Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2016; 7(14):2067-2076. doi:10.7150/jca.15786 This issue Cite
Research Paper
Hedgehog Signaling Non-Canonical Activated by Pro-Inflammatory Cytokines in Pancreatic Ductal Adenocarcinoma
Yuqiong Wang1,2*, Gang Jin3*, Quanjiang Li1,4*, Zhiping Wang2, Weimin Hu2, Ping Li1, Shude Li1, Hongyu Wu1, Xiangyu Kong1, Jun Gao1 ![]() , Zhaoshen Li1
, Zhaoshen Li1 ![]()
1. Department of Gastroenterology, Changhai Hospital, Second Military Medical University, Shanghai 200433, China.
2. No. 411 Hospital of PLA, Shanghai 200081, China.
3. Department of Hepatobiliary Pancreatic Surgery, Changhai Hospital, Second Military Medical University, Shanghai 200433, China.
4. Department of Oncology, No. 150 Central Hospital of PLA, Luoyang, Henan Province 471000, China.
*YQ. W., G.J. and QJ. L. contributed equally to this work as co-first authors.
Received 2016-4-8; Accepted 2016-9-4; Published 2016-10-22
Abstract
Hedgehog(HH) pathway is found to be activated through a manner of canonical, or the non-canonical HH pathways. Distinct hyperplasia stroma around tumor cells is supposed to express pro-inflammatory cytokines abundantly, such as tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β), etc. in pancreatic ductal adenocarcinoma (PDAC) tissues. In this study we observed the effects of TNF-α and IL-1β on HH pathway activation in PDAC cells, and explored their activation manners. Our results showed that pro-inflammatory cytokines, TNF-α and IL-1β, could up-regulate the expression of GLI1 gene, increase its nuclear protein expression and promote malignant cell behaviors including migration, invasion, epithelial-mesenchymal transition (EMT) and drug resistance as well. Moreover, GLI1 promoter-reporter assay in combination with blocking either NF-κB or Smoothened (SMO) suggested that TNF-α and IL-1β could transcriptionally up-regulate expression of GLI1 completely via NF-κB, whereas ablation of SMO could not completely attenuate the regulation effects of TNF-α and IL-1β on GLI1 expression. Collectively, our results indicated that TNF-α and IL-1β in hyperplasia stroma can promote the PDAC cell development by activating HH pathway, through both the canonical and non-canonical HH activation ways.
Keywords: pancreatic ductal adenocarcinoma, hedgehog signaling, hyperplasia stroma, TNF-α, IL-1β.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) remains one of the most lethal malignancies, with limited therapeutic options and poor prognosis1-4. Desmoplasia with abundant fibrotic stroma and local inflammatory reaction is a hallmark feature of PDAC5. Dissecting the possible mechanisms underlying crosstalk between cancer cells and strom may propose novel therapeutic strategies with high efficacy.
Stromal activation commonly starts around precancerous lesions6, 7, and chronic pancreatitis has been proved to be an important risk factor for PDAC8. Chronic inflammation can promote pancreatic cancer progression through extensive crosstalk between malignant epithelial cells and the surrounding microenvironment9-11. Inflammation cells contribute to build up a tumor microenvironment full of deranged proliferative signaling networks orchestrated mainly by tumor-associated macrophages(TAMs)12. Some key pro-inflammatory cytokines, such as tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β), may be secreted by TAMs, and contribute to tumor cell growth, migration, invasion, and epithelial-mesenchymal transition (EMT)13. However, the specific mechanisms involved in their promotion to PDAC are still unknown.
In 1980 Nusslein-Volhard and Wieschaus first discovered the Hedgehog (HH) gene, and in the early 1990s, three HH gene homologs, Sonic Hedgehog (SHH), Indian Hedgehog (IHH) and Desert Hedgehog (DHH), were detected in vertebrates14-17. SHH is the most potent and widely expressed ligands18, 19, while IHH and DHH have been shown to play important roles in normal tissue development, including pancreas and testis organogenesis, and bone formation 20-23. HH pathway plays an essential role in embryonic development and is critical for maintenance of tissue polarity. However, it also is one of the driver pathways engaged in the tumor microenvironment reconstruction in tumorigenesis3. Recently it has been proved that HH pathway can be activated through both canonical and non-canonical pathways in PDAC tumorigenesis. The canonical HH pathway is initiated by aberrant expression of SHH, which mechanistically bind to receptor Patched1 (PTCH1) on receiving cells24, 25, and then release Smoothened (SMO), which ultimately block SUFU's function. The processing of cytoplasmic sequestration and degradation of GLIs protein is then shut down, leading to the accumulation of transcriptional activator forms (GLIA)24-27. In vertebrates GLIs family, there are three GLI transcription factors (GLI1, GLI2 and GLI3). GLI1 is the only transcriptional activator, whereas GLI2 and GLI3 act as either a positive or negative regulators. In response to ligand binding, GLI2 drives transcriptional activation, overwhelmingly the negative regulation by GLI328. GLIs transcription factors can activate the oncogene target genes, such as Cyclin-D1, Myc, Bcl-2, Ang1/2, Snai1, Nanog, Sox2, and the HH pathway feedback genes such as GLI1, PTCH1, Hhip29-31. So, GLI1 expression leads to a positive feed-back loop, and it acts as a constitutive activator24. In most cases, GLI1 overexpression is considered as a symbol of HH pathway activation32. Other than canonical HH pathway, the non-canonical activation pathways are defined as any form of signaling relating to HH pathway components that differs from the usual HH pathway paradigm27. Non-canonical activation pathways refer to either: (1) activation of signaling from PTCH1/SMO but independent of GLI transcription factors; or (2) activation of GLI transcription factors independent of SHH ligand or PTCH1/SMO. It was proposed that the latter can increase GLI activity, and be mostly oncogenic33.
