Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2016; 7(14):2035-2044. doi:10.7150/jca.15200 This issue Cite
Research Paper
Breast Cancer Malignant Processes are Regulated by Pax-5 Through the Disruption of FAK Signaling Pathways
Sami Benzina1,2, Jason Harquail1,2, Roxann Guerrette1,2, Pierre O'Brien1,2, Stéphanie Jean1,2, Nicolas Crapoulet1, Gilles A. Robichaud1,2 ![]()
1. Department of Chemistry and Biochemistry, Université de Moncton, Moncton, NB, Canada E1A 3E9.
2. Atlantic Cancer Research Institute, Moncton, NB, Canada E1C 8X3.
Received 2016-2-4; Accepted 2016-8-3; Published 2016-10-22
Abstract
The study of genetic factors regulating breast cancer malignancy is a top priority to mitigate the morbidity and mortality associated with this disease. One of these factors, Pax-5, modulates cancer aggressiveness through the regulation of various components of the epithelial to mesenchymal transitioning (EMT) process. We have previously reported that Pax-5 expression profiles in cancer tissues inversely correlate with those of the Focal Adhesion Kinase (FAK), a potent activator of breast cancer malignancy. In this study, we set out to elucidate the molecular and regulatory relationship between Pax-5 and FAK in breast cancer processes. Interestingly, we found that Pax-5 mediated suppression of breast cancer cell migration is dependent of FAK activity. Our mechanistic examination revealed that Pax-5 inhibits FAK expression and activation. We also demonstrate that Pax-5 is a potent modulator of FAK repressors (p53 and miR-135b) and activator (NFκB) which results in the overall suppression of FAK-mediated signaling cascades. Altogether, our findings bring more insight to the molecular triggers regulating phenotypic transitioning process and signaling cascades leading to breast cancer progression.
Keywords: FAK, Pax-5, Breast cancer, EMT-MET, NFκB, migration, metastasis.
Introduction
Breast cancer is the most common cancer diagnosed in women worldwide. Specifically, metastasis is responsible for 90% of deaths in patients suffering from this disease. Despite major efforts in metastasis research, specific details on how cancer cells actually initiate the metastatic pathway are still lacking. In general, breast cancer cells encompass phenotypic changes between epithelial and mesenchymal states. In the epithelial-mesenchymal transition (EMT), tumor cells acquire motility and invasive properties to spread into distant organs via the circulatory system. Inversely, the mesenchymal-epithelial transition (MET) allows metastatic cancer cells to revert to an epithelial program to form secondary tumors in new tissue environments (reviewed in [1]).
One of the hallmarks of EMT in carcinoma and a critical signaling molecule involved in tumor malignancy is the Focal Adhesion Kinase 1 (FAK). FAK is a non-receptor protein tyrosine kinase that localizes to cellular focal adhesions or cell contacts with the extracellular matrix to regulate cancer aggressiveness and progression. In response to cell adhesion, FAK is initially activated by autophosphorylation of Tyr397 which promotes the interaction with Src homology 2 (SH2) domain containing proteins, such as Src family (reviewed in [2, 3]). The binding of Src family kinases onto FAK induces the phosphorylation of several other tyrosine residues located on FAK which results in FAK kinase activity and signaling via the mitogen-activated protein kinases/MAPKs (i.e. ERK1/2, JNK and p38) [4-7].
FAK regulates cell spreading, adhesion, migration, proliferation and angiogenesis; processes that are all involved in cancer progression [3, 8]. Aberrant FAK-mediated signaling has been correlated with a number of cancer types including brain [9, 10], breast [11-13], thyroid [14], prostate [15] and colon [12-16]. FAK also plays a key role in EMT through focal adhesion turnover which allows the tumor cell to increase both its migratory and invasive properties [17, 18]. As a result, FAK expression correlates with breast tumor invasion, metastatic potential and poor disease outcome [12, 13, 19, 20]. Although the effects of FAK activation and signaling in cancer malignancy are well described, very few studies have elucidated upstream mechanisms regulating FAK activity during cellular phenotypic transitioning processes leading to breast cancer progression.
Recently, we and others have reported roles for the Pax-5 transcription factor in breast cancer epithelial features that are reminiscent of MET [21, 22]. Pax-5 is a member of the Paired Box (PAX) gene family which control gene expression programs pivotal in cellular processes such as proliferation, differentiation and apoptosis [23-25]. Pax-5 is normally found in, and required for the development of B cells, embryonic central nervous system and adult testis [26-28]. In addition to its potent role as an oncogene in lymphoid cancer lesions, Pax-5 has been increasingly linked to other types of cancer including breast cancer [21, 29-33]. Intriguingly, Pax-5 seems to confer an anti-proliferative effect in most carcinomas studied in contrast to its stimulatory effect on B cell proliferation [21, 34]. Most importantly, we and others show that Pax-5 expression reduces mesenchymal marker expression, colony formation and migratory capabilities while concomitantly inducing epithelial characteristics in invasive breast carcinoma cells [21, 22, 35].
It is still unclear how phenotypic transitioning processes are initiated and coordinated during the metastatic cascade of breast cancer cells. However, we have previously reported that the expression profiles of pro-epithelial factor Pax-5 inversely correlate with those from pro-malignant FAK [22]. We therefore set out to establish and elucidate a possible regulatory mechanism between the Pax-5 transcription factor and FAK in breast cancer processes. In this study, we demonstrate that Pax-5 is a potent inhibitor of FAK expression and activity leading to the concomitant suppression of aggressive features in breast cancer cells. Our findings bring new insight into the regulatory triggers controlling FAK expression and activities during the phenotypic transitioning processes of breast cancer malignancy.
Materiel and Methods
Cell culture and reagents
The MDA-MB-231 (MB231) (mammary ductal carcinoma, HTB-26); MCF7 (mammary ductal carcinoma, HTB-22); and BT549 (mammary ductal carcinoma, HTB-122) cells lines were obtained from the American Type Culture Collection (Rockville, MD, USA). MB231 cell lines were cultured in DMEM high glucose medium supplemented with 10% fetal bovine serum (FBS) and L-glutamine (2 mM). BT549 were cultured in RPMI 1640 medium supplemented with 10% FBS, L-glutamine (2mM) and bovine insulin (0.01 mg/mL). MCF7 cells were maintained in DMEM low glucose medium supplemented with 10% FBS and L-glutamine (2 mM). Cell culture media and reagents were obtained from HyClone (ThermoFisher Scientific, Burlington, ON, Canada) except for the FBS which was provided by PAA Laboratories (Dartmouth, MA, USA). The FAK chemical inhibitor, FAK inhibitor 14 (CAS No. 4506-66-5), was purchased from Cayman chemicals (Ann Arbor, Michigan 48108 USA); TNFα from ThermoFisher; and pifithrin-α (a p53 inhibitor) from Sigma-Aldrich (Oakville, ON, Canada).
