancer differed dramatically. All of the datasets from TCGA and GEO showed significant hypermethylation in the promoter CpG sites of lung adenocarcinoma tissues when compared with normal tissues. However, none of the CpG sites was significantly differential methylated between lung squamous cell carcinoma tissues and normal tissues. Furthermore, we also conducted logistic regression model to evaluate the diagnosis ability of CDH13 methylation status in the lung adenocarcinoma and lung squamous cell tissues as well. Unsurprisingly, the AUCs of the former (AUC: 0.828-0.936) was much better than the latter (AUC: 0.596-0.744). Moreover, the expression data from TCGA level 3 RNA-Seq data was also concordant with this conclusion. The expression level of CDH13 was significantly lower in lung adenocarcinoma tissues but not in lung squamous cell carcinoma tissues when compared with normal tissues. This result was partially confirmed when we focused on the studies not aiming at diagnosis, the OR was found to be largely reduced in Ad%<65% subgroup (7.56, 95% CI: 4.56 to 12.52) than in Ad%>=65% subgroup (14.98, 95% CI: 6.64 to 33.81).
In summary, different results were drawn from meta-analysis and microarray datasets for the association of CDH13 promoter methylation and lung squamous cell carcinoma. As for the relatively inconsistent result, firstly, qMSP is the semi-quantitative method especially being used in low dose methylation. Secondly, the sparseness of CpG sites in HumanMethylation 450K array may be another key factor. The HumanMethylation 450K array only covers less than 2% of the whole genome CpG sites and thus can't explore all the promoter region of CDH13 and therefore might be misleading [44]. Besides, no method comparison between qMSP and HumanMethylation 450K has been conducted to our knowledge. As a result, more comprehensive and advanced methods like WGBS (whole genome bisulfite sequencing) and RRBS (restricted region bisulfite sequencing) are needed to draw a more robust conclusion [45-47].
To summarize, according to the previous results drawn from meta-analysis and microarray data analysis, CDH13 may be a powerful potential biomarker for the diagnosis of lung adenocarcinoma while the association of CDH13 methylation and lung squamous cell carcinoma needs more data to draw a robust conclusion. In addition, due to the different methylation profile of CDH13 in NSCLC subtypes, CDH13 methylation test could also be a promising biomarker to distinguish the lung adenocarcinoma and lung squamous cell carcinoma which might provide evidence for accurate chemotherapy and targeted therapy.
Conclusion
This integrated analysis of the pooled data provides strong evidence that the methylation status of the CDH13 promoter is significantly associated with lung adenocarcinoma. The aberrant CDH13 methylation could be a promising diagnostic biomarker for non-invasive lung adenocarcinoma detection.
Methods
Search strategy, selection of studies and data extraction
This pooled study involved searching a range of computerized databases, including PubMed, Cochrane Library, OVID Medline and Web of Science for articles published in English by December 2014. The study used a subject and text word strategy with (CDH13 OR CDHH OR P105 OR H-cadherin OR Cdht OR T-cadherin OR Tcad OR CH211-122A20.1 OR BOS_16969 OR cdhh) AND (lung or non-small) as the primary search terms. Wildcard character of star, dollar or some other truncations were applied according to the rules of the databases to allow effective article collection.
Two independent reviewers (Geng, Guo) screened the titles and abstracts derived from the literature search to identify relevant studies. The following types of studies were excluded: animal and cell experiments, case reports, reviews or meta-analyses and studies of non-case-control studies or studies with insufficient data or those inaccessible after making contacts with the authors. The remaining articles were further examined to see if they met the inclusion criteria: 1) the patients had to be diagnosed with NSCLC (Ad and Sc), 2) the studies had to contain CDH13 gene promoter methylation data from tissue, blood or serum, 3) the studies had to be case-control studies which included tissue-tissue, blood-blood or serum-serum in case and controls respectively. The reference sections of all retrieved articles were searched to identify further relevant articles. Potentially relevant papers were obtained and the full text articles were screened for inclusion by two independent reviewers (Geng, Guo). Disagreements were resolved by discussion with LXT, SDC, and LJ. Included studies were summarized in data extraction forms. Paper quality was assessed using Quality Assessment Tool for Diagnostic Accuracy Studies (QUADAS) criteria. Authors were contacted when relevant data was missing. The name of the first author, year of publication, sample size, age (mean or median), gender proportion (Male%), the proportion of TNM stage I samples (proportion of early stage of NSCLC samples), the percentage of adenocarcinoma (Ad%), publication aim (for diagnosis or not), analyzing multiple genes or not (one or more genes detected simultaneously in studies design), control type (autogenous or heterogeneous counterpart) and methylation status of the CDH13 promoter in human NSCLC and normal or control tissues were extracted.