Recent publications identified that besides the canonical HH activity by the increasing binding of the HH ligands signal in an autocrine or paracrine manner generate from tumor-stroma crosstalk in PDAC tumorigenesis34, non-canonical HH activation, such as non-classical activation (HH ligands and receptor independent) also plays critical roles in this process34. In current study, we aim to find out whether the key pro-inflammatory cytokines TNF-α and IL-1β in tumor microenvironment could facilitate HH pathway activation in pancreatic cancer cells through the non-canonical HH activation pathway.
Materials and Methods
Tissue samples
A total of 40 PDAC tissue samples were obtained in the period 2012~2014 at the time of surgery from Changhai Hospital, Shanghai, China, with approval of Shanghai Changhai Hospital Ethics Committee and patients' informed consent. All the samples were confirmed by pathology.
Cell culture
PDAC cells PaTu 8988T (a kind gift from Professor Thomas Elsaesser of Philipps University, Marburg, Germany) and AsPC-1 (bought from ATCC) were maintained in Dulbecco modified Eagle medium (DMEM, Gibco, USA) with 10% fetal bovine serum (FBS, Gibco, USA) and penicillin-streptomycin solution (100 U/mL penicillin/100 ng/mL streptomycin, Hyclone, USA), at 37°C with 5% CO2. Recombined human TNF-α and recombined human IL-1β were purchased from R&D Systems, USA. Cells were seeded in T 25 dishes (Corning, USA). When adherent cells reached 50% density, they were treated with TNF-α (10 ng/mL) or IL-1β (5 ng/mL), IL-1β/TNF-α with 5 μM BAY11-7082 (Beyotime Institute of Biotechnology, China), or with 10 μM cyclopamine (Sigma, USA) for 48 h, and the medium were changed every 12 h.
Nuclear and cytoplasmic protein extraction
The cells were collected with 0.25% trypsin (Gibco, USA) and centrifuged at 12,000 rpm for 5 min. After washing three times with phosphate-buffered saline (PBS), nuclear and cytoplasmic proteins were extracted with NE-PER nuclear and cytoplasmic extraction reagents (Thermo, USA) according to the manufacturer's instructions seperately. Protein concentration was measured by the bicinchoninic acid (BCA) protein assay kit (Thermo, USA) following the protocol. The protein was at last diluted to 1 μg/μL.
Western blot
Both nuclear and cytoplasmic extracted proteins were separated using sodium dodecyl sulfate(SDS)-polyacrylamide gels and then transferred to polyvinylidenefluoride (PVDF) membrane (Bio-Rad, USA). Immunoblot was performed using rabbit anti-SHH antibody (1:3000, Abcam, USA), rabbit anti-GLI1 antibody (1:1000, Abcam, USA), rabbit anti-NF-κB p65 antibody (1:200, Santa Cruz, USA), rabbit anti-SMO antibody (1:1000, Abcam, USA), rabbit anti-SUFU antibody (1:1000, Abcam, USA), rabbit anti-E-cadherin antibody (1:1000, Abcam, USA), rabbit anti-SNAI1 antibody (1:1000, Proteintech, USA), mouse anti-GAPDH antibody (1:2000, Abcam, USA), and mouse anti-Histone H3 antibody (1:2000, Abcam, USA) separately overnight at 4°C. GAPDH and Histone H3 were used as control of cytoplasmic or nuclear protein separately. The blots were detected using enhanced chemiluminescence reagent (ECL, Thermo, USA). The amount of each target gene was normalized by the level of GAPDH or Histone H3 in each sample. The experiments were repeated at least three times.
Quantitative Real-time Reverse Transcription Polymerase Chain Reaction (qRT-PCR)
Total RNA was prepared from cells or tissues, using Trizol reagent (TAKARA, Japan) according to the protocol. 1 µg of total RNA was mixed in 20 µL volume of RT reaction mixture (TAKARA, Japan). For qRT-PCR, GLI1, NF-κB p65, SHH, SMO, SUFU, and GAPDH Taqman probes were purchased from Life Technologies Company, USA. Taqman system (20 μL) (Life Technology, USA) was used. For E-cadherin, SNAI1, TNF-α, IL-1β, and GAPDH, SYBR (20 μL) (TAKARA, Japan) system was used. Each experiment was run for 50 cycles. The amount of each target gene was normalized by the level of GAPDH in each sample. The experiments were run in triplicate. E-cadherin, SNAI1, TNF-α, IL-1β, and GAPDH primers were as follows.
E-cadherin:
F 5'-AATCTGAAAGCGGCTGATACTG-3',
R 5'-TTGCCCCATTCGTTCAAGTA-3';
SNAI1:
F 5'-TCGGAAGCCTAACTACAGCG-3',
R 5'-AGATGAGCATTGGCAGCGA-3';
GAPDH:
F 5'-ATGACATCAAGAAGGTGGTG-3',
R 5'-CATACCAGGAAATGAGCTTG-3';
TNF-a:
F 5'-ATGAGCACTGAAAGCATGATC-3',
R5'-AGTGTCCCGTTACTGGGTTTCATCTGGA CGGG-3';
IL-1β:
F 5'-TGAACTGAAAGCTCTCCACCT-3',
R 5'-TGACCCGTTCAGTTTAAGGT-3'.