Plasmids and transfections
Transient overexpression of Pax-5 and FAK were performed using transfections with the human Pax-5 recombinant gene cloned into pcDNA3.1 as previously described [36] and cloned recombinant FAK [37] generously provided by Dr. Vita Golubovskaya (Roswell Park Cancer Institute, Buffalo, NY). BT549 cells stably expressing Pax-5 (BT549-Pax5) were generated using the Gateway cloning system (ThermoFisher Scientific) Briefly, the full-length Pax-5 cDNA devoid of a stop codon was generated by PCR (forward: 5'-GGGGACAAGTTTGTACAAAAAAGCAGGCTTCAAAATGGATTTAGAGAAAAATTATCCGAC-3' and reverse: 5'-GGGGACCACTTTGTACAAGAAAGCTGGGTCTCTGTGACGGTCATAGGCAGTGGCGGC-3') and integrated into the pDONR™221 vector to form the entry clones. The correct entry clones were confirmed by sequencing and then subcloned by recombination into the pLenti4/TO/V5-DEST lentiviral-expressing vector. BT549-Pax5 Zeocin-resistant clones were then selected, expanded and characterized for stable recombinant of Pax-5 using Western blot and qRT-PCR. Transfections of DNA plasmids were performed with the XtremeGene 9 reagent (Roche, Branford, CT, USA) as previously described [38]. Briefly, cells were seeded in six-well plates 24 h pre-transfection at a density of 5×105 cells/well. Cells were then incubated with a DNA-reagent complex (ratio of 1 μg of DNA/3 µL of reagent) for 24 h in media with reduced serum without antibiotics. We also made use of luciferase-based reporter gene constructs such as the renilla-luciferase (pRL) (Promega, Madison,WI, USA), pNFκB-luciferase (Promega), the p53 responsive p21 promoter luciferase construct (p21-luc) (generously provided by Dr. G. Matlashewski, McGill University, Montreal, Canada) [39], the Pax-5 responsive CD19 promoter (CD19-luc) [40] and the FAK-luciferase construct[41] (generously provided by Dr. Vita Golubovskaya, Roswell Park Cancer Institute, NY).
Gene suppression experiments were accomplished using a transfection mixture RNA-mediated suppression systems. Cells were seeded in six-well plates (5×105 cells/well), grown for 24 h, and transfected with either 100 pmol of siRNA targeting Pax-5 (ON-TARGET plus, Dharmacon, Rockford, IL, USA); or, 18 pmol/L of hsa-anti-miR-135b (ThermoFisher Scientific) using Lipofectamine 2000 (ThermoFisher Scientific) in OPTI-MEM with reduced serum. Controls include treatments with a non-specific (non-silencing) siRNA (Dharmacon) or scrambled non-targeting anti-miRs (ThermoFisher Scientific) respectively.
Western blot analysis
Cells (106) were lysed using 0.1 mL of 2X whole cell lysate (WCL) buffer (0.125M Tris pH 6.8, Glycerol 0.2 g/mL, 4.0% SDS) along with freshly added 10 µL phenylmethylsulfonyl fluoride (PMSF, 10 mg/mL), 5 µL/ml of protease inhibitor cocktail set III (Millipore, Billerica, MA. USA), 10 μL/ml sodium orthovanadate (100 mM) and 10 µL/mL phosphatase inhibitor cocktail 1 (Sigma-Aldrich). Protein content was measured using the Bicinchoninic acid protein assay (ThermoFisher Scientific). Equal amounts of protein were then submitted to a 12% SDS-polyacrylamide gel electrophoresis. Proteins were then transferred onto a polyvinylidene fluoride membrane (PVDF) (Millipore, Billerica, MA, USA), blocked with 5% non-fat milk and probed with either anti-Pax-5 (New England Peptide), anti-FAK (#05-537, Millipore, Billerica, MA, USA), anti-phospho-FAK (#AF4528, R&D Systems, Minneapolis, MN, USA), anti-p38 (#9212, Cell Signaling, Boston, MA, USA), anti-Paxilin (#2542, Cell Signaling, Boston, MA, USA), anti-JNK (#9252, Cell Signaling, Boston, MA, USA), anti-phospho-JNK (#9251, Cell Signaling, Boston, MA, USA), anti-Src (#05-184, Millipore), anti-phospho-Src (Tyr418) (#07-909, Millipore), IKKα (#ab32041), Abcam, Cambridge, MA, USA), IKKβ (#ab32135), Abcam), phospho-IKK(α and β) (#ab194528, Abcam), or anti-G3PDH (#2275-PC-100, Trevigen, Gaithersburg, MD, USA) antibodies. After washings, membranes were incubated with Horseradish peroxydase (HRP)-conjugated antibodies (ThermoFisher Scientific) and visualized by chemiluminescence according to the manufacturer's protocol (SuperSignal West Dura, Thermo Scientific).
Migration assays
Transwell migration assays were performed as previously described [38]. Briefly, cells were starved in DMEM containing only 0.1% FBS for 16 hours previous to the seeding into tissue culture transwell inserts (Greiner Bio-One, NC, USA). The inserts were first coated with 250 μl of gelatin 0.1% (Sigma-Aldrich). Following an incubation at 37°C for 2 hours, excess coating was then removed and 200 μl of suspended cells (50 000 cells/well) in DMEM 0.1% FBS were added to the inserts. The transwells were then placed in a well containing DMEM 0.1% FBS and incubated at 37°C for 1 hour. Next, they were moved into wells containing DMEM 20% and incubated for 9 hours at 37°C. The negative control inserts were moved into wells containing DMEM 0.1%. After incubation, cells that passed through the membrane were harvested with trypsin and submitted to a cell viability with CellTiter-Blue (Promega) for quantification and comparison.