Meta-analysis and SROC analysis
Data were analyzed and visualized mainly using R Software (R version 3.1.0) including meta [48], metafor[49] and mada packages[50]. The strength of association was expressed as pooled odds ratio (OR) with corresponding 95% confidence intervals (95% CI). Data were extracted from the original studies and recalculated if necessary. Heterogeneity was tested using the I2 statistic with values over 50% and Chi-squared test with P ≤ 0.1 indicating strong heterogeneity between the studies [21]. Tau-squared (τ2) was used to determine how much heterogeneity was explained by subgroup differences. The data were pooled using the DerSimonian and Laird random effects model (I2 > 50%, P ≤ 0.1) or fixed effects model (I2 < 50%) according to heterogeneity statistic I2 [51]. A two-sided P ≤ 0.05 was considered significant without special annotation. With a lack of heterogeneity among included studies, the pooled odds ratio estimates were calculated using the fixed effects model [52]. Otherwise, the random-effects model was used [53]. Sensitivity analysis was performed to assess the contributions of single studies to the final results with the abandonment of one article each time. Publication bias was analyzed by funnel plot with mixed-effects version of the Harbord test. If bias was suspected, the conventional meta-trim method was used to re-estimate the effect size.
Compared with traditional SNP association studies, methylation-associated research might be involved with different methylation-definition thresholds. In these cases, traditional weighted averages (pooled sensitivity and specificity) would not reflect the overall accuracy of the test, because the extremes of threshold criteria could skew the distribution, known as the threshold effect [54]. Thus, SROC analysis was applied to meta-analysis of diagnostic tests [54, 55]. The SROC curve showed the performance of the diagnostic ability of CDH13 methylation to NSCLC. Each study produced values for sensitivity, specificity and therefore true positive rate (TPR) and false positive rate (FPR), and the plots were placed over the TPR and FPR points to form a smooth curve. A linear regression model was selected to fit the SROC curve where sensitivity and (1-specificity) were transformed into complex logarithmic variables. The exact AUC for the SROC function was used to assess the accuracy of the test.
TCGA and GEO data extraction and analysis
TCGA DNA methylation dataset which included 23 lung adenocarcinoma and 40 lung squamous cell carcinoma tissues as well as 63 paired adjacent tissues, were collected from TCGA project [http://cancergenome.nih.gov/]. And GEO datasets including GSE39279 and GSE52401 and GSE56044 were downloaded from Gene Expression Omnibus [http://www.ncbi.nlm.nih.gov/geo/], including a sum of 568 NSCLC tissues and 256 adjacent or normal lung tissues. Illumina HumanMethylation450K beadchip was used to detect the methylation level for all of the above datasets. The estimation of methylation for each CG probe was calculated between methylated (M) and unmethylated (U) alleles. Specifically:
beta =
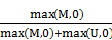
Both M and U represented mean signal intensities for about 30 replicates on the array. Beta value of the CpG sites were used as the measurement of methylation. CpG site would be immediately omitted when it was missing in one or more samples. The number of CpG sites of CDH13 gene in TCGA dataset and GEO datasets was not completely the same due to the quality control procedure previously mentioned. 6 or 7 CpG sites located in the same CpG island with the primers mentioned in the meta-analysis were the signatures for the methylation status of CDH13 (Table S1). Wilcoxon rank sum test or Wilcoxon signed-rank test along with logistic regression were conducted and generated a p-value for each comparison. Multiple comparison of the differential methylation was conducted with Benjamini and Hochberg at 5% FDR as the threshold. The diagnosis model was conducted using logistic regression along with 5-fold cross-validation. The statistical analysis was performed using R version 3.1.0 [56].
RNA-Seq data extraction and analysis
Level 3 RNA-Seq dataset was obtained from TCGA database, which includes 114 lung adenocarcinoma and 104 lung squamous cell carcinoma tissues as well as 218 normal tissues. Reads per kilo base per million mapped reads (RPKM) was regarded as the measurement for gene expression quantification. We assessed the significance of the differential gene expression by comparing the tumor tissues with paired adjacent normal tissues using Wilcoxon signed-rank test. For identification of differentially expression genes, p-value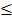 0.05 and fold change
0.05 and fold change 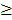 2.0 or
2.0 or 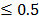 was set as the criteria. All the data analysis procedures were conducted with open-source R software (version 3.1.0).
was set as the criteria. All the data analysis procedures were conducted with open-source R software (version 3.1.0).