Transwell cell migration and invasion assay
A total of 2× 104 cells were seeded in 200 μL of no-serum DMEM in the upper chamber of a 24-well transwell system (Migration Chamber (BD 3422) for migration and Matrigel Invasion Chamber (BD 354480) for Invasion). 600 μL of DMEM with 10% FBS was added in the lower chamber. Inserts were removed 20 h (migration) or 36 h (invasion) later, and the cells on the upper surface were washed with PBS and scraped using cotton swaps. The cells were fixed in 100% ethanol for 20 min, followed by crystal violet staining. The stained inserts were cut and mounted on microscope slides. Six fields were counted from each insert, NC (negative control siRNA group) of each cell line (PaTu 8988T or AsPC-1) were used as the calibration. Images of migrating or invading cells were taken using an inverted microscope (Olympus). Three independent experiments were performed.
Drug sensitivity assay
5 × 103 cells incubated in the presence of NC, si-GLI1, TNF-α, IL-1β, TNF-α+si-GLI1, or IL-1β+si-GLI1 were seeded in 96-well plates overnight and later were incubated with different concentrations of gemcitabine (0.25, 0.5, 1, 2, 4, and 8 μM). Untreated cells (DMSO) served as control for the experiment. The medium of all samples were changed every 12 hours. Cell viability was measured 48 h later by cell counting kit (CCK)-8 (Beyotime, China). The cells were incubated for 3 h with the 10 μl of WST -8 reagents. The optical density was measured at 450 nm by microplate reader (Thermo). IC 50 (half maximal (50%) inhibitory concentration) was calculated by SPSS.
siRNA transfection
GLI1 siRNA (sc-37911), negative control siRNA (sc-37007), and siRNA transfection reagent (sc-29528) were purchased from Santa Cruz Biotechnology, USA. The cells were seeded in six-well plates before transfection. When cell density reached 60%, the cells were transfected with siRNA according to the instructions. The cells were treated with TNF-α (10 ng/mL)/IL-1β (5 ng/mL) or DMEM alone 6 h later for 48 h.
Luciferase assays
The cells were seeded in six-well plates (2×105 cells per well) for 12 h and then co-transfected with 10 ng pRL-TK (Promega, USA) and 500ng pGL3-8×GLI1 binding site (BS) luciferase report plasmids (wild or mutant) by FuGENE HD transfection (Promega, USA) according to the instructions. Six hours later, the cultures were changed. Luciferase assays were performed 48 h later with the dual luciferase assay kit (Promega, USA) according to the protocol. The luciferase activities were normalized to the Renilla luciferase activity. Mutant pGL3-8×GLI1 BS luciferase report plasmids were used as a negative control.
Statistical analysis
The calculations were performed using the Statistical Package for the Social Sciences (SPSS 17.0). The correlation between IL-1β/TNF-α and GLI1 expression was analyzed using Pearson correlation. The data were analyzed by ΔΔCT. An independent Student T test or one-way ANOVA was conducted to evaluate the one-way layout data. The data were expressed as mean ± standard deviation (SD). P<0.05 for a two-tailed test was considered to be statistically significant.
Results
TNF-α/IL-1βstimulation promoted HH pathway activation with a characteristic profile
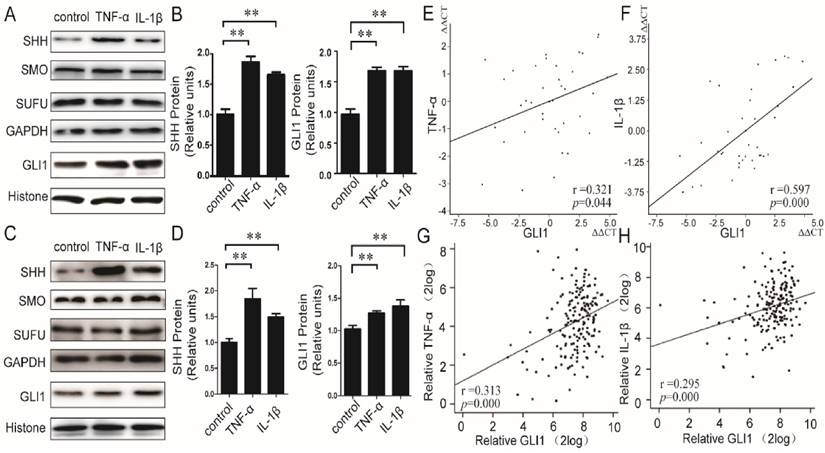
We first evaluated the effects of TNF-α/IL-1β on HH pathway in PDAC cells. Recombined human TNF-α/IL-1β protein was used to stimulate PaTu 8988T and AsPC-1 cells, and both of mRNA and protein expressions of principal members in the HH pathway, including SHH, SMO, SUFU, and GLI1, were then detected. Our results showed that the mRNA (Fig.S1) expression levels of GLI1 and SHH, but not SMO and SUFU, were significantly up-regulated by TNF-α/IL-1β. The SHH protein in membrane and cytoplasm and the GLI1 protein in nucleus were also up-regulated (Fig.1A-D). Furthermore, in PDAC tissues (40 cases), the mRNA expressions of TNF-α and IL-1β were significantly positively correlated with GLI1 (between TNF-α and GLI1, r=0.321, P=0.044; between IL-1β and GLI1, r=0.597, P=0.000) (Fig.1E-F). Meanwhile, we also got a consistency test that there existed both positive relationships of GLI1 with TNF-α and IL-1β mRNA expression after the analysis of the 183 PDAC samples from the TGCA set data (https://tcga-data.nci.nih.gov/docs/publications/tcga/ Pancreatic adenocarcinoma) (Fig.1 G-H).These results indicated that pro-inflammatory cytokines of TNF-α/IL-1β could activate HH pathway with a profile of the up-regulated expression of GLI1 and SHH expression, but little effect on SMO and SUFU.
