Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2016; 7(14):2124-2131. doi:10.7150/jca.16399 This issue Cite
Research Paper
Co-expression of Achaete-Scute Homologue-1 and Calcitonin Gene-Related Peptide during NNK-Induced Pulmonary Neuroendocrine Hyperplasia and Carcinogenesis in Hamsters
Xu Naizhen1, R. Ilona Linnoila1, Shioko Kimura2 ![]()
1. Cell and Cancer Biology Branch, National Cancer Institute, National Institutes of Health, Bethesda, Maryland 20892, USA;
2. Laboratory of Metabolism, National Cancer Institute, National Institutes of Health, Bethesda, Maryland 20892, USA.
Received 2016-6-6; Accepted 2016-8-14; Published 2016-10-23
Abstract
Achaete-scute homologue-1 or ASCL1 (MASH1, hASH1) plays roles in neural development and pulmonary neuroendocrine (NE) differentiation, and it is expressed in certain lung cancers. This study was aimed to assess whether and/or how ASCL1 plays a role in 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK)-induced pulmonary NE hyperplasia and carcinogenesis in hamsters. Hamsters were injected 3 times weekly with either NNK or solvent alone (control) for treatment periods of 6 and 24 weeks, both without and with 6-week recovery. Immunohistochemical analysis was carried out to examine the expressions of ASCL1, CGRP (calcitonin gene-related peptide), secretoglobin SCGB1A1 (club [Clara] cell specific 10 kD protein, CC10, CCSP), synaptophysin (SYP), and PCNA (proliferating cell nuclear antigen). The number of ASCL1-expressing NE foci per airway increased from 0.8 in controls to 1.6 and 2.0 during NNK exposure for 6 and 24 weeks, respectively, and the number of cells per foci doubled after NNK exposure. Most ASCL1-expressing cells in NEBs (neuroepithelial bodies) were also CGRP immunoreactive; NNK enhanced this co-expression with CGRP, a NE marker with known proliferation-promoting properties. NNK also increased PCNA expression within NE foci. NNK-induced tumors showed no immunoreactivity for NE markers. This study confirms ASCL1 as an excellent marker for pulmonary NE cells and demonstrates CGRP co-expression in ASCL1-positive NEB cells participating in NNK-induced NE hyperplasia.
Keywords: lung, ASCL1, CGRP, neuroendocrine hyperplasia, NNK.
Introduction
Neuroendocrine (NE) tumors arise from cells of the endocrine and nervous systems, including those occurring in the intestine, pancreas, lung, and elsewhere. They share common features such as similar structural and morphological appearance and presence of secretory granules containing various peptides and hormones [1, 2]. NE features are seen in approximately 25% of lung cancers including small cell lung carcinoma (SCLC, comprising 20%), the commonest NE cancer of the lung [3]. SCLC is thought to be directly derived from differentiated pulmonary NE cells [4]. SCLC is aggressively metastatic, and poorly differentiated NE features may contribute to the virulent behavior of this tumor.
NE cells in the normal lung airway lining include scattered individual pulmonary neuroendocrine cells (PNECs) and well-defined cell clusters called neuroepithelial bodies (NEBs); a NEB is often overlaid by a “cap” of non-ciliated epithelial cells similar to club cells (formerly Clara cells) [5]. Among gene products characteristically expressed by NEBs is achaete-scute homologue-1 or ASCL1 (MASH1, hASH1), a basic helix-loop-helix transcription factor, conserved from the Drosophila achaete-scute complex. In mammals, Ascl1 gene operates in fetal nervous system development [6]; during neurogenesis, Ascl1 gene expression is largely restricted to mitotically active precursor cells and is silenced before terminal differentiation [7, 8]. Ascl1 plays a pivotal role in normal as well as neoplastic pulmonary NE cell differentiation [9, 10]. Ascl1-null mice had no detectable pulmonary neuroendocrine cells [9], while constitutive expression of Ascl1 gene in transgenic mouse lung enhanced SV40-induced tumorigenesis and NE differentiation in transformed epithelial cells [11]. Inhibition of Ascl1 expression by RNA interference suppressed growth of lung cancer cells through cell cycle arrest and induced apoptosis in Ascl1 expression-dependent manner [12]. ASCL1 was suggested to be involved in lineage-specific survival and growth of lung cancers with NE features [12, 13] and to play a critical role in regulating tumor-initiating capacity of SCLC [14]. ASCL1 is expressed in greater than 50% of SCLC and large-cell neuroendocrine lung carcinomas [15].