Luciferase reporter gene assays
Luciferase-based reporter gene assays were performed in cells using the Dual-Glo luciferase system (Promega). Transfections included 200 ng of renilla-luciferase (pRL) (Promega) as an internal control in addition to 1.8 μg of firefly-based luciferase plasmid. 48 h post-transfection, cells were lysed and analyzed for luciferase activity using a luminometer (BMG Fluostar, ThermoFisher Scientific). Relative reporter activity was calculated and normalized based on renilla-luciferase activity which reflected transfection efficiency. At least three separate experiments were performed, each in triplicate.
PCR and qRT-PCR assays
Reverse transcriptions were performed on purified total RNA retrieved from live cells by Trizol Reagent (ThermoFisher Scientific) according to manufacturer's instructions. Levels of gene expression were verified by Taqman PCR as previously described [42]. Reaction mixtures were composed of 20 nM of Taqman PCR primers (ThermoFisher Scientific) and cDNA, diluted in water to a total of 12.5 µL where 12.5 µL of 2X iQ SYBR Green Supermix (BioRad, Mississauga, Ontario, Canada) was added. Reactions were run in an Eppendorf Realplex real time PCR apparatus (Eppendorf, Ontario, Canada) with the following parameters: 2-minute initial denaturation, 35 cycles of 30 sec at 95°C, 30 sec at 60°C (optics on), 30 sec at 72°C. Each PCR assay was performed in triplicates on at least three separate biological replicates. Comparative expression levels were calculated using the ΔΔCt method of Livak and Schmittgen, (2001) [43], using the hypoxanthine ribosyltransferase (HPRT) transcript as a normalizing control.
Taqman PCR on non-coding miRNA was performed on miRNA enriched lysates using the mirVanaTM miRNA isolation kit (ThermoFisher Scientific). MiRNAs were reverse transcribed using the TaqMan® microRNA Reverse Transcription Kit (ThermoFisher Scientific) followed by Taqman assays (ThermoFisher Scientific) for the respective miRNA studied using the Realplex PCR apparatus. Relative expression levels from each miRNA were normalized using the expression levels of RNU48 as an internal control by Taqman (Life Technologies).
Results
Pax-5 regulates FAK activation and FAK-mediated signal transduction
The roles of Pax-5 (pro-epithelial) and FAK (pro-mesenchymal) in breast cancer phenotype identity have been well established. We have previously shown that Pax-5 expression levels inversely correlate with those from FAK in cancer cells [22]. We thus set out to establish the effects of Pax-5 overexpression on FAK expression and phosphorylation levels (active form). MCF7 and MB231 breast cancer cells were transiently transfected with Pax-5 or the empty vector alone pcDNA3.1 and submitted to Western blot analysis for the evaluation of total and phosphorylated forms of FAK (Figure 1A). Interestingly, Pax-5 overexpression suppressed total FAK expression in both MCF7 and MB231 breast cancer cell lines. In addition, Pax-5 transfected cells also displayed attenuated phosphorylated forms of FAK. Control samples were also performed which include Pax-5 recombinant expression and GAPDH as an internal loading control. These results strongly suggest that Pax-5 is a modulator of FAK expression and activation in breast cancer cells.
Pax-5 suppresses FAK protein levels and FAK-mediated cascades. Western blot was performed on MCF7 and MB231 cells either non-transfected (NT); or, transfected with the control empty pcDNA vector (pCT) and Pax-5. (A) Protein expression levels were studied for Pax-5, FAK, phosphorylated-FAK (P-FAK) and GAPDH used as an internal control. (B) Expression levels from downstream signaling components of FAK were also evaluated by Western blot for p38, JNK, phosphorylated-JNK (P-JNK), paxillin, AKT, HEF-1 and GAPDH used as a loading control. The presented data is the calculated mean of three independent samples and is representative of three different experiments.
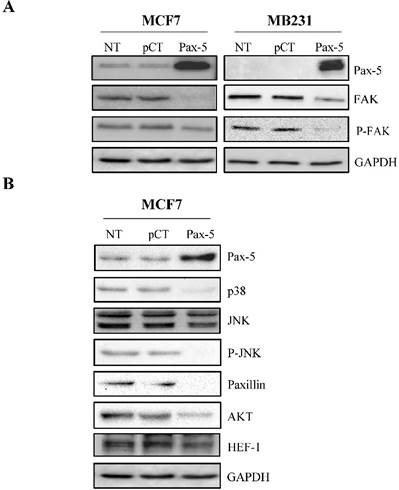
To further extend our studies of Pax-5-mediated regulation of FAK, we examined whether Pax-5-mediated suppression of FAK would also affect commonly known downstream effectors of the FAK cascade. We thus made use of the MCF7 breast cancer cell model transfected with Pax-5 and evaluated the expression levels of: p38, JNK, paxillin, PI3K, AKT and HEF-1 by Western blot (Figure 1B). As expected, we found that the expression levels from the majority of signaling components located downstream of FAK were attenuated by Pax-5. More precisely, Pax-5 slightly suppressed the expression of Src, JNK, AKT and HEF-1. On the other hand, Pax-5 completely abrogated the levels of p38, phosphorylated JNK, and paxillin. We were unable to detect any PI3K levels in our cell models (data not shown). Altogether, our results further support a role for Pax-5 as a repressor of the FAK-induced signaling cascade in breast cancer cells.
To elucidate the regulatory mechanisms of Pax-5-mediated suppression of FAK expression, we examined the capacity of Pax-5 to regulate FAK gene transcription in breast cancer cells. Using Taqman assays, we analyzed FAK mRNA expression in MCF7 cells transfected with Pax-5. Surprisingly, no significant differences in FAK transcript levels were observed between Pax-5 or vector-transfected cells (Figure 2A). We then proceeded with luciferase-based reporter gene under the control of the human FAK promoter region (FAK-luc). We found that Pax-5 transfected cells only displayed a mild decrease in FAK reporter activities (figure 2B).
Pax-5-modulates FAK protein levels by indirect mechanisms. MCF7 cells transfected with either the empty vector (pcDNA) or Pax-5 were analyzed for (A) FAK mRNA expression using Taqman PCR assays and standardized against the HPRT housekeeping gene. The presented data is the calculated mean and representative of three different experiments in relation to the non-transfected parental cell line. (B) FAK promoter transactivation potential was assessed using dual reporter gene assays (Promega) with the FAK promoter region cloned upstream from the firefly luciferase gene [41]. Normalization of luciferase activity was performed using non-inducible renilla luciferase and plotted in relative light units (RLU) to each respective control sample. (C) Western blot was performed on transfected or non-transfected (NT) MCF7 and MB231 cells. Protein expression levels were studied for Pax-5, calpain (CAPN), Src, phosphorylated-Src (P-Src) and GAPDH as an internal control. Results are representative of triplicate experiments.
