Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2016; 7(13):1780-1790. doi:10.7150/jca.15792 This issue Cite
Research Paper
Diet- and Genetically-induced Obesity Produces Alterations in the Microbiome, Inflammation and Wnt Pathway in the Intestine of Apc+/1638N Mice: Comparisons and Contrasts
Wei Liu1,2, Jimmy W. Crott3, Lin Lyu1, Anna C. Pfalzer3, Jinchao Li1, Sang-Woon Choi1, 4, Yingke Yang5, Joel B. Mason3,6, Zhenhua Liu1,3 ![]()
1. Nutrition and Cancer Prevention Laboratory, School of Public Health and Health Sciences, University of Massachusetts, Amherst, USA.
2. Institute of Food Science and Technology, Shandong Academy of Agricultural Sciences, Jinan, China.
3. Jean Mayer USDA Human Nutrition Research Center on Aging, Tufts University, Boston, USA.
4. CHA Cancer Research Center, CHA University, Seoul, Korea.
5. Department of Molecular Medicine, Hunan University, Changsha, China.
6. Tufts Medical Center Cancer Center, Tufts University, Boston, USA.
Received 2016-4-10; Accepted 2016-6-29; Published 2016-8-12
Citation:
Liu W, Crott JW, Lyu L, Pfalzer AC, Li J, Choi SW, Yang Y, Mason JB, Liu Z. Diet- and Genetically-induced Obesity Produces Alterations in the Microbiome, Inflammation and Wnt Pathway in the Intestine of Apc+/1638N Mice: Comparisons and Contrasts. J Cancer 2016; 7(13):1780-1790. doi:10.7150/jca.15792. https://www.jcancer.org/v07p1780.htm
Other stylesAbstract
Obesity is an established risk factor for colorectal cancer (CRC). Our previous study indicated that obesity increases activity of the pro-tumorigenic Wnt-signaling. Presently, we sought to further advance our understanding of the mechanisms by which obesity promotes CRC by examining associations between microbiome, inflammation and Wnt-signaling in Apc+/1638N mice whose obesity was induced by one of two modalities, diet- or genetically-induced obesity. Three groups were employed: Apc+/1638NLepr+/+ fed a low fat diet (10% fat), Apc+/1638NLepr+/+ fed a high fat diet (60% fat, diet-induced obesity), and Apc+/1638NLeprdb/db fed a low fat diet (genetically-induced obesity). All animals received diets for 16 weeks from 8 to 24 weeks of age. The abundance of 19 bowel cancer-associated bacterial taxa were examined by real-time PCR. The abundance of Turicibacter and Desulfovibrio decreased, but F. prausnitizii increased, in diet-induced obese mice (p < 0.05). In contrast, in genetically-induced obese mice, Bifidobacterium, A. muciniphila and E. rectale decreased, but Peptostrptococcus, and E. coli increased (p < 0.05). Both diet- and genetically-induced obesity altered the expression of genes involved in bacterial recognition (MyD88) and increased inflammation as indicated by elevated levels of cytokines (IFNγ and TNF-α for genetically-induced obesity, and IL-6 for diet-induced obesity). The elevated inflammation was associated with altered expression of genes that are integral components of the Wnt-signaling cascade in a fashion indicating its activation. These findings demonstrate that the composition of the small intestinal microbiome is affected differently in diet- and genetically-induced obesity, but both are associated with elevated intestinal inflammation and alterations of the Wnt pathway towards enhancing tumorigenesis.
Keywords: Obesity, Microbiome, Inflammation, Wnt pathway, Intestinal cancer.
Introduction
In recent decades, there has been a striking increase of obesity in both developed countries and urban regions of developing countries. Obesity is now a major global public health issue and a further increase of ~50% by 2030 is predicted (1, 2). Epidemiological studies have suggested obesity is an important risk factor for colorectal cancer (CRC), with 50-100% increased risks for women and men who have a BMI ≥ 30 (3). Animal studies are in agreement with both diet- and genetically-induced obesity promoting intestinal tumorigenesis (4, 5). CRC is a major cause of death: ~50,000 individuals annually in the US and ~700,000 worldwide (6, 7), thus understanding all of the risk factors for CRC and identifying means to combat them is of the utmost importance.
It is well-known that obesity induces chronic low-grade inflammation in the adipose tissue as well as systemically (8, 9), and recent animal studies—including our own—indicate that an inflammatory milieu resides in the colon as well (10, 11). Our prior study demonstrates that obesity-driven inflammation, exemplified by an increased production of inflammatory cytokine TNF-α, is associated with alterations of several key components (GSK3β and β -catenin) of the Wnt-signaling pathway, (10) the over-activation of which is thought to be an early integral step in 90% of human CRCs (12, 13).