In addition to ASCL1, pulmonary CGRP (calcitonin gene-related peptide) is of particular interest. CGRP is a 37 amino acid neuropeptide occurring in two closely-related forms, α and β, differing by only three amino acid residues. αCGRP and βCGRP are products derived from different splice combinations of exons of CALC1 and CALC2 genes on human chromosome 11 [16]. αCGRP is the main form expressed in both central and peripheral nervous systems, while βCGRP is usually expressed in enteric tissues [17]. These two forms, antigenically very close, are thought to possess similar biological activities. CGRP is recognized as a biomarker for sensory nerve endings and pulmonary NE cells [4]. Among 16 SCLC cell lines, α and β CGRP mRNAs were expressed in 8 and 13 cell lines respectively, while 12 cell lines expressed CGRP protein by radioimmunoassay [16].
CGRP has mitogenic properties and stimulated proliferation of guinea pig tracheal epithelial cells in vitro [18]. Cultured A549 human lung carcinoma cells showed enhanced proliferation when exposed to CGRP, and rats intratracheally exposed to recombinant adenovirus expressing nuclear-targeted CGRP exhibited proliferation of lung alveolar cells [19]. Further, when Lewis lung carcinoma cells were implanted in Cgrp(-/-) and wild-type mice, tumor neovascularization and growth were depressed in knockout mice compared with wild-type mice, demonstrating that endogenous host-derived CGRP may stimulate angiogenesis [20]. Thus, CGRP could play a significant role in pulmonary epithelial hyperplasia and tumor growth.
NNK (4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone) exposure has been associated with lung tumors in mice [21] and distinct pulmonary NE cell hyperplasia and tumors in hamsters [22]. In the current study, we employed a NNK carcinogenesis animal model with an aim to understand the adult expression pattern of ASCL1 with relation to other relevant biomarkers under these conditions. In particular, we wished to assess effects, if any, of NNK-induced hyperplasia on expression within NEBs of CGRP, a recognized NE marker possessing the interesting biological properties remarked above. Other antigens examined were SCGB1A1 (club cell [Clara cell] specific 10-kD protein, CC10, CCSP), SYP (synaptophysin, another NE marker), and PCNA (proliferating cell nuclear antigen), the last included to assay the extent of hyperplastic proliferation.
Materials and Methods
Animal studies
Syrian golden hamsters (male, 8-9 weeks, N=59, 6-10 animals/group) were given three subcutaneous injections per week of either 25 mg/kg NNK or solvent alone (trioctanoin) as control. The animals comprised 8 treatment groups: 6 weeks NNK exposure; 6 weeks' solvent-only control; 6 weeks NNK exposure with 6 weeks' recovery; 6 weeks' solvent-only control with 6 weeks' recovery; 24 weeks NNK exposure; 24 weeks' solvent-only control; 24 weeks NNK exposure with 6 weeks' recovery; 24 weeks' solvent-only control with 6 weeks' recovery. The number of animals was determined to obtain statistically meaningful results. At the end of each treatment period, the animals were killed by lethal injection, and two lung lobes from each animal were fixed in neutral buffered 4% formaldehyde, paraffin embedded, and serially sectioned (5 µm). All animals were housed in a temperature and humidity controlled specific pathogen-free facility under a 12-hour light/dark cycle with free access to water and food, and handled in a humane manner in an AAALAC-accredited facility in accordance with the standards established in the NIH Guideline. Animal studies were carried out under protocols approval by the National Cancer Institute Animal Care and Use Committee.
Immunohistochemical analysis
Sections were deparaffinized, rehydrated, and microwaved for 10 min in 0.05 M citrate buffer, pH 6, for antigen retrieval. Sections were then incubated with primary antibody overnight at 4°C; primary antibodies and dilutions used are summarized in Supplementary Table 1. For brightfield single visualization of ASCL1, CGRP, PCNA, or SYP, biotinylated secondary antibody was applied and its localization was detected by an avidin-biotin complex (ABC) staining method, using peroxidase and DAB (diaminobenzidine, Vector Laboratories, Burlingame, CA). Nickel intensification was achieved by immersing the slides in a substrate solution containing 0.075% DAB, 0.007% hydrogen peroxide, and 2% nickel sulfate in 0.1 M acetate buffer, pH 6, for 10 min at room temperature [23]. Sections were counterstained with light green SF. For brightfield double staining, sections were first incubated with mouse primary antibody against ASCL1, followed by biotinylated secondary antibody, ABC peroxidase and DAB (without intensification). Sections were then incubated with rabbit primary antibody against CGRP, followed by biotinylated secondary antibody, ABC alkaline phosphatase and Vector® Red (Vector Laboratories), with no subsequent counterstaining. For fluorescence studies, Alexa® Fluor 488 coupled goat anti-rabbit IgG and Alexa® Fluor 594 coupled goat anti-mouse IgG (Molecular Probes, Eugene, OR) were used as secondary antibodies (1:250) applied in solution together for 45 min at room temperature. Representative sections were also stained with hematoxylin and eosin for morphological study. Sections were evaluated visually by brightfield or epi-fluorescence microscopy, and digital images were captured using Metamorph® software (Universal Imaging Corporation, Downingtown, PA). The number of immunopositive cells was compared within a group by unpaired t-test for statistical differences.