Relationship between TNF-α/IL-1β and HH pathway key members in both PDAC cells and tissues. The protein expression changes (A-B for PaTu 8988T, C-D for AsPC-1) of nuclear GLI, and SHH, SMO, as well as SUFU after TNF-α or IL-1β stimulation, are presented. The statistical plots were calculated from the results of three independent experiments. The data are expressed as the mean ± SD. **, P<0.01, vs. control. The relationship between GLI1 and TNF-α or IL-1β mRNA expression were also showed above (E, G for TNF-α, and F, H for IL-1β; E and F from our data, G and H from the TGCA set data).The coefficient of association r was calculated using Pearson correlation method.
TNF-α/IL-1β stimulation induced EMT phenotypes, malignant behaviors and drug resistance through modulating GLI1 expression
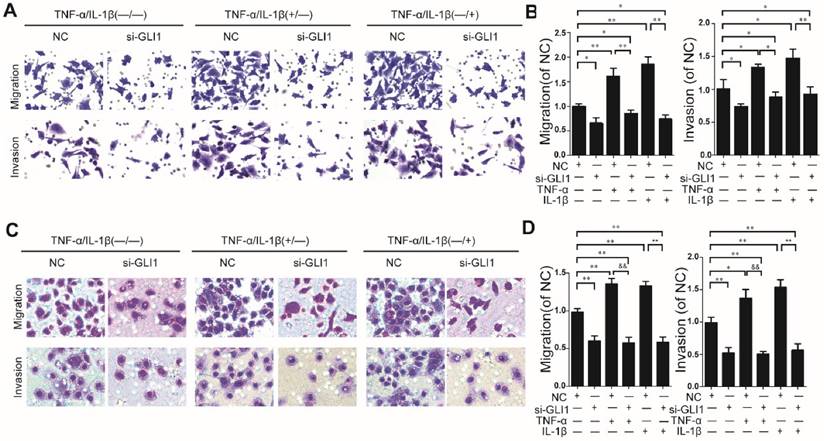
Knockdown efficiency against GLI1 by siRNA was evaluated through qRT-PCR and western blots. (Fig.S2). It is observed that TNF-α/IL-1β could enhance cell migration and invasion abilities, which could be blocked by GLI1 knockdown (Fig.2 A-D).
Enhanced cell migration/invasion abilities induced by TNF-α and IL-1β stimulation was diminished by GLI1 knock-down in PDAC cells. The typical photograph of the cell migration and invasion changes (A for PaTu 8988T, C for AsPC-1) are presented after the stimulation of TNF-α (+) or IL-1β (+) combined with the si-GLI1 transfection (si-GLI1) or the negative control si-RNA (NC) containing a scrambled sequence. The statistical plots (B for PaTu 8988T and D for AsPC-1) showed the stimulation effects by TNF-α or IL-1β were significantly diminished by the si-GLI1. The data are expressed as the mean ± SD. *P<0.05, **, &&P<0.01.
Then, to explore the effects of TNF-α/IL-1β on EMT, we tested expression of two EMT markers, E-cadherin and SNAI1, to explore the effects of TNF-α/IL-1β on EMT. Our data showed that TNF-α/IL-1β could down-regulate E-cadherin and up-regulate Snai1, both in mRNA (Fig.S3) and in protein levels (Fig.3 A-H). Such effects were completely diminished in GLI1-deficient cells. The above results indicated that GLI1 may play critical roles in TNF-α/IL-1β's promoted effects on EMT.
The effects of TNF-α and IL-1β on EMT key bio-markers and chemotherapeutics resistance were diminished by GLI1 knock-down in PDAC cells. The EMT key bio-markers (E-cadherin and SNAI1) expression changes (A-D for PaTu 8988T, and E-H for AsPC-1) of PDAC cells after TNF-α or IL-1β stimulation, are presented. NC was used as control. The coefficient of association r was calculated using Pearson correlation method. The changes of gemcitabine IC50 were observed in different treatment groups (I for PaTu 8988T, and J for AsPC-1). The data are expressed as the mean ± SD. *P<0.05, **P<0.01.