While increasing evidence indicates that obesity-driven chronic intestinal inflammation plays an important role in colorectal tumorigenesis, the origin of this inflammation remains elusive. Recent studies suggest that alterations in the composition and diversity of intestinal microbiome is an important characteristic of obesity (14). Animal studies indicate that both diet- (15) and genetically-induced (16) obesity alters the composition of microbiome characterized by an expansion of Firmicutes and reduction in Bacteroidetes at the phylum level. A disruption of the 'normal' microbial community is thought to increase the risk of pathogen infection and to promote inflammation (17).
To understand the role of gut microbiome in linking obesity with inflammation, Wnt pathway and tumorigenesis, we sought to examine how high fat diet and obesity per se drive gut dysbiosis, promote intestinal inflammation, activate the Wnt pathway, and thereby enhance intestinal tumorigenesis using the Apc+/1638N mouse model, the tumorigenic phenotype of which is particularly sensitive to dietary modifications (18, 19).
Experimental Methods
Animal study
The protocol for this study was approved by the Institutional Animal Care and Use Committee of the Jean Mayer USDA Human Nutrition Research Center on Aging at Tufts University. A detailed description of the protocol has previously been reported (20). Briefly, in order to study how obesity influences the intestinal microbiome and tumorigenesis, we utilized both high fat diet-induced (60% kcal fat) and genetically-induced (Leprdb/db) obese models (21). To study intestinal tumorigenesis, we utilized the Apc+/1638N model (22). Though this model develops tumors predominantly in the small intestine rather than the colorectum, it is the nature of other genetically-engineered rodent models of CRC such as the widely used ApcMin mouse. Nevertheless, the Apc+/1638N model has a mildly tumorigenic phenotype that sensitively responds to dietary modification and the predilection to developing these small intestinal tumors responds to dietary perturbations in the same manner that diet modifies CRC risk in the human, underscoring the relevance of this model to human colonic tumorigenesis (18, 19). Since the tumors predominantly occur in the small intestine in this mouse model, the small intestinal microbial constitution is more reflective of the tumor microenvironment, and therefore in this paper we focused on the microbial composition, inflammatory status and expression of Wnt pathway-specific genes in the small intestine as opposed to the large intestine.
Three groups with 2 different genotypes and 2 dietary treatments were used in this study: Apc+/1638NLepr+/+ fed a low fat diet (10% kcal fat, Apc-LF), Apc+/1638NLepr+/+ fed a high fat diet (60% kcal fat, Apc-HF), and Apc+/1638N Leprdb/db fed a low fat diet (Apc-DB). Animals (9-12 mice/group) received diets for 16 weeks from 8 to 24 weeks of age. The compositions of experimental diets were previously described. (20) Mice were housed in a controlled environment (inverted 12-h daylight cycle) with ad libitum access to food and water.
After 16 weeks on diet, mice were sacrificed, the abdomen was opened, and small intestines were removed onto ice-cold glass plates. Samples of the intestinal contents were harvested, frozen in liquid nitrogen, and stored at -80°C for further microbial abundance analysis. After rinsing thoroughly with ice-cold PBS with protease inhibitors (Roche, Indianapolis, IN), the small intestinal tumors were characterized under a dissecting microscope. The remaining normal-appearing small intestinal mucosa was harvested by gentle scraping with microscope slides at 0oC, frozen in liquid nitrogen, and stored at -80 °C for later analysis of inflammatory cytokines and expression of Wnt pathway-specific genes.
Quantification of small intestinal microbiome
The relative abundance of 19 bacterial taxa, which have been reported to be associated with the development of colonic adenomas and cancers in mice and/or humans (23), were determined by real-time PCR. Most of primers used in this study have been previously validated; the remaining primers were designed on the basis of 16S rDNA gene sequences available from the GenBank database and Ribosomal Database Project database (http://rdp.cme.msu.edu/). Primers were synthesized commercially (Invitrogen Life Technologies, Carlsbad, CA), and sequences are listed in Supplementary Data (Table S1).
Bacterial DNA was extracted from small intestinal contents using the QIAamp DNA Stool Mini Kit (Qiagen, Valencia, CA) according to the manufacturer's instructions. The concentration and purity of extracted DNA was determined using the NanoDrop-2000 spectrophotometer (Thermo Scientific, Waltham, MA). Real-time PCR assays were on the ViiA™ 7 Real-Time PCR System (Applied Biosystems, Foster City, CA). PCR conditions were 15 min at 95°C, followed by 40 cycles of 95°C for 1 min, 30s at the annealing temperature, and then 72°C for 1 min.