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This research was partially supported by the grants from the 111 Project (B13016). The computations involved in this study were supported by Fudan University High-End Computing Center.
Authors' contributions
JW, SG and XC contributed to the conception, design and final approval of the submitted version. XG, SG, ZL, AW, YT, JW, WP contributed to the meta-analysis and interpretation of data, WP, SG, XG, SC, LT, YX contributed to TCGA and GEO NSCLC data analysis. All authors read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA: a cancer journal for clinicians. 2015;65:5-29
2. Chen W, Zhang S, Zou X. Evaluation on the incidence, mortality and tendency of lung cancer in China. Thoracic Cancer. 2010;1:35-40
3. Jemal A, Center MM, DeSantis C, Ward EM. Global patterns of cancer incidence and mortality rates and trends. Cancer epidemiology, biomarkers & prevention: a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2010;19:1893-907
4. Devesa SS, Bray F, Vizcaino AP, Parkin DM. International lung cancer trends by histologic type: male: female differences diminishing and adenocarcinoma rates rising. International journal of cancer. 2005;117:294-9
5. Hankey BF, Ries LA, Edwards BK. The surveillance, epidemiology, and end results program: a national resource. Cancer epidemiology, biomarkers & prevention: a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 1999;8:1117-21
6. van Rens MT, de la Riviere AB, Elbers HR, van Den Bosch JM. Prognostic assessment of 2,361 patients who underwent pulmonary resection for non-small cell lung cancer, stage I, II, and IIIA. Chest. 2000;117:374-9
7. He Y, Cui Y, Wang W, Gu J, Guo S, Ma K. et al. Hypomethylation of the hsa-miR-191 locus causes high expression of hsa-mir-191 and promotes the epithelial-to-mesenchymal transition in hepatocellular carcinoma. Neoplasia. 2011;13:841-53
8. Flores K, Wolschin F, Corneveaux JJ, Allen AN, Huentelman MJ, Amdam GV. Genome-wide association between DNA methylation and alternative splicing in an invertebrate. Bmc Genomics. 2012:13
9. Gokul G, Khosla S. DNA methylation and cancer. Sub-cellular biochemistry. 2013;61:597-625
10. Hulpiau P, van Roy F. Molecular evolution of the cadherin superfamily. The international journal of biochemistry & cell biology. 2009;41:349-69
11. Shamay M, Krithivas A, Zhang J, Hayward SD. Recruitment of the de novo DNA methyltransferase Dnmt3a by Kaposi's sarcoma-associated herpesvirus LANA. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:14554-9
12. Riener MO, Nikolopoulos E, Herr A, Wild PJ, Hausmann M, Wiech T. et al. Microarray comparative genomic hybridization analysis of tubular breast carcinoma shows recurrent loss of the CDH13 locus on 16q. Human pathology. 2008;39:1621-9
13. Sato M, Mori Y, Sakurada A, Fujimura S, Horii A. The H-cadherin (CDH13) gene is inactivated in human lung cancer. Hum Genet. 1998;103:96-101
14. Sato M, Mori Y, Sakurada A, Fukushige S, Ishikawa Y, Tsuchiya E. et al. Identification of a 910-Kb region of common allelic loss in chromosome bands 16q24.1-q24.2 in human lung cancer. Gene Chromosome Canc. 1998;22:1-8
15. Toyooka KO, Toyooka S, Virmani AK, Sathyanarayana UG, Euhus DM, Gilcrease M. et al. Loss of expression and aberrant methylation of the CDH13 (H-cadherin) gene in breast and lung carcinomas. Cancer research. 2001;61:4556-60
16. Qian ZR, Sano T, Yoshimoto K, Asa SL, Yamada S, Mizusawa N. et al. Tumor-specific downregulation and methylation of the CDH13 (H-cadherin) and CDH1 (E-cadherin) genes correlate with aggressiveness of human pituitary adenomas. Modern Pathol. 2007;20:1269-77
17. Ogama Y, Ouchida M, Yoshino T, Ito S, Takimoto H, Shiote Y. et al. Prevalent hyper-methylation of the CDH13 gene promoter in malignant B cell lymphomas. Int J Oncol. 2004;25:685-91