Results
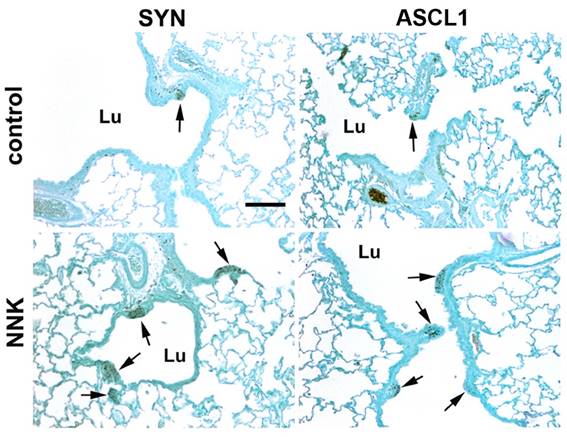
The pulmonary NE cell population, as defined by immunoreactivity to CGRP, is represented by foci of both solitary cells and cell clusters [5]; foci of three or more cells are described as neuroepithelial bodies (NEBs). In the case of normal control hamster airways, the great majority of NEBs had a typical size of about five cells, although some NEBs had appreciably more while some had fewer. Both small and large NEBs found in control and NNK-exposed hamsters also demonstrated positive immunostaining for SYP and ASCL1 (Fig. 1). The number of solitary NE cells (PNECs) was relatively small, representing only 6% of the total NE cell population in control animals, while in NNK-treated animals they numbered less than 1%. For this reason, data presented here mainly reflect foci of the multiple-cell type. However, even though small, solitary PNECs were recognizable by their positive CGRP or ASCL1 staining.
Focal immunohistochemical reactivity for synaptophysin (SYP) and ASCL1 in neuroepithelial bodies (NEBs). NEBs (arrows) visibly protrude into lung airway lumen (Lu) and tend to be larger in NNK-treated animals. Sections were stained using a standard avidin-biotin complex (ABC) method with peroxidase and DAB, followed by counterstaining with light green SF. Scale bar: 100 µm.
NE hyperplasia following NNK exposure was evidenced by both an increased ratio of foci per airway (Fig. 2A) and an increased number of ASCL1-immunoreactive cells per foci, the latter considered to define foci size (Fig. 2B). The mean number of NE foci per airway expressing ASCL1 increased from 0.8 in control hamsters to 1.6 and 2.0 in hamsters exposed to NNK for 6 and 24 weeks, respectively (Fig. 2A). This was associated with significant increases in numbers of ASCL1-positive cells per foci, and the effect was more profound for the groups exposed to NNK for 24 weeks with and without 6 weeks' recovery. The number of ASCL1-positive cells per foci in the “24wk+6wk rec” group was more than 2.8 times that observed in the respective control (Fig. 2B). Such increases mainly reflected larger sizes of NEBs seen in NNK-exposed animals (Fig. 1).
Quantitative analysis of ASCL1-expressing foci among different groups of control and NNK-exposed hamsters. (A) Comparison of number of ASCL1-expressing foci per airway. Statistical significance is between solvent-only (control) vs. NNK-treated groups. (B) Number of ASCL1-expressing cells per foci (equivalent to foci size). Foci size was also determined by CGRP-positive cell numbers. 6wk: 6 weeks' solvent only (control) or NNK treatment; 6wk+6wk rec: 6 weeks' solvent only or NNK treatment, followed by recovery period of 6 weeks, denoted by “rec”; 24wk: 24 weeks' solvent only or NNK-treatment; and 24wk+6wk rec: 24 weeks' solvent only or NNK-treatment, followed by recovery period of 6 weeks. Mean ± SD is shown from N=6-10 animals per group. *: P<0.05, **: P<0.01, ***: P<0.001. Statistical analysis by unpaired t-test.