Inflammatory cytokine assay
The small intestine mucosa scrapings (50~100 mg) were mixed with 400 μL lysis buffer (150 mM NaCl, 20 mM Tris, 1 mM EDTA, 1% Triton X-100, pH 7.5) and phosphatase and protease inhibitors (Sigma, St. Louis, MO) were added to inhibit degradation. Samples were homogenized and then centrifuged at 4℃ at 12000 rpm for 30min, the protein fraction was collected, and the total protein concentrations were measured in the extract using the Bradford assay. Concentrations of IL-1β, TNF-α, IL-6 and IFNγ were determined using the Proinflammatory Panel 1 (mouse) V-PLEXTM Kit and QuickPlex SQ 120 (Mesoscale Discovery, Rockville, MD) according to manufacturer's protocols. Cytokines are expressed as ng/mg protein.
Gene expression profile
The relative expressions were determined for 6 genes involved in bacterial recognition and inflammatory processes, and 16 genes selected from the Wnt pathway cascade including genes of Wnt ligands and Wnt antagonists, receptors and signaling transduction genes, and the downstream target genes. Briefly, total RNA was extracted from the small intestine with Trizol (Invitrogen, Carlsbad, CA), and cDNA was synthesized with SuperScript III (Invitrogen, Carlsbad, CA, USA). Real-time PCR was performed on the ViiA™ 7 Real-Time PCR System (Applied Biosystems, Foster City, CA). Primer sequences were listed in supplementary Data (Table S2).
Data analysis
Data are expressed as means ± SEM. Data analysis was performed using SAS (Version 9.4, SAS Institute, Cary, NC) or GraphPad PrismTM (Version 6, GraphPad Software, La Jolla, CA). Comparisons between groups were made with a T-test or ANOVA and associations were assessed by Pearson's Correlation. Significance was accepted when p < 0.05, and a false discovery rate cutoff of q < 0.2 was used when multiple comparisons were conducted. Heatmaps were created based on transformed z-score values using HemI software (24).
For the bacterial data analysis, the abundance of specific bacterial taxa was normalized to total bacteria (ΔCt = Ctspecific-Cttotal), and for the gene expression data analysis, the expression of each gene was normalized to the housekeeping gene GAPDH (Cttarget gene-CtGAPDH). Statistical analyses were performed based on ΔCt. The relative abundance of specific bacterial taxa or relative gene expression in diet- and genetically-induced obese groups (Apc-HF and Apc-DB, respectively) comparing to the control group (Apc-LF) were reported as 2-ΔΔCt, where ΔΔCt=ΔCtApc-HF-ΔCtApc-LF or ΔΔCt=ΔCtApc-DB-ΔCt Apc-LF.
Results
Physiological characteristics and intestinal tumors
Physiological and tumor data have been previously reported (20). Briefly, the lack of leptin receptor in the genetically-induced obese Leprdb/db mouse model (Apc-DB) resulted in significantly increased body weight in both males (64.8%) and females (113%) when compared to the wildtype control animals (Apc-LF) even though both groups were fed a low fat diet (10% kcal fat) (p < 0.01). The high fat diet (60% kcal fat, Apc-HF) also induced significant increases (p < 0.05) in body weight when compared to the low fat diet group (Apc-LF), although the magnitude in both males (44.3%) and females (29.2%) were lower than genetically-induced obesity. Fat mass was also significantly higher in Apc-DB mice compared to Apc-HF and Apc-LF mice, and Apc-HF mice had significantly greater body fat than Apc-LF mice (20). Tumor incidence, the proportion of animals in each group possessing tumors, was 33%, 67% and 100% in Apc-LF, Apc-HF and Apc-DB mice, respectively (p < 0.01). A similar significant stepwise increase in tumor multiplicity (number of tumors harbored by each animal) and tumor burden (total volume of tumors in each animal) was also observed (20).
Small intestinal microbiome in diet- or genetically-induced obese mice
Among the 19 cancer-related bacterial genera or species studied, the abundance of 4 taxa was altered by diet-induced obesity, while 5 different taxa were altered by genetically-induced obesity (p < 0.05) (Fig. 1). Specifically, the genera Desulfibrio and Turicibacter and species C.leptum were reduced while the species F.prausnitzii was elevated by high fat intake. The genus Bifidobacterium, and the species A. municiphila and E. rectale were decreased while genus Peptostreptococcus and species E.coli were increased by genetically-induced obesity. Very interestingly, none of these taxa were significantly altered by both modalities of obesity (Fig. 2). When a Pearson's correlation analysis was performed between the abundance of identified bacterial taxa and the body weight, statistically significant correlations (p < 0.05) were observed mainly for those bacterial taxa whose abundance was altered by genetically-induced obesity, but not for diet-induced obesity (Table 1).