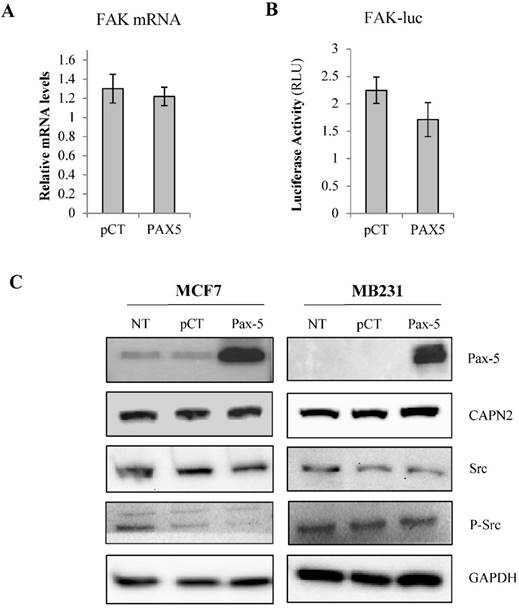
To assess the role of Pax-5 in FAK protein stabilization, we examined the expression levels of Src, phosphorylated Src and calpain, two known regulators of FAK protein stability [44, 45]. Using Western blot on Pax-5 transfected MCF7 and MB231 cells, we found that Pax-5 slightly attenuates Src and phosphorylated-Src levels in MCF7 with no apparent changes to calpain levels when compared to the GAPDH loading control (Figure 2C).
Pax-5 regulates FAK modulators
Given that FAK transcriptional control was very moderate following Pax-5 transfection, we expanded our studies to examine the potential of Pax-5 to affect other commonly known regulators of FAK gene expression. We next evaluated the effects of Pax-5 on two potent transcriptional factors previously shown to bind and regulate the FAK promoter, p53 (repressor) and NFκB (activator) [41]. Using reporter gene assays, we found that Pax-5 transfected MCF7 cells displayed greater p21-Luc activity and repressed NFκB-Luc activity respectively in comparison to vector-transfected control cells (Figure 3A). We also repeated these experiments in the malignant MB231 breast model and observed the same (Pax-5 induced p21-Luc activity and repressed NFκB-Luc activity respectively) (Figure 3A).
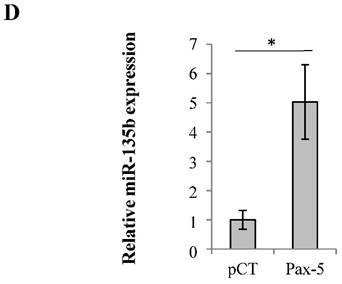
Pax-5 regulates FAK expression modulators p53, NFκB and miR135b. (A) Breast cancer cells transfected with either the empty vector (pCT) or Pax-5 were analyzed for p53 (using the p53 responsive p21 promoter element/p21-luc) [39], NFκB (NFκB-luc) and Pax-5 (using the Pax-5 responsive CD19 promoter/CD19-luc) [40] transactivation potential in MCF7 (left panel) and MB231 (right panel) using dual reporter gene assays (Promega). Normalization of firefly luciferase activity was performed using non-inducible renilla luciferase and plotted in relative light units (RLU) to each respective control. (B) Western blot was performed on MB231 and MCF7 cells which were either non transfected (NT); or, transfected with the pcDNA control vector (pCT) and Pax-5 to reveal the expression levels of Pax-5, IKK-α, IKK-β and phosphorylated-IKK-α/β (P-IKK-α/β) and GAPDH as an internal control. (C) MB231 cells transfected with Pax-5 or the empty vector (pCT) were either left untreated (-) or treated with TNFα (20ng/ml) or pifithrin (PFT, 30μM) for 24h and examined for FAK, Pax-5 and GAPDH expression by Western blot. Expression ratios (FAK/GAPDH) were determined using pixel densities using ImageJ software. (D) MCF7 cells were also examined for miR-135b expression levels using Taqman PCR which was standardized against the RNU48 small nucleolar RNA used as a housekeeping gene. The presented data is the calculated mean of three independent experiments in triplicates samples and plotted in relation to non-treated parental cells (*p <0.05).
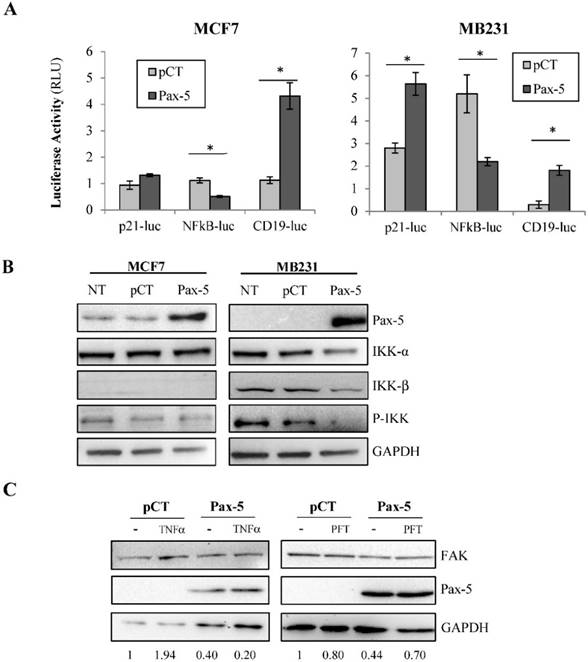
Given the marked suppression of NFκB-dependent reporter activity by Pax-5, we further examined the effects of Pax-5 on the NFκB signaling cascade. More specifically, we assessed the ability of Pax-5 to modulate the expression and activation of IκB inhibitors (IKK) in breast cancer cells (MCF7 and MB231) by Western blot. We found that Pax-5 inhibited total IKKα and IKKβ in addition to phosphorylated-IKK in MB231 cells (Figure 3B). On the other hand, Pax-5 did not significantly alter IKKα expression in MCF7 where IKKβ was undetectable. Our results support a role for Pax-5-mediated suppression of IKK expression and activity (notably in aggressive breast cancer cells) which consequently leads to inhibition of NFκB activity in breast cancer cells.