18. Sun D, Zhang Z, Van DN, Huang GW, Ernberg I, Hu LF. Aberrant methylation of CDH13 gene in nasopharyngeal carcinoma could serve as a potential diagnostic biomarker. Oral Oncol. 2007;43:82-7
19. Kim DS, Kim MJ, Lee JY, Kim YZ, Kim EJ, Park JY. Aberrant methylation of E-cadherin and H-cadherin genes in nonsmall cell lung cancer and its relation to clinicopathologic features. Cancer. 2007;110:2785-92
20. Zhao QT, Guo T, Wang HE, Zhang XP, Zhang H, Wang ZK. et al. Diagnostic value of SHOX2 DNA methylation in lung cancer: a meta-analysis. Oncotargets Ther. 2015;8:3433-9
21. Guo SC, Tan LX, Pu WL, Wu JJ, Xu K, Wu JH. et al. Quantitative assessment of the diagnostic role of APC promoter methylation in non-small cell lung cancer. Clin Epigenetics. 2014:6
22. Huang YZ, Wu W, Wu K, Xu XN, Tang WR. Association of RASSF1A Promoter Methylation with Lung Cancer Risk: a Meta-analysis. Asian Pac J Cancer P. 2014;15:10325-8
23. Pu W. et al. Aberrant methylation of FHIT can be a diagnostic biomarker for NSCLC in Asian population. Submitted. 2016
24. Fang N. et al. [A meta-analysis of Association between MGMT gene promoter methylation and non-small cell lung cancer]. Zhongguo Fei Ai Za Zhi. 2014;17(8):601-5
25. Liang YL, He LP, Yuan H, Jin YL, Yao YS. Association between RUNX3 promoter methylation and non-small cell lung cancer: a meta-analysis. J Thorac Dis. 2014;6:694-705
26. Hua F, Fang NZ, Li XB, Zhu SW, Zhang WS, Gu JD. A Meta-Analysis of the Relationship Between RAR beta Gene Promoter Methylation and Non-Small Cell Lung Cancer. PloS one. 2014:9
27. Zeng Y, Liu R, Zhang H. [Meta-analysis of association between E-cadherin promoter methylation and lung cancer risk]. Zhongguo Fei Ai Za Zhi. 2013;16(7):353-8
28. Gu JD, Wen YJ, Zhu SW, Hua F, Zhao H, Xu HR. et al. Association between P-16INK4a Promoter Methylation and Non-Small Cell Lung Cancer: A Meta-Analysis. PloS one. 2013:8
29. Zhong YH, Peng H, Cheng HZ, Wang P. Quantitative assessment of the diagnostic role of CDH13 promoter methylation in lung cancer. Asian Pacific journal of cancer prevention: APJCP. 2015;16:1139-43
30. Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207-10
31. Toyooka S, Maruyama R, Toyooka KO, McLerran D, Feng Z, Fukuyama Y. et al. Smoke exposure, histologic type and geography-related differences in the methylation profiles of non-small cell lung cancer. International journal of cancer Journal international du cancer. 2003;103:153-60
32. Feng Q, Hawes SE, Stern JE, Wiens L, Lu H, Dong ZM. et al. DNA methylation in tumor and matched normal tissues from non-small cell lung cancer patients. Cancer epidemiology, biomarkers & prevention: a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2008;17:645-54
33. Hsu HS, Chen TP, Hung CH, Wen CK, Lin RK, Lee HC. et al. Characterization of a multiple epigenetic marker panel for lung cancer detection and risk assessment in plasma. Cancer. 2007;110:2019-26
34. Jin M, Kawakami K, Fukui Y, Tsukioka S, Oda M, Watanabe G. et al. Different histological types of non-small cell lung cancer have distinct folate and DNA methylation levels. Cancer science. 2009;100:2325-30
35. Tsou JA, Galler JS, Siegmund KD, Laird PW, Turla S, Cozen W. et al. Identification of a panel of sensitive and specific DNA methylation markers for lung adenocarcinoma. Molecular cancer. 2007;6:70
36. Ulivi P, Zoli W, Calistri D, Fabbri F, Tesei A, Rosetti M. et al. p16INK4A and CDH13 hypermethylation in tumor and serum of non-small cell lung cancer patients. Journal of cellular physiology. 2006;206:611-5
37. Wang Y, Zhang D, Zheng W, Luo J, Bai Y, Lu Z. Multiple gene methylation of nonsmall cell lung cancers evaluated with 3-dimensional microarray. Cancer. 2008;112:1325-36
38. Zhai X, Li S-J. Methylation of RASSF1A and CDH13 Genes in Individualized Chemotherapy for Patients with Non-small Cell Lung Cancer. Asian Pacific Journal of Cancer Prevention. 2014;15:4925-8