Table 1
Correlations between the abundance of bacteria and body weight.
| Bacteria | Relative Coefficience (R) | Bacteria | Relative Coefficience (R) |
|---|---|---|---|
| At the genus level | At the species level | ||
| Bifidobacterium | -0.408 * | A.muciniphila | -0.390 * |
| Desulfovibrio | -0.165 | C.leptum | -0.021 |
| Pepstreptococcus | 0.428 * | E.rectale | -0.435 * |
| Turicibacter | 0.104 | E.coli | 0.439 * |
| F.prausnitizii | -0.225 | ||
* Indicates a significant correlation (p < 0.05). Negative coefficient values indicate a reverse correlation, whereas positive coefficient values indicate a positive correlation. The correlation analyses were performed on all experimental animals.
Figure 1
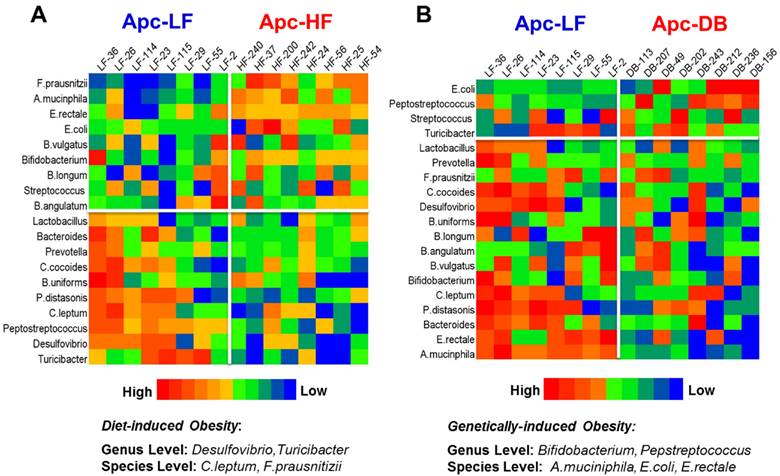
Heatmaps of microbiome in the small intestine of mice. The microbial taxa are ranked based on the relative abundance from the high to the low scale. Microbial taxa with increased abundance appear in the upper boxes (above the white line) and taxa with decreased abundance are in the lower boxes (below the white line). Those taxa whose relative abundance were significantly different in the obese state compared to the lean are listed at the bottom. A) Heatmap for the comparison between the Apc+/1638NLepr+/+ fed a high fat diet (60% kcal fat, Apc-HF) and the Apc+/1638NLepr+/+ fed a low fat diet (10% kcal fat, Apc-LF). B) Heatmap for the comparison between the Apc+/1638NLeprdb/db fed a low fat diet (10% kcal fat, Apc-DB) and the Apc+/1638NLepr+/+ fed a low fat diet (10% kcal fat, Apc-LF).
Figure 2
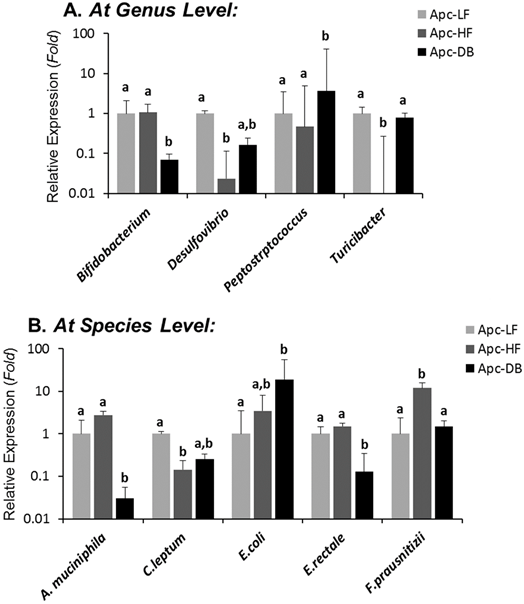
Differential influences of high fat- (60% kcal fat, Apc-HF) and genetically-induced (Apc-DB) obesity on the intestinal cancer-related microbiome compared to the low fat control group (10% kcal fat, Apc-LF). Altered abundances of Turicibacter and F. prausnitzii are present in high fat diet-induced obesity, and altered abundances for Bifidobacterium, Peptostreptococcus, A. mucinphila and E. rectale are present in genetically-induced obesity. A) At the genus level. B) At the species level.