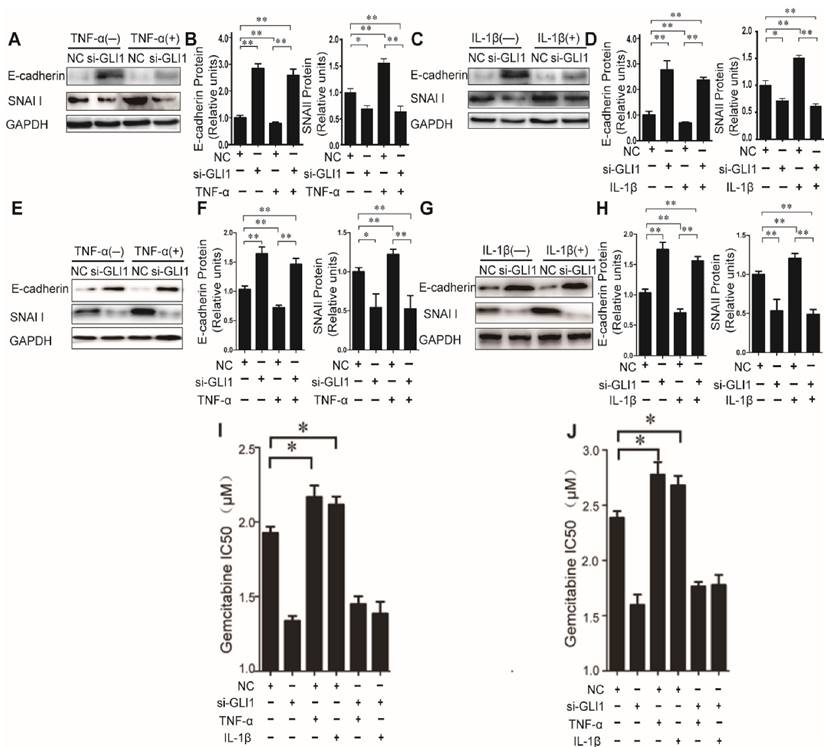
As known, EMT in cancer cells helpeddrug resistance.So, we further evaluated the effects of TNF-α/IL-1β on the gemcitabine drug resistance in different treatment groups by testing the IC50. Similar to the above results, our data showed that TNF-α/IL-1β stimulation could help reduce sensitivity of cancer cells to gemcitabine, which also significantly attenuated by GLI1 knockdown (Fig. 3I-J).
Taken together, these results indicated that the effects of TNF-α and IL-1β could induce PDAC cells to undergo EMT, enhance malignant phenotypes, and stimulation on EMT phenotypes, malignant behaviors and drug resistance could be rescued by GLI1 knock-down.
The stimulation of TNF-α/IL-1β on GLI1 expression was dependent on NF-κB nuclear translocation
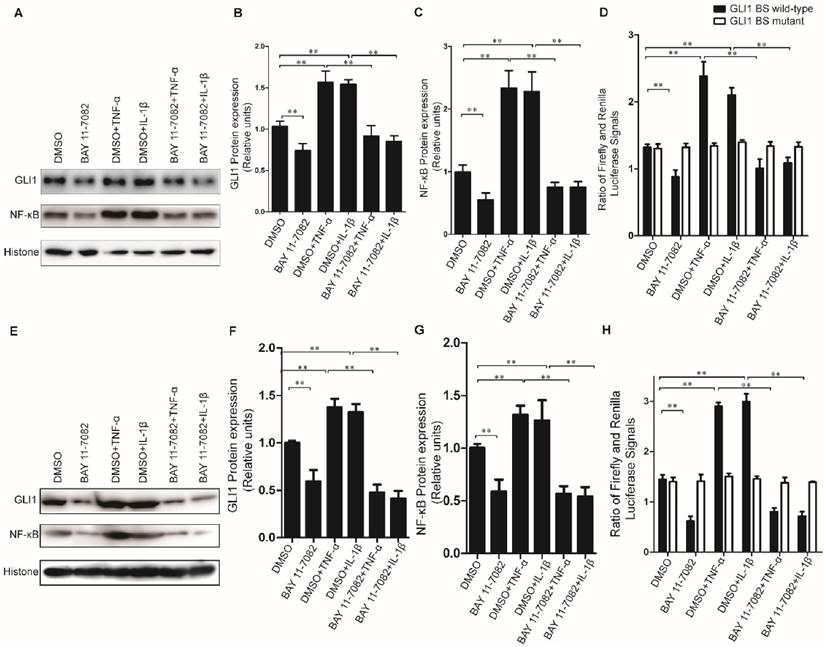
To further explore the regulatory mechanism for the alteration of GLI1 expression by TNF-α/IL-1β stimulation, the GLI1 promoter reporter assays and BAY 11-7082 (an NF-κB inhibitor) were used to test its dependency on NF-κB nuclear translocation. Our data showed that the nuclear NF-κB was significantly reduced by BAY 11-7082 and the regulatory effects of TNF-α/IL-1β against GLI1 would be significantly diminished after BAY 11-7082 treatment (Fig.4A-C, E-G). Moreover, the GLI1 promoter reporter assays showed that the effect of TNF-α/IL-1β on GLI1 mainly occurs at the transcriptional level, and that this regulation was completely dependent on modulation of the NF-κB pathway (Fig.4D, H).