To further examine whether Pax-5-induced suppression of FAK is dependent upon NFκB or p53 activities, we performed rescue experiments using MB231 cells transfected with Pax-5 and treated with either TNFα (NFκB activator) or pifithrin (p53 inhibitor). As expected, Pax-5 transfection resulted in a 60% decrease of FAK expression in Western blot (Figure 3C). However, although TNFα treatments alone induced FAK expression (1.94 fold), TNFα did not rescue FAK expression in Pax-5 transfected cells. On the other hand, when Pax-5-transfected cells were treated with pifithrin (p53 inhibitor), FAK expression was increased 38 % (from 0.44 to 0.70) in Pax-5 bearing cells. These results suggest that p53 is an important modulator of Pax-5-induced suppression of FAK in breast cancer cells.
To pursue our elucidation of Pax-5-mediated effects on FAK regulators, we also studied the possible Pax-5-induced modulation of non-coding RNAs regulating FAK expression. Recent studies have reported that the human micro-RNA 135b (miR-135b) targets FAK mRNA for inhibition [46]. Cells were thus transfected with Pax-5 where miR-135b was assessed by Taqman PCR and standardized according to RNU48 levels used as an internal control. We found that Pax-5 induced miR-135b expression levels 5 fold in comparison to vector transfected controls (Figure 3D). Globally, we show that Pax-5 is an important modulator of FAK regulators such as p53, NFκB and miR-135b leading to the suppression of FAK expression and activation.
Pax-5 regulates aggressiveness of breast cancer through FAK
We have previously shown that the expression levels of Pax-5 (pro-epithelial) and FAK (pro-mesenchymal) inversely correlate during breast cancer progression. To determine whether Pax-5 suppresses breast cancer invasiveness through the mediated inhibition of FAK, we conducted several rescue experiments using conditional expression of both Pax-5 and FAK on breast cancer migration properties. First, we evaluated cell migration in the epithelial-dominant MCF7 cell line which were knocked-down for Pax-5 endogenous expression using a pool of siRNAs (siPax-5) in addition to concomitantly inhibiting FAK using the FAK-specific chemical inhibitor 14. As expected, control treatments which consisted of untouched cells; scrambled siRNA transfection; and FAK inhibitor 14-treated cells did not migrate (Figure 4A). In contrast, MCF7 with suppressed levels of Pax-5 (siPax-5) increased cell migration 2.4-fold over scrambled siRNA transfected controls. More importantly, the observed induction of migration in Pax-5 attenuated cells was completely abolished when FAK was inhibited by chemical treatment (Figure 4A). These findings suggest that the induction of cell migration in Pax-5 downregulated cells is FAK dependant.
Pax-5-mediated suppression of breast cancer migration is FAK dependent. (A) Migration assays were performed on MCF7 cells transfected with Pax-5-targeting siRNAs (siPax-5) or non-targeting control siRNAs (CT-siRNA) and treated with the FAK inhibitor (inhibitor 14) (FAK inhib.) using transwell chambers. Results are the calculated mean of raw fluorescence and representative of triplicate experiments. (B) Migration assays were also performed on BT549 cells stably expressing the empty control pLenti4 vector (BT549-CT) or Pax-5 (BT549-Pax-5) which were transiently transfected for FAK recombinant expression (pFAK) [37] or the control vector (pCT). The data is presented in fold change relative to control cells and is the calculated mean of three independent samples represented from three different experiments (*p <0.05).
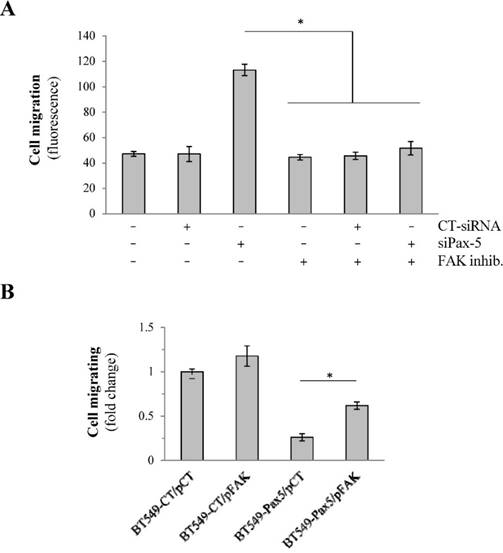
To confirm our observations, we also studied cell migration in mesenchymal-dominant BT549 cells stably transfected with either the Pax-5 gene (BT549-Pax5) or the empty vector alone as a control (BT549-CT). As expected, invasive BT549-CT control cells demonstrated cell migration which was slightly enhanced (31%) by FAK transfection (pFAK) (Figure 4B). In contrast, cells transfected with recombinant Pax-5 (BT549-Pax5) decreased cell migration by 67%. More interestingly, FAK transfection in BT549-Pax5 cells partially rescued migration properties. Overall, our results suggest that Pax-5 mediated suppression of breast cancer cell migration is dependent of FAK activity.
Discussion
Breast cancer metastasis is a multistep process, which consists of a cancer cell acquiring new biological processes and transitioning between phenotypic identities (EMT-MET) to colonize new environments. At the present time, it is still not entirely known how and when EMT and MET programs are coordinated. We and others have recently reported that the expression of Pax-5 in cancer cells suppresses mesenchymal properties and concomitantly promotes epithelial features reminiscent of MET [21, 22, 35]. Moreover, we have shown that the expression levels of pro-epithelial factor Pax-5 inversely correlate with those from pro-malignant FAK in cancer cells [22] thus suggesting a molecular relationship between these genes which could lead to the modulation of EMT-MET processes. In this study, we demonstrate that Pax-5 acts as a potent inhibitor of FAK expression and activity leading to the concomitant suppression of aggressive features in breast cancer cells.
In an attempt to elucidate the molecular mechanisms supporting Pax-5-mediated suppression of FAK, we first assessed the potential of the Pax-5 transcription factor to modulate FAK gene expression. This hypothesis was supported by chromatin immunoprecipitation (ChIP) sequencing data provided by the ENCODE project (HudsonAlpha Institute for Biotechnology) [47] which found that the human FAK promoter region is co-immunoprecipitated using Pax-5 antibodies. These observations thus suggested that the FAK gene promoter is targeted for Pax-5 binding and transactivation (data not shown). In addition, these findings correlate with the JASPAR CORE database which also demonstrates the presence of multiple Pax-5 binding motifs within the FAK promoter region [48]. Surprisingly, despite the significant downregulation of FAK protein levels by Pax-5, we were unable to detect any significant changes in neither FAK transcriptional levels; nor FAK reporter gene activity following Pax-5 conditional expression in the breast cancer model tested.