Transcriptional expression of genes involved in bacterial recognition and pro-inflammatory processes
Among the 6 genes involved in bacterial recognition and pro-inflammatory processes, the transcriptional expression of myeloid differentiation factor 88 (MyD88), a cytoplasmic molecule which plays a central role in immune response to many bacterial cell components including lipopolysaccharide, peptidoglycan, and lipopeptide (25, 26), was significantly elevated in both diet-and genetically-induced obesity groups when comparing to the low fat diet control group (p < 0.05). In addition, the expression of NFκB1, the encoded protein of which plays a central role in determining the specificity of NFκB signaling, was significantly higher in the Apc-HF group whereas the expression of Nod2, which encodes a protein that recognizes bacterial peptidoglycans and links recognition to the appropriate immune response, was low in the Apc-DB group compared to the Apc-HF group (Fig. 3A). When a Pearson's correlation analysis was performed for those genes involved in bacterial recognition and pro-inflammatory processes including inflammatory cytokines and microbial taxa, we detected a number of correlations among them, and the correlations for the two genera, Bifidobacterium and Turicibacter, were shown in Table 2.
Table 2
The correlation of transcriptional expression between genes involved in bacterial recognition and pro-inflammatory process with the abundance of microbial taxa.
| Bifidobacterium | Turicibacter | ||
|---|---|---|---|
| Genes involved in bacterial Recognition | Nod2 | r= 0.61 (p = 0.044) | NS |
| MyD-88 | NS | r= -0.56 (p = 0.020) | |
| Genes involved in pro- inflammation | NF-κB1 | NS | r= -0.61 (p = 0.007) |
| TNF-α | r= -0.69 (p = 0.013) | NS | |
Figure 3
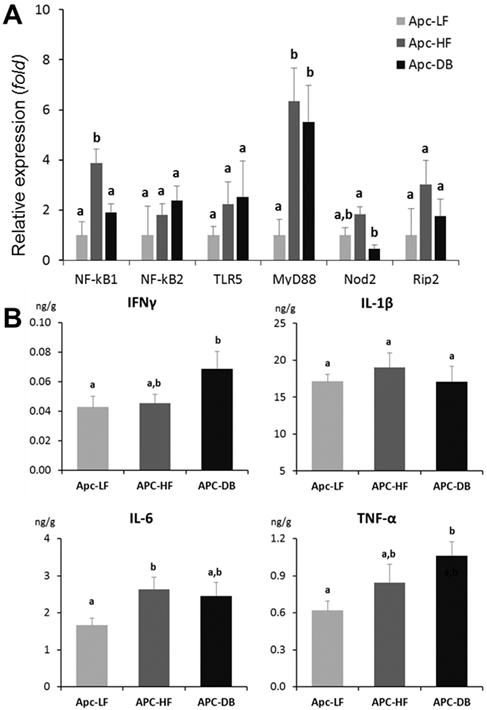
Impact of obesity on inflammation in the small intestine of mice. A) Impact of obesity on the expression of genes involved in bacterial recognition and pro-inflammatory processes in the small intestine of mice. B) Impact of obesity on inflammatory cytokines in the small intestine of mice. Apc-LF: Apc+/1638NLepr+/+ mice fed a low fat diet (10% kcal fat). Apc-HF: Apc+/1638NLepr+/+ mice fed a high fat diet (60% kcal fat) and Apc-DB: Apc+/1638NLeprdb/db mice fed a low fat diet (10% kcal fat).
Intestinal inflammatory cytokine profile
Among the 4 inflammatory cytokines examined (IFNγ IL-1β, IL-6 and TNF-α), IFNγ and TNF-α were significantly increased (72% and 60% respectively, p < 0.05) in the genetically-induced obesity group (Apc-DB) when comparing to the Apc-LF group, whereas IL-6 was significantly increased (58%, p < 0.05) in the diet-induced obesity group (Apc-HF). Based on the inflammatory cytokine profile (Fig. 3B), both diet- and genetically-induced obesity resulted in a low-grade inflammation in the small intestine as indicated by numerical or statistically significant elevattions of inflammatory cytokine protein levels. The magnitudes of cytokine elevation for the genetically-induced obesity group were relatively higher than for the diet-induced group.
Wnt pathway-specific gene expression profile
We assessed the expression of 16 genes selected from Wnt pathway cascade including genes of Wnt ligands and Wnt antagonists, receptors and signaling transduction genes and the downstream target genes (Fig. 4A). Among the 16 genes examined, we observed 7 genes whose expressions were significantly elevated (p < 0.05) by either diet- or genetically-induced obesity or both (Supplementary Materials, Table S2). Since the expression patterns between the diet- and genetically-induced groups are not significantly different when compared to the low fat diet control group, we combined the Apc-HF and Apc-DB groups to generate the heatmap. Among the 7 genes identified, three are the Wnt pathway downstream target genes, three are receptors and signaling transduction genes, and only 1 gene (Wnt5A) is among the Wnt ligands and Wnt antagonists group (Fig. 4B).