Inhibition of nuclear NF-κB expression could completely prevent GLI1 nuclear expression from up-regulation by TNF-α and IL-1β stimulation in PDAC cells. The typical Western blot results of GLI1 and NF-κB nuclear protein expression (A for PaTu 8988T, E for AsPC-1) were shown above, after TNF-α/IL-1β treatments with or without NF-κB inhibitor BAY 11-7082. The statistical plots showed GLI1 (B for PaTu 8988T and F for AsPC-1) and NF-κB nuclear protein (C for PaTu 8988T and G for AsPC-1) expression. GLI1 promoter reporter plasmid (GLI1 BS wild-type) was used to test active GLI1 expression after TNF-α/IL-1β treatments with or without BAY 11-7082, and GLI1 BS mutant plasmid (GLI1 BS mutant) was used as a negative control (D for PaTu 8988T, H for AsPC-1.). The data are shown as the mean ± SD. *, P<0.05, **, P<0.01.
The effect of TNF-α/IL-1β stimulation on GLI1 expression couldn't be completely diminished by cyclopamine treatment
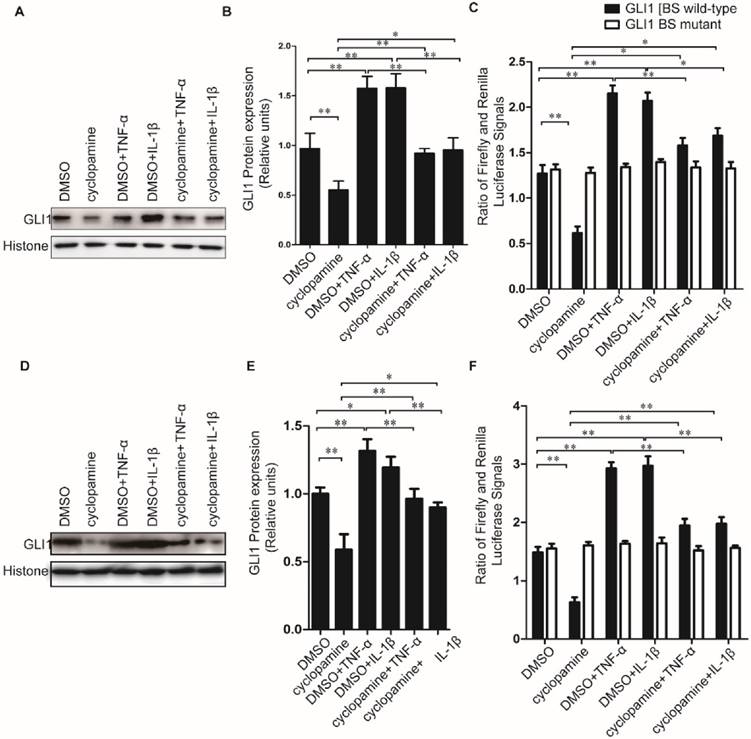
Cyclopamine has long been established an inhibitor against canonical HH activation by SMO inhibition. It was used here to further explore whether the up-regulated nuclear GLI1 expressions by TNF-α/IL-1β was totally dependent on the upstream of SMO in HH canonical pathway. Different from the results of NF-κB inhibitor BAY 11-7082 treatment, when cells were treated by cyclopamine, the up-regulated nuclear GLI1 expressions by TNF-α/IL-1β could not be thoroughly fallen back to the same level as treated only by cyclopamine treatment (Fig.5A-E), but in the middle between the cyclopamine treatment and TNF-α/IL-1β treatment only. The GLI1 promoter reporter plasmid also demonstrated that the up-regulation of GLI1 by TNF-α/IL-1β stimulation was incompletely dependently on the upstream of SMO in HH canonical pathway, suggesting that non-canonical HH activation pathways involved in the effects of TNF-α/IL-1β on HH pathway activation in PDAC cells.
Cyclopamine could incompletely block HH activation induced by TNF-α and IL-1β stimulation in PDAC cells. Typical Western blot results of GLI1 nuclear protein expression (A for PaTu 8988T, D for AsPC-1.) were shown above, after TNF-α/IL-1β treatments with or without HH inhibitor cyclopamine. The statistical plots of GLI1 nuclear protein expression were shown above (B for PaTu 8988T, E for AsPC-1). GLI1 promoter reporter plasmid was used to test active GLI1 expression after TNF-α/IL-1β treatments with or without HH inhibitor cyclopamine, and GLI1 BS mutant plasmid was used as a negative control (C for PaTu 8988T, F for AsPC-1). The data are shown as the mean ± SD. *, P<0.05, **, P<0.01.
Discussion
The pathogenesis of PDAC development remains unclear13. Inflammation, characterized by a number of TAMs in the tumor tissue microenvironment, is now believed to be involved in the etiology of PDAC. Amount of pro-inflammatory cytokines such as TNF-α and IL-1β and a variety of signaling pathways ligands such as transforming growth factor-beta (TGF-β) and platelet derived growth factor (PDGF)13 were aberrantly and abundantly expressed in tumor microenvironment. On the other aspect, it was proved that HH signaling pathway was activated both in tumor and the adjacent stroma cells during the PDAC development. Recently, more researches indicated that HH ligands secreted by PDAC cells activated mesenchymal cells in a paracrine manner, while less effect on PDAC cells themselves in autocrine manner35. Therefore, a dual directional regulation mechanism between the inflammation in adjacent mesenchymal cells and HH pathway in PDAC cells may be involved in tumorigenesis10.
Previous studies showed that TNF-α/IL-1β could increase GLI1 nuclear expression in tumor cells by the canonical HH activity way36. However, recent researches suggested some non-canonical HH activity ways, which were independent on the typical ligand-receptor interaction, also played important roles in tumorigenesis34, 37, such as the inflammatory cytokine osteopontin (OPN)38. So we hypothesized the effects of TNF-α/IL-1β on HH activation, may involve both canonical and non-canonical HH activation pathways.