We extended our study to examine the impact of Pax-5 on previously reported modulators of FAK expression. The most significant effects of Pax-5 were observed on the promoter activities of p53 and NFκB, two potent transcription factors known to bind and regulate the FAK promoter [41]. First, we found that Pax-5 induces p53 which has previously been reported in breast cancer cells [21]. Rescue experiments also validate that p53 may be a prominent means for Pax-5 to inhibit FAK expression. On the other hand, we also observed that Pax-5 is a potent inhibitor of NFκB which has previously been shown to induce FAK expression. Although we were unable to validate NFκB as a direct mediator of Pax-5-induced suppression of FAK, these findings are interesting given that NFκB-targeted genes are associated with EMT [49] and breast cancer malignancy [50-52]. Mechanistically, NFκB is usually sequestered in the cytoplasm by its natural inhibitor (IκB) which is regulated by the IκB kinase (IKK) complex composed of the catalytic kinases IKKα and IKKβ (reviewed in [53, 54]). Upon cell activation, IKKs phosphorylate IκB which is degraded and allows nuclear import of NFκB to exert its role as a transcription factor on mesenchymal cancer genes. Accordingly, we found that recombinant expression of Pax-5 significantly suppressed both IKKα and IKKβ expression and phosphorylation events in the mesenchymal-dominant MB231 breast cancer cell line. These results further validate the suppressive role of Pax-5 on NFκB activation. Studies have also shown that FAK and NFκB can mutually activate each other in cancer processes [41, 55, 56]. In this case, the suppressive effects of Pax-5 on each component of the NFκB/FAK cascade would greatly enhance breast cancer cell epithelialisation and inhibit tumor malignancy. Indeed, these results concur with reports showing that elevated Pax-5 expression in primary breast tumor sites correlates with lower risk of disease progression and relapse [22]. Our findings thus strongly support a beneficial role for Pax-5 expression in breast primary tumors.
We also investigated possible Pax-5-regulated miRNAs capable of targeting FAK inhibition. MiRNAs are small non-coding RNAs that bind the 3'UTR of mRNAs, which result in the degradation or translation inhibition of the target mRNAs [57]. Aberrantly expressed miRNAs are frequently associated with a variety of cancers including breast cancers [58-60]. Among these miRNAs, the miR-135 family (miR-135a and miR-135b) is remarkably regulated in carcinogenesis. MiR-135a has been identified to increase apoptosis and decrease proliferation in classic Hodgkin lymphoma by targeting JAK2 [61]. On the other hand, miR-135a and miR-135b promote colon cancer by targeting the tumor suppressor APC (Adenoma Polyposis Coli) [62]. Recently, miR-135b was reported to target FAK resulting in the suppression of cancer cell invasion [46]. In our study, we found that Pax-5 induced miR-135b expression in breast cancer cells which would again increase Pax-5's capacity to downregulate FAK protein expression and suppress breast cancer progression.
Altogether, our study describes a new role for Pax-5 as a FAK inhibitor and tumor suppressor of breast cancer processes. Mechanistically, we demonstrate that Pax-5 inhibits FAK expression and activation indirectly through the regulation of FAK modulators (notably through p53). These findings are striking given that Pax-5 is a potent oncogene in lymphoid cancers [63]. The study herein suggests that Pax-5-mediated suppression of breast cancer aggressiveness is largely due to FAK inhibition. These finding are of particular interest given the association of FAK in invasive cancers and the interest in FAK targeting as a promising anticancer strategy [64, 65].
Acknowledgements
Research funds were provided by grants from the New Brunswick (NB) Innovation Foundation, the Canadian Breast Cancer Foundation-Atlantic Chapter, the Canadian Breast Cancer Society/QEII Foundation, and the NB Health Research Foundation (NBHRF). GAR is supported by a Canadian Institutes of Health Research (CIHR) New Investigator Award; SB is supported by the Beatrice Hunter Cancer Research Institute (BHCRI) with funds provided by the Terry Fox Strategic Health Research Training Program in Cancer Research at CIHR in partnership with the NB Health Research Foundation and The Roses of Hope Foundation (La Vie en Rose Foundation); JH is supported by a trainee award from the BHCRI with funds provided by the Canadian Breast Cancer Foundation, Atlantic Region and the NBHRF as part of The Terry Fox Strategic Health Research Training Program in Cancer Research at CIHR; and RG is a trainee in the Cancer Research Training Program of the BHCRI, with funds provided by the Canadian Breast Cancer Foundation - Atlantic Region and the NBHRF.
Conflict of interest
We have no potential conflicts of interest to disclose.