Pearson's correlations of transcriptional expression were then performed between MyD88, NFκB1 and Nod2, the 3 genes involved in bacterial recognition and pro-inflammatory processes and, with genes within the Wnt pathway cascade. We observed that multiple Wnt pathway genes correlated with MyD88 and NFκB1 (Table 3), and this was particularly true for downstream target genes and signaling transduction genes. Correlations between MyD88 and NFκB1 with C-Myc and Cyclin D1, the 2 critical Wnt pathway downstream oncogenes are shown in Fig. 4C.
Figure 4
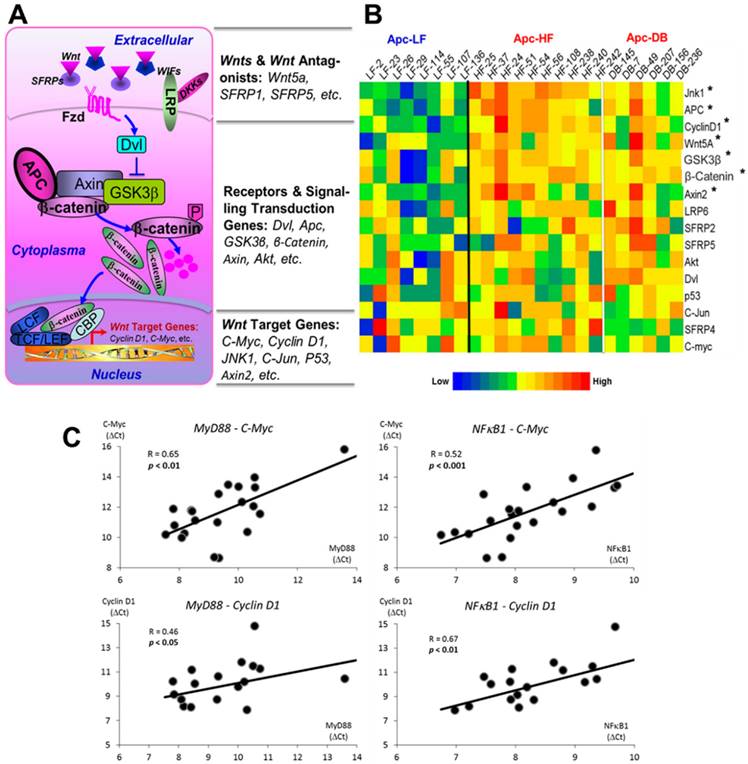
Effects of obesity on Wnt pathway-specific gene expression. A) Wnt-signaling pathway. 16 genes were selected, including Wn ligands and Wnt antagonists, receptors, signaling transduction genes and downstream target genes. B) Heatmap for the relative expression of Wnt pathway-specific genes examined in the small intestine of mice. The genes are ranked based on the relative expression, from the high to the low. C) Correlations between gene expression of MyD88 and NFκB1, genes involved in bacterial recognition and inflammation, with C-Myc and Cyclin D1, downstream targets of the Wnt pathway. The expression patterns between the diet- and genetically-induced groups are not significantly different when comparing to the low fat diet control group. Therefore, the Apc-HF and Apc-DB groups were combined to generate the heatmap. Apc-LF: Apc+/1638NLepr+/+ mice fed a low fat diet (10% kcal fat). Apc-HF: Apc+/1638NLepr+/+ mice fed a high fat diet (60% kcal fat, Apc-HF) and Apc-DB: Apc+/1638NLeprdb/db mice fed a low fat diet (10% kcal fat).
Table 3
Correlations of expression between genes involved in bacterial recognition and inflammation with genes integral to the Wnt pathway cascade.
| Genes involved in bacterial recognition & pro-inflammatory process | Genes within Wnt Pathway Cascade | ||
|---|---|---|---|
| Wnts and Wnt Antagonists | Wnt Receptors and Signaling Transduction Genes | Wnt Target Genes | |
| MyD88 | NS | LRP6*, Apc**, GSK3β**, β-catenin**, Akt** | C-myc**, Cyclin D1*, Jnk1**, C-Jun* |
| NFκB1 | SFRP5* | Dvl*, LRP6**, Apc**, GSK3β**, β-catenin**, Akt** | C-myc**, Cyclin D1**, Axin 2*, Jnk1**, C-Jun** |
| Nod2 | NS | NS | NS |
* p < 0.05; ** p < 0.01; NS: No statistically significant correlations identified.