In this study, we first observed a significant positive correlation between GLI1 and TNF-α, and GLI1 and IL-1β, in PDAC tissues, although with different r values due to factors such as intrinsic property of cytokines and samples size. Secondly, TNF-α/IL-1β can significantly enhance the expression of nuclear factor GLI1 in PDAC cells, elevating malignant cell behaviors including migration, invasion, EMT and drug resistance. Finally, our rescue experiments demonstrated that the enhanced GLI1 nuclear expression by TNF-α/IL-1β was entirely dependent on the NF-κB pathway, but not completely dependent on HH canonical pathway. Taken together, we proposed that TNF-α/IL-1β in hyperplasia stroma, facilitated PDAC development by promoting both canonical and non-canonical HH activation pathways.
The hyperplasia stroma around the tumor cells is the PDAC distinct pathological feature. HH pathway is a critical mediator among the sophisticated tumor-stroma interplays in PDAC development6, 38. Recently the ligands concentration-dependent paracrine manner, which was proved to regulate the proliferation, migration, and differentiation of target cells in as partial and temporal way during embryonic development, was verified to still act on the hyperplasia stroma in PDAC tissues27, 33-35. However, on the other aspect, it also proved that the growth of tumor cells dependent on both autocrine or paracrine manner of HH ligands27, 34. HH ligands secreted by tumor cell stimulated itself growth (autocrine) and diffuse locally to the tumor stroma as well, promoting fibroblast growth, angiogenesis and the secretion of stroma-derived growth factors (paracrine). Meanwhile, tumor cells respond to stroma-derived HH ligands directly in reverse paracrine signaling27. Among these complex interaction nets, our results in this study added a new clue that the hyperplasia stroma also supported that the growth of tumor cells through a new path that the pro-inflammatory cytokines mainly secreted by TAMs activate the HH pathway in both the canonical and non-canonical HH activation pathways.
To sum up, tumor-stroma inter-activation is critical for tumorigenesis. TNF-α and IL-1β, as important elements of tumor stroma, could activate the HH pathway, and further accelerate the development and progression of PDAC. However, the mechanisms underlying are complicated that both canonical and non-canonical HH activation pathways were involved in this process. Here, we pave the way in the knowledge of pro-inflammation/HH pathway interaction in tumor-stroma interplay. Further studies are still needed to illuminate the specific underlying molecular mechanisms.
Supplementary Material
Supplementary figures.
Acknowledgements
This study was supported by grants from National Natural Science Foundation of China (No. 81272663 and 81472279 to Jun Gao, No.30910103911 to Zhaoshen Li), Shanghai Science and Technology Committee Major Project on Basic Research (No. 11441901800 to Jun Gao), and Shanghai Municipal Bureau of Health Research Topic (No.2012-2015 to Shude Li).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Guerra C, Collado M, Navas C. et al. Pancreatitis-induced inflammation contributes to pancreatic cancer by inhibiting oncogene-induced senescence. Cancer cell. 2011;19:728-39
2. Damhofer H, Medema JP, Veenstra VL. et al. Assessment of the stromal contribution to Sonic Hedgehog-dependent pancreatic adenocarcinoma. Molecular oncology. 2013;7:1031-42
3. Vincent A, Herman J, Schulick R, Hruban RH, Goggins M. Pancreatic cancer. Lancet (London, England). 2011;378:607-20
4. Gao J, Li Z, Chen Z. et al. Antisense SMO under the control of the PTCH1 promoter delivered by an adenoviral vector inhibits the growth of human pancreatic cancer. Gene therapy. 2006;13:1587-94
5. Ene-Obong A, Clear AJ, Watt J. et al. Activated pancreatic stellate cells sequester CD8+ T cells to reduce their infiltration of the juxtatumoral compartment of pancreatic ductal adenocarcinoma. Gastroenterology. 2013;145:1121-32
6. Erkan M, Hausmann S, Michalski CW. et al. The role of stroma in pancreatic cancer: diagnostic and therapeutic implications. Nature reviews Gastroenterology & hepatology. 2012;9:454-67
7. Wang W, Gao J, Man XH, Li ZS, Gong YF. Significance of DNA methyltransferase-1 and histone deacetylase-1 in pancreatic cancer. Oncology reports. 2009;21:1439-47
8. Lowenfels AB, Maisonneuve P, Cavallini G. et al. Pancreatitis and the risk of pancreatic cancer. International Pancreatitis Study Group. The New England journal of medicine. 1993;328:1433-7
9. Guerra C, Schuhmacher AJ, Canamero M. et al. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer cell. 2007;11:291-302
10. Baumgart S, Ellenrieder V, Fernandez-Zapico ME. Oncogenic transcription factors: cornerstones of inflammation-linked pancreatic carcinogenesis. Gut. 2013;62:310-6
11. Greer JB, Whitcomb DC. Inflammation and pancreatic cancer: an evidence-based review. Current opinion in pharmacology. 2009;9:411-8
12. Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860-7