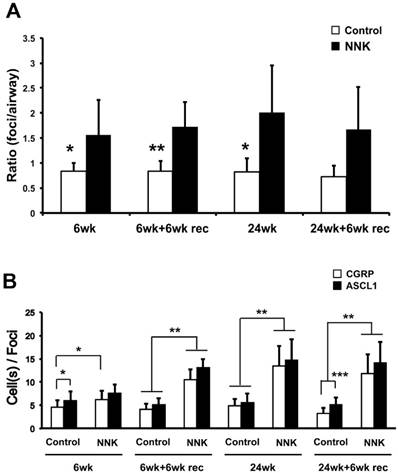
Most ASCL1-positive cells in NEBs were also positive for CGRP (Fig. 2B). The number of CGRP-expressing cells per foci tended to be lower than ASCL1-expressing cells in all groups; in particular, “6wk” and “24wk+6wk rec” control groups showed statistically significantly lower numbers of CGRP-expressing cells per foci than ASCL1-expressing cells, while no statistically significant differences were found in respective NNK-treated groups. This suggests that some cells were immunoreactive for ASCL1 only, and NNK exposure caused a distinct decrease of this fraction. Thus, NNK exposure resulted in a larger proportion of cells co-expressing CGRP and ASCL1. Localization of CGRP was usually strongest in the basal cytoplasm of cells expressing this antigen (Fig. 3A, left panel). Expression of SCGB1A1 was observed fairly consistently within NEBs of both control and NNK-exposed hamsters (Fig. 3A, middle panel); SCGB1A1 often assumed an apical position near the airway lumen. Many ASCL1-positive cells of NEBs in NNK-exposed animals were also positive for SCGB1A1. As expected, SCGB1A1 was widely present in normal airway club cell epithelium as well.
Double immunofluorescence staining of NEBs in control and NNK-treated animals. (A) Left panel: Red for ASCL1 and green for CGRP. Middle panel: Red for ASCL1 and green for SCGB1A1. Right panel: Red for PCNA and green for CGRP. Representative NEBs that co-express indicated markers are shown. In control lungs, NEB cells co-express ASCL1 and CGRP (left), but most cells do not co-express ASCL1 and SCGB1A1 (middle) or PCNA and CGRP (right). Both ASCL1 and PCNA localizations are nuclear, whereas SCGB1A1 and CGRP are cytoplasmic. Note that CGRP tends to be distributed basally in cells, while SCGB1A1 appears more abundant at the apical region, near the luminal free surface; however, club cells expressing SCGB1A1 sometimes overlie NEB cells. In NNK-treated animals, NEBs are larger and exhibit evident co-expressions. Lu: airway lumen. Scale bar: 25 µm. (B) Comparisons of focal immunoreactivities for ASCL1, PCNA, and CGRP in control and NNK-exposed hamsters. Serial sections obtained from 24-week treatment groups were separately stained with ABC peroxidase and nickel-intensified DAB, counterstained with light green SF. In the control NEB, all central cells express ASCL1 but are essentially unreactive for PCNA. In the NNK-stimulated NEB, both ASCL1 and PCNA are strongly expressed; tailed arrows indicate apparent same-cell nuclear localization of both antigens in adjacent serial sections. However, some PCNA-expressing cells (arrowheads) do not express ASCL1. In both control and NNK-exposed NEBs, cytoplasmic CGRP is abundantly evident, often basally-distributed. Scale bar: 25 µm.
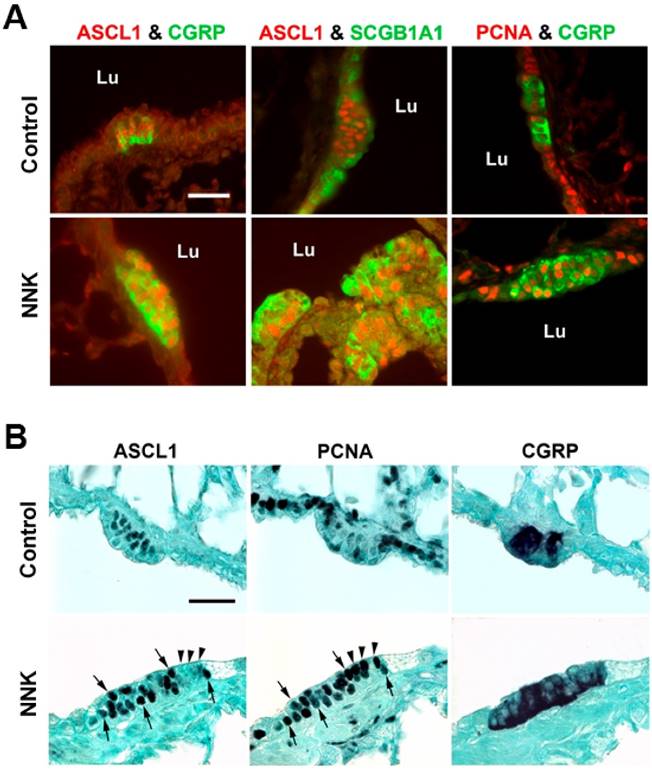
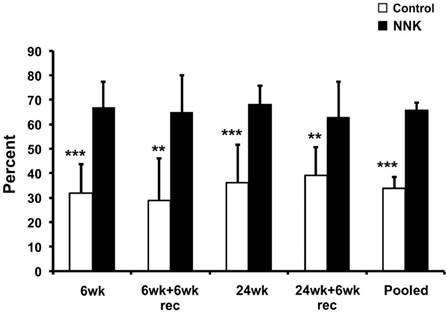
In control animals, within NE foci (defined by CGRP staining), PCNA was infrequently detectable, and cell nuclei often appeared completely unlabeled (Fig. 3A, right panel). Interestingly, PCNA was widely labeled in nuclei of ordinary airway epithelium. In contrast to controls, PCNA was clearly evident in many NE cell nuclei of hamsters exposed to NNK. Brightfield studies of adjacent serial sections separately stained for ASCL1, PCNA, and CGRP further substantiated these observations; PCNA reactivity was much more prominent in foci of NNK-treated animals (Fig. 3B). Same cell nuclear co-localization of ASCL1 with PCNA appeared demonstrable in NEBs of NNK-treated animals, but much less often in normal controls. Counts of PCNA-positive foci (containing one or more positive cells) versus negative foci, using fluorescence images as shown in Fig. 3A, showed that NNK-treated animals had twice as many PCNA-positive foci compared to controls (overall, 67% and 34%, respectively; Fig. 4).