Discussion
Although obesity is an established risk factor for the development of several malignancies (27, 28), including CRC (29-31), the detailed mechanisms mediating this relationship require further elucidation. We previously demonstrated that obesity elevates Wnt-signaling in the colon (10). By directly comparing two modalities by which obesity is produced the current study expands on prior observations by examining the effects produced by a high fat diet versus those produced by obesity per se (i.e.: genetically-induced obesity). Further, the present study also begins to examine the potential roles that members of the intestinal microbiome might play in this process. Findings from this study indicated that diet- and genetically-induced obesity resulted in distinct bacterial patterns, but both of them leads to an elevated state of intestinal inflammation as indicated by increased inflammatory cytokines, and in parallel with that are the alterations of the Wnt-signaling pathway and increased tumorigenesis in the obese Apc+/1638N model.
The current study demonstrated that several bacteria are differentially altered by diet- and genetically-induced obesity. Of note, the abundance of genus Turicibacter was decreased and species F. prausnitizii increased in the small intestine of the Apc-HF, but not the Apc-DB group (Fig. 2). In line with our observation, Everard et al showed that Turicibacter decreased markedly in mice fed a high fat diet (32). Data from a couple of studies showing a depletion of Turicibacter in animal models of inflammatory bowel disease (33, 34) raises the question whether this taxa might have anti-inflammatory properties. The species F. prausnitizii is generally considered an anti-inflammatory commensal bacterium that can secrete metabolites that block NFκB activation (35). Contrary to our expectations and findings from others (36, 37), an increased abundance of F. prausnitizii was observed in our study. But our data is not unique in this regard: Faval et al (38) reported that a 24-week intervention with a high saturated fat diet significantly increased F. prausnitizii in 88 subjects 'at risk' of metabolic syndrome. This inconsistency underscores the complexity of intestinal microbiota, and suggests that there may be additional as of yet unidentified factors that interact with F. prausnitizii in determining its effects on the intestinal environment.
A. mucinphila, E. rectale and Bifidobacterium decreased whereas Peptostreptococcus increased only in the genetically-induced Apc-DB obese group. The mucin-degrading bacterium, A.muciniphila, and butyrate-producing bacterium, E.rectale, are thought to protect against colorectal cancer and colitis (39, 40). Members of the Bifidobacterium genus are widely used as probiotics, and its abundance has been shown to be significantly reduced in colon cancer patients (41). Peptostreptococcus is a commensal bacterium that can cause a wide variety of infections in multiple sites, including small intestine mucosa. It is also abundant in the gut microbiota of colon cancer patients (40). The changes in these genera and species are therefore all consistent with what one might expect in the Apc-DB group, which displays enhanced tumorigenesis.
Interestingly, when we examined correlations between the altered intestinal microbiota with mouse body weight, the results showed that significant associations were mainly observed for those bacteria altered by genetic-induced obesity (Table 1). This result may simply be due to the fact that a greater magnitude of obesity was achieved with the genetically-induced obesity and therefore correlations might be easier to detect, but might also indicate that the dysbiosis observed in Apc-HF mice might be more determined by aspects of the diet rather than the obese state per se.
Despite the differences in microbial profiles between the two modes of obesity, there was a commonality of inflammation. Among the genes involved in bacterial recognition and inflammation, myeloid differentiation factor 88 (MyD88) protein--a cytoplasmic molecule that plays a central role in the immune response to bacterial cell components, including lipopolysaccharide, peptidoglycan, and lipopeptide (25, 26)--was significantly increased in both diet- and genetically-induced obesity (Fig. 3A). MyD88 has a very complex role in tumorigenesis: its expression has often been observed to have a pro-tumorigenic effect in the liver (42) and colorectum (43), but it also assumes a tumor suppressive role in some inflammation-associated cancer models (44, 45). The findings from these studies reiterate the need for further elucidation of this gene's roles in explaining the inter-relationships between the intestinal microbiome, inflammation and tumorigenesis in the setting of obesity. No significant differences were observed in the patterns of elevated inflammatory cytokines between diet- and genetically-induced obesity. The incremental increase in TNF-α protein concentration across the three groups paralleled the incremental increases in both body weight and tumor incidence (20) (Fig. 3B), underscoring prior observations that have established causative links between obesity, intestinal TNF-α, and tumorigenesis (4, 10).