39. Zhang Y, Wang R, Song H, Huang G, Yi J, Zheng Y. et al. Methylation of multiple genes as a candidate biomarker in non-small cell lung cancer. Cancer letters. 2011;303:21-8
40. Nikolaidis G, Raji OY, Markopoulou S, Gosney JR, Bryan J, Warburton C. et al. DNA methylation biomarkers offer improved diagnostic efficiency in lung cancer. Cancer research. 2012;72:5692-701
41. Hanabata T, Tsukuda K, Toyooka S, Yano M, Aoe M, Nagahiro I. et al. DNA methylation of multiple genes and clinicopathological relationship of non-small cell lung cancers. Oncology reports. 2004;12:177-80
42. Whiting PF, Weswood ME, Rutjes AW, Reitsma JB, Bossuyt PN, Kleijnen J. Evaluation of QUADAS, a tool for the quality assessment of diagnostic accuracy studies. BMC medical research methodology. 2006;6:9
43. Cancer Genome Atlas Research N. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511:543-50
44. Sun ZF, Cunningham J, Slager S, Kocher JP. Base resolution methylome profiling: considerations in platform selection, data preprocessing and analysis. Epigenomics-Uk. 2015;7:813-28
45. Bibikova M, Fan JB. Genome-wide DNA methylation profiling. Wires Syst Biol Med. 2010;2:210-23
46. Bock C, Tomazou EM, Brinkman AB, Muller F, Simmer F, Gu HC. et al. Quantitative comparison of genome-wide DNA methylation mapping technologies. Nat Biotechnol. 2010;28:1106-U196
47. Li N, Ye MZ, Li YR, Yan ZX, Butcher LM, Sun JH. et al. Whole genome DNA methylation analysis based on high throughput sequencing technology. Methods. 2010;52:203-12
48. Schwarzer G. Meta: An R package for meta-analysis. R news. 2007;7:40-5
49. Viechtbauer W. Conducting meta-analyses in R with the metafor package. J Stat Softw. 2010;36:1-48
50. Doebler P, Holling H. Meta-Analysis of Diagnostic Accuracy with mada. 2012.
51. DerSimonian R, Laird N. Meta-analysis in clinical trials. Controlled clinical trials. 1986;7:177-88
52. Mantel N, Haenszel W. Statistical aspects of the analysis of data from retrospective studies of disease. Journal of the National Cancer Institute. 1959;22:719-48
53. Huizenga HM, Visser I, Dolan CV. Testing overall and moderator effects in random effects meta-regression. The British journal of mathematical and statistical psychology. 2011;64:1-19
54. Midgette AS, Stukel TA, Littenberg B. A meta-analytic method for summarizing diagnostic test performances: receiver-operating-characteristic-summary point estimates. Medical decision making: an international journal of the Society for Medical Decision Making. 1993;13:253-7
55. Jones CM, Athanasiou T. Summary receiver operating characteristic curve analysis techniques in the evaluation of diagnostic tests. The Annals of thoracic surgery. 2005;79:16-20
56. Team R C. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria, 2012[J]. 2014
Author contact
![]() Corresponding authors: Shicheng Guo, Department of Bioengineering, University of California at San Diego, 9500 Gilman Drive, MC0412, La Jolla, CA 92093-0412. Telephone: 281-685-5882, Fax: 858-534-5722, Email: scguoedu. Jiucun Wang, National Ministry of Education Key Laboratory of Contemporary Anthropology, School of Life Sciences, Fudan University, Shanghai 200433, China, Phone: +86-21-55665499, Fax: +86-21-556648845, E-mail: jcwangedu.cn. Xiaofeng Chen, Department of Cardiothoracic Surgery, Huashan Hospital, Fudan University, Shanghai 200032, China, Phone: 52888299, E-mail: cxf3166com
Corresponding authors: Shicheng Guo, Department of Bioengineering, University of California at San Diego, 9500 Gilman Drive, MC0412, La Jolla, CA 92093-0412. Telephone: 281-685-5882, Fax: 858-534-5722, Email: scguoedu. Jiucun Wang, National Ministry of Education Key Laboratory of Contemporary Anthropology, School of Life Sciences, Fudan University, Shanghai 200433, China, Phone: +86-21-55665499, Fax: +86-21-556648845, E-mail: jcwangedu.cn. Xiaofeng Chen, Department of Cardiothoracic Surgery, Huashan Hospital, Fudan University, Shanghai 200032, China, Phone: 52888299, E-mail: cxf3166com