Increased percentages of PCNA-positive foci following NNK exposure. The analysis was carried out using digitally recorded epi-fluorescence images of sections double-stained for PCNA and CGRP. A neuroendocrine focus (identified by CGRP reactivity) was regarded as positive if it contained one or more PCNA-reactive nuclei. 6wk: 6 weeks' solvent only (control) or NNK treatment; 6wk+6wk rec: 6 weeks' solvent only or NNK treatment, followed by recovery period of 6 weeks, denoted by “rec”; 24wk: 24 weeks solvent only or NNK-treatment; and 24wk+6wk rec: 24 weeks solvent only or NNK-treatment, followed by recovery period of 6 weeks. Mean ± SD is shown from N=6-10 animals per group. **: P<0.01, ***: P<0.001 between control vs. NNK treatment groups. Statistical analysis by unpaired t-test
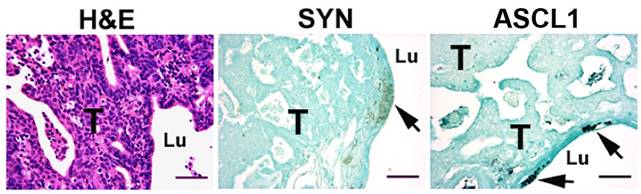
Finally, lungs were examined for tumor development. Control lung sections examined did not have any tumors, and no tumors were found in hamsters after 6 weeks NNK exposure (data not shown). However, a spectrum of tumors which appeared to be NSCLCs (non-small cell lung cancers) were seen following 24 weeks NNK exposure plus 6 weeks' recovery (Fig. 5). No immunohistochemical staining of NE markers such as SYP and ASCL1 was detected in tumor areas, and there was no morphological evidence that the tumors were related to NE foci.
Lack of neuroendocrine (NE) differentiation in NNK-induced tumors, following 24 weeks NNK exposure plus 6 weeks recovery. Left panel: Tumor mass (T) is stained with hematoxylin and eosin (H&E). Middle and right panels: Immunohistochemical reactivity is found only in NEBs (arrows) for synaptophysin (SYP) and ASCL1, respectively, whereas tumor (T) shows no NE immunoreactivity. Sections were stained using ABC peroxidase and DAB, followed by counterstaining with light green SF (ASCL1 staining intensified with nickel sulfate). Lu: airway lumen. Scale bar: 50 µm.
Discussion
The concordance between ASCL1 and the recognized NE markers CGRP and SYP establishes ASCL1 as an excellent marker for pulmonary NE cells. Its nuclear expression, as seen in this study, is consistent with the fact that ASCL1 is a transcription factor. Unlike the situation in central nervous system development, ASCL1 continues to be expressed in normal adult NE cells of the lung. However, NNK exposure resulted in the emergence and/or proliferation of increased numbers of ASCL1-positive cells; these cells were mostly confined to foci (NEBs), which increased in number and size during exposure.
Whether the apparent NE hyperplasia was due to proliferation or simply to differentiation of preexisting “silent” precursor cells is unclear; however, prominent PCNA immunoreactivity of many cells within NEBs following NNK stimulation (compared with the frequent PCNA negativity of small normal NEBs in unexposed controls) suggests that such cells had entered S phase and would proliferate. Examples of same-cell nuclear co-expression of ASCL1 and PCNA were observed in adjacent serial sections of NEBs from NNK-exposed animals, suggesting that some ASCL1-expressing cells were actively proliferating. Nevertheless, while it is often observed that PCNA levels rise during late G1 and S phases [24], the role of PCNA in relation to cell cycle and DNA synthesis is complex [25]. Often, 5-bromodeoxyuridine incorporation and PCNA immunoreactivity do not correlate well [26]. Those considerations aside, however, it does seem likely that proliferation had occurred, because many NEBs were physically much larger in NNK-exposed animals, as compared to typical NEBs in controls.