References
1. Yao D, Dai C, Peng S. Mechanism of the mesenchymal-epithelial transition and its relationship with metastatic tumor formation. Mol Cancer Res. 2011;9:1608-20
2. McLean GW, Carragher NO, Avizienyte E, Evans J, Brunton VG, Frame MC. The role of focal-adhesion kinase in cancer - a new therapeutic opportunity. Nat Rev Cancer. 2005;5:505-15
3. Golubovskaya VM, Kweh FA, Cance WG. Focal adhesion kinase and cancer. Histol Histopathol. 2009;24:503-10
4. Zhao J, Guan JL. Signal transduction by focal adhesion kinase in cancer. Cancer Metastasis Rev. 2009;28:35-49
5. Bouchard V, Demers MJ, Thibodeau S, Laquerre V, Fujita N, Tsuruo T. et al. Fak/Src signaling in human intestinal epithelial cell survival and anoikis: differentiation state-specific uncoupling with the PI3-K/Akt-1 and MEK/Erk pathways. Journal of cellular physiology. 2007;212:717-28
6. Huang C, Rajfur Z, Borchers C, Schaller MD, Jacobson K. JNK phosphorylates paxillin and regulates cell migration. Nature. 2003;424:219-23
7. Horowitz JC, Rogers DS, Sharma V, Vittal R, White ES, Cui Z. et al. Combinatorial activation of FAK and AKT by transforming growth factor-beta1 confers an anoikis-resistant phenotype to myofibroblasts. Cell Signal. 2007;19:761-71
8. Luo M, Guan JL. Focal adhesion kinase: a prominent determinant in breast cancer initiation, progression and metastasis. Cancer Lett. 2010;289:127-39
9. Canel M, Secades P, Rodrigo JP, Cabanillas R, Herrero A, Suarez C. et al. Overexpression of focal adhesion kinase in head and neck squamous cell carcinoma is independent of fak gene copy number. Clin Cancer Res. 2006;12:3272-9
10. Gutenberg A, Bruck W, Buchfelder M, Ludwig HC. Expression of tyrosine kinases FAK and Pyk2 in 331 human astrocytomas. Acta Neuropathol. 2004;108:224-30
11. Cance WG, Craven RJ, Weiner TM, Liu ET. Novel protein kinases expressed in human breast cancer. Int J Cancer. 1993;54:571-7
12. Cance WG, Harris JE, Iacocca MV, Roche E, Yang X, Chang J. et al. Immunohistochemical analyses of focal adhesion kinase expression in benign and malignant human breast and colon tissues: correlation with preinvasive and invasive phenotypes. Clin Cancer Res. 2000;6:2417-23
13. Lark AL, Livasy CA, Dressler L, Moore DT, Millikan RC, Geradts J. et al. High focal adhesion kinase expression in invasive breast carcinomas is associated with an aggressive phenotype. Mod Pathol. 2005;18:1289-94
14. Kim SJ, Park JW, Yoon JS, Mok JO, Kim YJ, Park HK. et al. Increased expression of focal adhesion kinase in thyroid cancer: immunohistochemical study. J Korean Med Sci. 2004;19:710-5
15. Lacoste J, Aprikian AG, Chevalier S. Focal adhesion kinase is required for bombesin-induced prostate cancer cell motility. Mol Cell Endocrinol. 2005;235:51-61
16. Lark AL, Livasy CA, Calvo B, Caskey L, Moore DT, Yang X. et al. Overexpression of focal adhesion kinase in primary colorectal carcinomas and colorectal liver metastases: immunohistochemistry and real-time PCR analyses. Clin Cancer Res. 2003;9:215-22
17. Bailey KM, Liu J. Caveolin-1 up-regulation during epithelial to mesenchymal transition is mediated by focal adhesion kinase. J Biol Chem. 2008;283:13714-24
18. Cicchini C, Laudadio I, Citarella F, Corazzari M, Steindler C, Conigliaro A. et al. TGFbeta-induced EMT requires focal adhesion kinase (FAK) signaling. Exp Cell Res. 2008;314:143-52
19. Pylayeva Y, Gillen KM, Gerald W, Beggs HE, Reichardt LF, Giancotti FG. Ras- and PI3K-dependent breast tumorigenesis in mice and humans requires focal adhesion kinase signaling. J Clin Invest. 2009;119:252-66
20. Lightfoot HM Jr, Lark A, Livasy CA, Moore DT, Cowan D, Dressler L. et al. Upregulation of focal adhesion kinase (FAK) expression in ductal carcinoma in situ (DCIS) is an early event in breast tumorigenesis. Breast Cancer Res Treat. 2004;88:109-16
21. Vidal LJ, Perry JK, Vouyovitch CM, Pandey V, Brunet-Dunand SE, Mertani HC. et al. PAX5alpha enhances the epithelial behavior of human mammary carcinoma cells. Mol Cancer Res. 2010;8:444-56
22. Crapoulet N, O'Brien P, Ouellette RJ, Robichaud GA. Coordinated expression of Pax-5 and FAK1 in metastasis. Anticancer Agents Med Chem. 2011;11:643-9
23. Strachan T, Read AP. PAX genes. Curr Opin Genet Dev. 1994;4:427-38
24. Tremblay P, Gruss P. Pax: genes for mice and men. Pharmacol Ther. 1994;61:205-26
25. Robson EJ, He SJ, Eccles MR. A PANorama of PAX genes in cancer and development. Nat Rev Cancer. 2006;6:52-62
26. Hagman J, Wheat W, Fitzsimmons D, Hodsdon W, Negri J, Dizon F. Pax-5/BSAP: regulator of specific gene expression and differentiation in B lymphocytes. Curr Top Microbiol Immunol. 2000;245:169-94
27. Rolink AG, Schaniel C, Busslinger M, Nutt SL, Melchers F. Fidelity and infidelity in commitment to B-lymphocyte lineage development. Immunol Rev. 2000;175:104-11
28. Adams B, Dorfler P, Aguzzi A, Kozmik Z, Urbanek P, Maurer-Fogy I. et al. Pax-5 encodes the transcription factor BSAP and is expressed in B lymphocytes, the developing CNS, and adult testis. Genes Dev. 1992;6:1589-607
29. Vouyovitch CM, Vidal L, Borges S, Raccurt M, Arnould C, Chiesa J. et al. Proteomic analysis of autocrine/paracrine effects of human growth hormone in human mammary carcinoma cells. Adv Exp Med Biol. 2008;617:493-500
30. Palmisano WA, Crume KP, Grimes MJ, Winters SA, Toyota M, Esteller M. et al. Aberrant promoter methylation of the transcription factor genes PAX5 alpha and beta in human cancers. Cancer Res. 2003;63:4620-5
31. Ross DT, Scherf U, Eisen MB, Perou CM, Rees C, Spellman P. et al. Systematic variation in gene expression patterns in human cancer cell lines. Nat Genet. 2000;24:227-35
32. Ellsworth RE, Seebach J, Field LA, Heckman C, Kane J, Hooke JA. et al. A gene expression signature that defines breast cancer metastases. Clin Exp Metastasis. 2009;26:205-13
33. Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207-10
34. Liu W, Li X, Chu ES, Go MY, Xu L, Zhao G. et al. Paired box gene 5 is a novel tumor suppressor in hepatocellular carcinoma through interaction with p53 signaling pathway. Hepatology. 2011;53:843-53