13. Helm O, Held-Feindt J, Grage-Griebenow E. et al. Tumor-associated macrophages exhibit pro- and anti-inflammatory properties by which they impact on pancreatic tumorigenesis. International journal of cancer Journal international du cancer. 2014;135:843-61
14. Nusslein-Volhard C, Wieschaus E. Mutations affecting segment number and polarity in Drosophila. Nature. 1980;287:795-801
15. Echelard Y, Epstein DJ, St-Jacques B. et al. Sonic hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of CNS polarity. Cell. 1993;75:1417-30
16. Krauss S, Concordet JP, Ingham PW. A functionally conserved homolog of the Drosophila segment polarity gene hh is expressed in tissues with polarizing activity in zebrafish embryos. Cell. 1993;75:1431-44
17. Roelink H, Augsburger A, Heemskerk J. et al. Floor plate and motor neuron induction by vhh-1, a vertebrate homolog of hedgehog expressed by the notochord. Cell. 1994;76:761-75
18. Ingham PW, McMahon AP. Hedgehog signaling in animal development: paradigms and principles. Genes & development. 2001;15:3059-87
19. Pathi S, Pagan-Westphal S, Baker DP. et al. Comparative biological responses to human Sonic, Indian, and Desert hedgehog. Mechanisms of development. 2001;106:107-17
20. Hebrok M, Kim SK, St Jacques B, McMahon AP, Melton DA. Regulation of pancreas development by hedgehog signaling. Development (Cambridge, England). 2000;127:4905-13
21. Kawahira H, Ma NH, Tzanakakis ES, McMahon AP, Chuang PT, Hebrok M. Combined activities of hedgehog signaling inhibitors regulate pancreas development. Development (Cambridge, England). 2003;130:4871-9
22. St-Jacques B, Hammerschmidt M, McMahon AP. Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes & development. 1999;13:2072-86
23. Yao HH, Whoriskey W, Capel B. Desert Hedgehog/Patched 1 signaling specifies fetal Leydig cell fate in testis organogenesis. Genes & development. 2002;16:1433-40
24. Lauth M, Toftgard R. Non-canonical activation of GLI transcription factors: implications for targeted anti-cancer therapy. Cell cycle. 2007;6:2458-63
25. Robbins DJ, Fei DL, Riobo NA. The Hedgehog signal transduction network. Science signaling. 2012;5:re6
26. Thayer SP, di MaGLIano MP, Heiser PW. et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature. 2003;425:851-6
27. Marini KD, Payne BJ, Watkins DN, Martelotto LG. Mechanisms of Hedgehog signalling in cancer. Growth factors. 2011;29:221-34
28. Kim J, Kato M, Beachy PA. GLI2 trafficking links Hedgehog-dependent activation of SMOothened in the primary cilium to transcriptional activation in the nucleus. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:21666-71
29. Stecca B, Ruiz IAA. Context-dependent regulation of the GLI code in cancer by HEDGEHOG and non-HEDGEHOG signals. Journal of molecular cell biology. 2010;2:84-95
30. Scales SJ, de Sauvage FJ. Mechanisms of Hedgehog pathway activation in cancer and implications for therapy. Trends in pharmacological sciences. 2009;30:303-12
31. Hui CC, Angers S. GLI proteins in development and disease. Annual review of cell and developmental biology. 2011;27:513-37
32. Damhofer H, Veenstra VL, Tol JA, van Laarhoven HW, Medema JP, Bijlsma MF. Blocking Hedgehog release from pancreatic cancer cells increases paracrine signaling potency. Journal of cell science. 2015;128:129-39
33. Brennan D, Chen X, Cheng L, Mahoney M, Riobo NA. Noncanonical Hedgehog signaling. Vitamins and hormones. 2012;88:55-72
34. Shevde LA, Samant RS. Nonclassical hedgehog-GLI signaling and its clinical implications. International journal of cancer Journal international du cancer. 2014;135:1-6
35. Yauch RL, Gould SE, Scales SJ. et al. A paracrine requirement for hedgehog signalling in cancer. Nature. 2008;455:406-10
36. Nakashima H, Nakamura M, Yamaguchi H. et al. Nuclear factor-kappaB contributes to hedgehog signaling pathway activation through sonic hedgehog induction in pancreatic cancer. Cancer research. 2006;66:7041-9
37. Blotta S, Jakubikova J, Calimeri T. et al. Canonical and noncanonical Hedgehog pathway in the pathogenesis of multiple myeloma. Blood. 2012;120:5002-13
38. Das S, Samant RS, Shevde LA. Nonclassical activation of Hedgehog signaling enhances multidrug resistance and makes cancer cells refractory to SMOothened-targeting Hedgehog inhibition. The Journal of biological chemistry. 2013;288:11824-33
Author contact
![]() Corresponding authors: Jun Gao, MD, Ph.D., Department of Gastroenterology, Changhai Hospital, Second Military Medical University, Shanghai, China. E-mail: 13816012151com; Tel: +8613816012151 or Zhaoshen Li, MD, Ph.D., Department of Gastroenterology, Changhai Hospital, Second Military Medical University, Shanghai, China. E-mail: zhslinet.
Corresponding authors: Jun Gao, MD, Ph.D., Department of Gastroenterology, Changhai Hospital, Second Military Medical University, Shanghai, China. E-mail: 13816012151com; Tel: +8613816012151 or Zhaoshen Li, MD, Ph.D., Department of Gastroenterology, Changhai Hospital, Second Military Medical University, Shanghai, China. E-mail: zhslinet.