Cellular co-localization of ASCL1 and CGRP was unmistakable, with the majority of cells expressing both markers, suggesting a mature NE phenotype. The focal pattern of ASCL1 immunoreactivity, mainly associated with NEBs, may suggest the presence of a stem or progenitor population in this location that, especially when stimulated, matures into CGRP-expressing cells. With adult lung tissue injury, NE cells also possess the capacity to produce club cells and ciliated cells, with occasional co-expression of CGRP and SCGB1A1 [4]. In this study, we noted ASCL1-positive cells, particularly following NNK stimulation, that were also immunoreactive for SCGB1A1, the club cell specific protein. This is suggestive of bidirectional differentiation [5] of NEB cells after NNK exposure along either NE or epithelial pathways (i.e., toward expression of CGRP or SCGB1A1, respectively). Possibly, the uncommitted progenitor stage of this development is an ASCL1-expressing cell of fundamental NE lineage.
In this study, consistent with previous work [22], NNK induced both NE hyperplasia and non-NE neoplasia. Of numerous and varied tumors seen in the present study, none was immunoreactive for NE markers. Also, there was no morphological evidence for a direct developmental relationship between hyperplastic NEB tissue and nearby tumors. It would appear that the NNK dosage regimen applied in these experiments is hyperplasia-inducing for NE cells but insufficient to induce their neoplasia. However, insight might be gained from electron microscope studies [27-29] of lungs in hamsters treated with various nitrosamines (N-diethylnitrosamine, N-dibutylnitrosamine, N-nitrosomorpholine, nitrosoheptamethyleneimine). Those studies demonstrated nitrosamine-induced hyperplasia in both airway NE cells (described as APUD cells) and club (Clara) cells. Under prolonged treatment, club cells developed lamellated inclusions normally found in alveolar type II cells. On the other hand, NE cells tended toward squamous metaplasia while progressively losing their characteristic dense-cored neurosecretory granules (their ultrastructural hallmark) which sometimes became so scarce that NE-derived cells were difficult to identify. Adenoid and mixed lung tumors occurred, some apparently with cells of both lineages. Therefore, in our present study, NE-negative tumors may have contained some NE-derived cells with neurosecretory markers too reduced to be detectable by immunohistochemistry. That hyperplastic areas were often SCGB1A1 positive (see Figure 3A, NNK middle frame) also suggests contribution from a club cell lineage. Possibly the neoplastic origins of NNK-induced tumors included both club cells and NE cells, even though the resulting tumors appeared to be NE-immunonegative.
Whether and how the increased NEBs might relate to development of NNK-induced NSCLC-type tumors remains to be determined. ASCL1 and CGRP expressed in the lung, both by NEBs and by solitary PNECs, may play roles in supporting carcinogenesis, regardless what the actual cellular origin of a tumor might be. Among solitary PNECs, abundantly scattered within the airway epithelium, some co-express CGRP and the NE marker PGP9.5 [30]. Such PNECs might presumably also express ASCL1 as found in the present study. Regulation of CGRP synthesis is poorly understood, but it was suggested that ASCL1 may act as a transactivation factor for the calcitonin/CGRP gene complex [31, 32]. Further, CGRP synthesis appears upregulated in tissues undergoing inflammatory response [17]. NEB hypertrophy and proliferation occur following experimental exposure to bleomycin [33], naphthalene [34], or NNK as in the present study; these all induce damage and/or inflammation. CGRP is a mitogen and a pro-angiogenesis agent; it is also the most powerful microvascular dilator known [17]. These biological activities suggest that CGRP could play a role in promoting and supporting epithelial repair and/or pathological hypertrophy in damaged lung epithelium. With coincidental cancer-initiating gene mutations in lung epithelial cells, or perhaps even in NE cells themselves, ASCL1 and CGRP working in concert along with other cellular factors which doubtless may be present, such as VEGF (vascular endothelial growth factor) [20], might sustain and encourage a neoplastic growth. The actions of CGRP through stimulating cell proliferation, promoting angiogenesis, and enabling increased local nutritional supply through microvascular dilation might be ways in which this could occur. The influence of ASCL1 toward increasing the expressions of both CGRP and the anti-apoptotic regulator BCL2 [35] may also contribute to survival of a neoplasm and favor its growth, once initiated.