35. Schebesta A, McManus S, Salvagiotto G, Delogu A, Busslinger GA, Busslinger M. Transcription factor Pax5 activates the chromatin of key genes involved in B cell signaling, adhesion, migration, and immune function. Immunity. 2007;27:49-63
36. Robichaud GA, Nardini M, Laflamme M, Cuperlovic-Culf M, Ouellette RJ. Human Pax-5 C-terminal isoforms possess distinct transactivation properties and are differentially modulated in normal and malignant B cells. J Biol Chem. 2004;279:49956-63
37. Beviglia L, Golubovskaya V, Xu L, Yang X, Craven RJ, Cance WG. Focal adhesion kinase N-terminus in breast carcinoma cells induces rounding, detachment and apoptosis. Biochem J. 2003;373:201-10
38. Cormier K, Harquail J, Ouellette RJ, Tessier PA, Guerrette R, Robichaud GA. Intracellular expression of inflammatory proteins S100A8 and S100A9 leads to epithelial-mesenchymal transition and attenuated aggressivity of breast cancer cells. Anticancer Agents Med Chem. 2014;14:35-45
39. Ghosh A, Stewart D, Matlashewski G. Regulation of human p53 activity and cell localization by alternative splicing. Mol Cell Biol. 2004;24:7987-97
40. Nutt SL, Morrison AM, Dorfler P, Rolink A, Busslinger M. Identification of BSAP (Pax-5) target genes in early B-cell development by loss- and gain-of-function experiments. EMBO J. 1998;17:2319-33
41. Golubovskaya V, Kaur A, Cance W. Cloning and characterization of the promoter region of human focal adhesion kinase gene: nuclear factor kappa B and p53 binding sites. Biochim Biophys Acta. 2004;1678:111-25
42. Robichaud GA, Perreault JP, Ouellette RJ. Development of an isoform-specific gene suppression system: the study of the human Pax-5B transcriptional element. Nucleic Acids Res. 2008;36:4609-20
43. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402-8
44. Franco SJ, Rodgers MA, Perrin BJ, Han J, Bennin DA, Critchley DR. et al. Calpain-mediated proteolysis of talin regulates adhesion dynamics. Nat Cell Biol. 2004;6:977-83
45. Carragher NO, Westhoff MA, Fincham VJ, Schaller MD, Frame MC. A novel role for FAK as a protease-targeting adaptor protein: regulation by p42 ERK and Src. Curr Biol. 2003;13:1442-50
46. Golubovskaya VM, Sumbler B, Ho B, Yemma M, Cance WG. MiR-138 and MiR-135 directly target focal adhesion kinase, inhibit cell invasion, and increase sensitivity to chemotherapy in cancer cells. Anticancer Agents Med Chem. 2014;14:18-28
47. Consortium EP, Birney E, Stamatoyannopoulos JA, Dutta A, Guigo R, Gingeras TR. et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799-816
48. Mathelier A, Zhao X, Zhang AW, Parcy F, Worsley-Hunt R, Arenillas DJ. et al. JASPAR 2014: an extensively expanded and updated open-access database of transcription factor binding profiles. Nucleic Acids Res. 2014;42:D142-7
49. Maier HJ, Schmidt-Strassburger U, Huber MA, Wiedemann EM, Beug H, Wirth T. NF-kappaB promotes epithelial-mesenchymal transition, migration and invasion of pancreatic carcinoma cells. Cancer Lett. 2010;295:214-28
50. Chua HL, Bhat-Nakshatri P, Clare SE, Morimiya A, Badve S, Nakshatri H. NF-kappaB represses E-cadherin expression and enhances epithelial to mesenchymal transition of mammary epithelial cells: potential involvement of ZEB-1 and ZEB-2. Oncogene. 2007;26:711-24
51. Huber MA, Azoitei N, Baumann B, Grunert S, Sommer A, Pehamberger H. et al. NF-kappaB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. J Clin Invest. 2004;114:569-81
52. Min C, Eddy SF, Sherr DH, Sonenshein GE. NF-kappaB and epithelial to mesenchymal transition of cancer. J Cell Biochem. 2008;104:733-44
53. Shen RR, Hahn WC. Emerging roles for the non-canonical IKKs in cancer. Oncogene. 2011;30:631-41
54. Prasad S, Ravindran J, Aggarwal BB. NF-kappaB and cancer: how intimate is this relationship. Mol Cell Biochem. 2010;336:25-37
55. Sonoda Y, Matsumoto Y, Funakoshi M, Yamamoto D, Hanks SK, Kasahara T. Anti-apoptotic role of focal adhesion kinase (FAK). Induction of inhibitor-of-apoptosis proteins and apoptosis suppression by the overexpression of FAK in a human leukemic cell line, HL-60. J Biol Chem. 2000;275:16309-15
56. Wang X, Chen Q, Xing D. Focal adhesion kinase activates NF-kappaB via the ERK1/2 and p38MAPK Pathways in amyloid-beta25-35-induced apoptosis in PC12 cells. J Alzheimers Dis. 2012;32:77-94
57. Guo H, Ingolia NT, Weissman JS, Bartel DP. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature. 2010;466:835-40
58. Bloomston M, Frankel WL, Petrocca F, Volinia S, Alder H, Hagan JP. et al. MicroRNA expression patterns to differentiate pancreatic adenocarcinoma from normal pancreas and chronic pancreatitis. Jama. 2007;297:1901-8
59. Iorio MV, Ferracin M, Liu CG, Veronese A, Spizzo R, Sabbioni S. et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005;65:7065-70
60. Zhao G, Wang B, Liu Y, Zhang JG, Deng SC, Qin Q. et al. miRNA-141, downregulated in pancreatic cancer, inhibits cell proliferation and invasion by directly targeting MAP4K4. Molecular cancer therapeutics. 2013;12:2569-80
61. Navarro A, Diaz T, Martinez A, Gaya A, Pons A, Gel B. et al. Regulation of JAK2 by miR-135a: prognostic impact in classic Hodgkin lymphoma. Blood. 2009;114:2945-51
62. Nagel R, le Sage C, Diosdado B, van der Waal M, Oude Vrielink JA, Bolijn A. et al. Regulation of the adenomatous polyposis coli gene by the miR-135 family in colorectal cancer. Cancer Res. 2008;68:5795-802
63. O'Brien P, Morin P Jr, Ouellette RJ, Robichaud GA. The Pax-5 gene: a pluripotent regulator of B-cell differentiation and cancer disease. Cancer Res. 2011;71:7345-50
64. van Nimwegen MJ, van de Water B. Focal adhesion kinase: a potential target in cancer therapy. Biochem Pharmacol. 2007;73:597-609
65. Parsons JT, Slack-Davis J, Tilghman R, Roberts WG. Focal adhesion kinase: targeting adhesion signaling pathways for therapeutic intervention. Clin Cancer Res. 2008;14:627-32
Author contact
![]() Corresponding author: Gilles A. Robichaud, Université de Moncton, Moncton, NB, Canada, E1A 3E9. gilles.robichaudca; Tel: (506)858-4320; Fax: (506)858-4541.
Corresponding author: Gilles A. Robichaud, Université de Moncton, Moncton, NB, Canada, E1A 3E9. gilles.robichaudca; Tel: (506)858-4320; Fax: (506)858-4541.