In conclusion, this study demonstrated a consistent close association between expressions of ASCL1 and the NE marker CGRP in pulmonary NEBs. Individual NEB cells co-express these two biomarkers, and some cells express SCGB1A1 also. NNK exposure in a chemical carcinogenesis hamster model induces hyperplastic increases both in size and number of airway NEBs, presumably enhancing net pulmonary expressions of both ASCL1 and CGRP. That actual cell proliferation is responsible for these increases is supported by nuclear co-expression of ASCL1 and PCNA. In the current experiments, NNK induced a variety of lung tumors, all NSCLC types, which did not express detectable NE markers. Whether higher levels of NNK or a longer latency time is required to induce actual NE tumors, and to what extent ASCL1-expressing cells might proliferate and play a developmental role in such a process, remain to be seen. A tentative hypothesis is offered in which, regardless of the cellular origin of a lung neoplasm, both ASCL1 and CGRP may contribute to the survival and growth of an established tumor.
Supplementary Material
Supplementary table 1.
Acknowledgements
This work was supported by an Intramural Research Program of the National Cancer Institute, Center for Cancer Research (ZIA SC000167 for RIL and ZIA BC010449 for SK). We gratefully acknowledge Dr. Hanspeter Witschi (Institute of Toxicology and Environmental Health, University of California, Davis, CA) for providing the NNK hamster model, and we thank Dr. Franco J. DeMayo for SCGB1A1 antibody.
Conflict of Interest
The authors declare no conflict of interest.
References
1. Gustafsson BI, Kidd M, Chan A, Malfertheiner MV, Modlin IM. Bronchopulmonary neuroendocrine tumors. Cancer. 2008;113:5-21
2. Vinik AI, Chaya C. Clinical Presentation and Diagnosis of Neuroendocrine Tumors. Hematol Oncol Clin North Am. 2016;30:21-48
3. Rekhtman N. Neuroendocrine tumors of the lung: an update. Arch Pathol Lab Med. 2010;134:1628-38
4. Song H, Yao E, Lin C, Gacayan R, Chen MH, Chuang PT. Functional characterization of pulmonary neuroendocrine cells in lung development, injury, and tumorigenesis. Proc Natl Acad Sci U S A. 2012;109:17531-6
5. Linnoila RI. Functional facets of the pulmonary neuroendocrine system. Lab Invest. 2006;86:425-44
6. Guillemot F, Lo LC, Johnson JE, Auerbach A, Anderson DJ, Joyner AL. Mammalian achaete-scute homolog 1 is required for the early development of olfactory and autonomic neurons. Cell. 1993;75:463-76
7. Schuurmans C, Guillemot F. Molecular mechanisms underlying cell fate specification in the developing telencephalon. Curr Opin Neurobiol. 2002;12:26-34
8. Sommer L, Shah N, Rao M, Anderson DJ. The cellular function of MASH1 in autonomic neurogenesis. Neuron. 1995;15:1245-58
9. Borges M, Linnoila RI, van de Velde HJK, Chen H, Nelkin BD, Mabry M. et al. An achaete-scute homologue essential for neuroendocrine differentiation in the lung. Nature. 1997;386:852-5
10. Wang XY, Dakir el H, Naizhen X, Jensen-Taubman SM, DeMayo FJ, Linnoila RI. Achaete-scute homolog-1 linked to remodeling and preneoplasia of pulmonary epithelium. Lab Invest. 2007;87:527-39
11. Linnoila RI, Zhao B, DeMayo JL, Nelkin BD, Baylin SB, DeMayo FJ. et al. Constitutive achaete-scute homologue-1 promotes airway dysplasia and lung neuroendocrine tumors in transgenic mice. Cancer Res. 2000;60:4005-9
12. Osada H, Tatematsu Y, Yatabe Y, Horio Y, Takahashi T. ASH1 gene is a specific therapeutic target for lung cancers with neuroendocrine features. Cancer Res. 2005;65:10680-5
13. Osada H, Tomida S, Yatabe Y, Tatematsu Y, Takeuchi T, Murakami H. et al. Roles of achaete-scute homologue 1 in DKK1 and E-cadherin repression and neuroendocrine differentiation in lung cancer. Cancer Res. 2008;68:1647-55
14. Jiang T, Collins BJ, Jin N, Watkins DN, Brock MV, Matsui W. et al. Achaete-scute complex homologue 1 regulates tumor-initiating capacity in human small cell lung cancer. Cancer Res. 2009;69:845-54
15. Jiang SX, Kameya T, Asamura H, Umezawa A, Sato Y, Shinada J. et al. hASH1 expression is closely correlated with endocrine phenotype and differentiation extent in pulmonary neuroendocrine tumors. Mod Pathol. 2004;17:222-9
16. Kelley MJ, Snider RH, Becker KL, Johnson BE. Small cell lung carcinoma cell lines express mRNA for calcitonin and alpha- and beta-calcitonin gene related peptides. Cancer Lett. 1994;81:19-25
17. Russell FA, King R, Smillie SJ, Kodji X, Brain SD. Calcitonin gene-related peptide: physiology and pathophysiology. Physiol Rev. 2014;94:1099-142
18. White SR, Hershenson MB, Sigrist KS, Zimmermann A, Solway J. Proliferation of guinea pig tracheal epithelial cells induced by calcitonin gene-related peptide. Am J Respir Cell Mol Biol. 1993;8:592-6
19. Kawanami Y, Morimoto Y, Kim H, Nakamura T, Machida K, Kido T. et al. Calcitonin gene-related peptide stimulates proliferation of alveolar epithelial cells. Respir Res. 2009;10:8
20. Toda M, Suzuki T, Hosono K, Hayashi I, Hashiba S, Onuma Y. et al. Neuronal system-dependent facilitation of tumor angiogenesis and tumor growth by calcitonin gene-related peptide. Proc Natl Acad Sci U S A. 2008;105:13550-5
21. Hecht SS, Morse MA, Amin S, Stoner GD, Jordan KG, Choi CI, Chung FL. Rapid single-dose model for lung tumor induction in A/J mice by 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and the effect of diet. Carcinogenesis. 1989;10:1901-4
22. Sunday ME, Willett CG, Graham SA, Oreffo VIC, Linnoila RI, Witschi H. Histochemical characterization of non-neuroendocrine tumors and neuroendocrine cell hyperplasia induced in hamster lung by 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone with or without hyperoxia. Am J Pathol. 1995;147:740-52
23. Polak JM, Van Noorden S. Introduction to Immunocytochemistry. New York: Spring-Verlag. 1997
24. Kurki P, Vanderlaan M, Dolbeare F, Gray J, Tan EM. Expression of proliferating cell nuclear antigen (PCNA)/cyclin during the cell cycle. Exp Cell Res. 1986;166:209-19
25. Maga G, Hübscher U. Proliferating cell nuclear antigen (PCNA): a dancer with many partners. J Cell Sci. 2003;116:3051-60
26. Muskhelishvili L, Latendresse JR, Kodell RL, Henderson EB. Evaluation of cell proliferation in rat tissues with BrdU, PCNA, Ki-67(MIB-5) immunohistochemistry and in situ hybridization for histone mRNA. J Histochem Cytochem. 2003;51:1681-8
27. Reznik-Schüller H. Ultrastructural alterations of APUD cells during nitrosamine-induced lung carcinogenesis. J Pathol. 1977;121:79-82
28. Reznik-Schüller H. Sequential morphologic alterations in the bronchial epithelium of Syrian golden hamsters during N-nitrosomorpholine-induced pulmonary tumorigenesis. Am J Pathol. 1977;89:59-66
29. Reznik-Schüller H. Ultrastructure of nitrosoheptamethyleneimine-induced lung tumors in European hamsters. Am J Pathol. 1978;93:45-52
30. Weichselbaum M, Sparrow MP, Hamilton EJ, Thompson PJ, Knight DA. A confocal microscopic study of solitary pulmonary neuroendocrine cells in human airway epithelium. Respir Res. 2005;6:115
31. Tverberg LA, Russo AF. Cell-specific glucocorticoid repression of calcitonin/calcitonin gene-related peptide transcription. Localization to an 18-base pair basal enhancer element. J Biol Chem. 1992;267:17567-73
32. Ball DW, Azzoli CG, Baylin SB, Chi D, Dou S, Donis-Keller H. et al. Identification of a human achaete-scute homolog highly expressed in neuroendocrine tumors. Proc Natl Acad Sci U S A. 1993;90:5648-52
33. Liu J, Song N, Tian S, Yu J. Neuroepithelial body increases in bleomycin-treated mice. Respir Physiol Neurobiol. 2014;193:52-4
34. Peake JL, Reynolds SD, Stripp BR, Stephens KE, Pinkerton KE. Alteration of pulmonary neuroendocrine cells during epithelial repair of naphthalene-induced airway injury. Am J Pathol. 2000;156:279-86
35. Augustyn A, Borromeo M, Wang T, Fujimoto J, Shao C, Dospoy PD. et al. ASCL1 is a lineage oncogene providing therapeutic targets for high-grade neuroendocrine lung cancers. Proc Natl Acad Sci U S A. 2014;111:14788-93
Author contact
![]() Corresponding author: Shioko Kimura, Ph.D. Bldg. 37, Rm. 3106, NIH. Bethesda, MD 20892. Phone: 301-496-0958 Fax: 301-496-8419 Email: kimurasnih.gov.
Corresponding author: Shioko Kimura, Ph.D. Bldg. 37, Rm. 3106, NIH. Bethesda, MD 20892. Phone: 301-496-0958 Fax: 301-496-8419 Email: kimurasnih.gov.