Similarly, the expression patterns of genes in Wnt pathway were not differently altered between the diet- and genetically-induced obese groups when comparing them to the low fat group (Fig. 4B). When we correlated expressions of those significantly altered genes involved in bacterial recognition and pro-inflammatory processes (MyD88, NFκB1 and Nod2) with expressions of genes within the Wnt pathway, multiple genes were identified to be significantly associated with MyD88 and NFκB1, particularly the downstream target genes and signaling transduction genes (Table 3). Our prior study (10) reported that obesity-promoted inflammation (TNF-α) may phosphorylate GSK3β and thereby activate Wnt-signaling and increase expressions of Wnt pathway downstream genes. Therefore, it is not surprising a group of downstream target genes whose transcriptions were identified to be elevated in this study. Interestingly, elevated transcriptional levels of a number of signaling transduction genes, such as Apc, GSK3β and β-catenin, were observed in this study. Apc, GSKβ, and β-catenin operate as a complex that plays a critical role in the regulation of β-catenin degradation in the Wnt pathway (46). However, evidence is limited and inconsistent regarding how a high fat diet or obesity per se regulates expression of these genes. For instance, a transcriptional upregulation of β-catenin was reported in one study in response to high fat diet (47). whereas another study showed that β-catenin protein, but not the mRNA, is elevated in genetic obesity and reduced by calorie restriction (48). Further investigation into the regulation of this complex is needed. Nevertheless, significant correlations between MyD88 and NFκB1 with the 2 critical Wnt pathway downstream oncogenes, C-Myc and Cyclin D1, inform the potential mechanism that obesity mediates intestinal tumorigenesis via inflammation-driven activation of Wnt-signaling (Fig. 4C).
This study represented a logical extension of our previous publications (10, 20) and added significant insights into the mechanisms mediating obesity-associated intestinal cancer. Using the Apc+/1638N mouse model, this study clearly demonstrated that the inflammation-driven Wnt-signaling serves as a critical mechanism towards enhanced intestinal cancer in the obese state. In terms of the characterization of microbiome, in this work, we particularly focused on the small intestinal microbial constitution, as opposite the colonic microbial composition, since the tumors exclusively occur in the small intestine in this mouse model and the small intestinal microbiome is more reflective of the tumor microenvironment. We demonstrated several bacterial taxa exclusively associated with the two modalities of obesity, but both leads to similar alterations of genes involved in bacterial recognition (MyD88) and inflammatory cytokines (IFNγ, IL-6 and TNF-α) in parallel with alterations of genes that are integral components of the Wnt-signaling cascade in a fashion indicating its activation. In this work, we focused on inflammatory regulation of the critical Wnt pathway, alterations of which appear in up to 90% of CRC (12, 13). However, it is noteworthy that other pathways may also be altered, probably in a less degree, and thereby contribute to the development of CRC in the obese state. For instance, the MAPK pathway is also a critical pathway in tumorigenesis. The MAPK possesses the capability to phosphate C-myc, via which it can interact with the Wnt pathway and regulate cell proliferation and apoptosis. A defect of the MAPK pathway has been demonstrated to be able to promote uncontrolled growth, a necessary step for the development of many types of cancers including CRC. The MAPK pathway also plays a critical role in the regulation of a number of the inflammatory regulators, such as NF-κB and AP-1, and in turn leads to tumorigenesis (49, 50).
In summary, this study has examined how obesity alters the microenvironment of the small intestine of the Apc+/1638N mouse. We have demonstrated that, although high fat diet-induced and genetically-induced obesity alter the intestinal microbiome differently, both lead to similar states of biochemical inflammation and similar alterations in gene expression among members of the Wnt pathway towards enhancing intestinal tumorigenesis. These salient findings not only demonstrate inflammation-driven Wnt-signaling as a novel mechanism mediating obesity-associated intestinal cancer, the information provided by this work also implies that new strategies to suppress inflammation-driven Wnt-signaling can be utilized to attenuate intestinal cancer regardless the two modalities of obesity, but different approaches should be used if targeting to mediate gut microbiome.
Supplementary Material
Supplementary tables.
Acknowledgements
This project is supported by USDA grants (2014-67017-21762 & MAS00454 to ZL), SPHHS Deans Incentive Grant (ZL), and the Agricultural Research Service of the United States Department of Agriculture (Cooperative Agreements #58-1950-0-014 and #58-1950-4-003 to JBM).
Author Contributions
Conception and design of the experiments: ZL S-WC JWC JBM. Conduction of the experiments and acquisition of data: WL JWC ACP LL JL ZL. Analysis and interpretation of the data: ZL WL JWC JBM. Writing of the manuscript: ZL WL JWC JBM.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Kelly T, Yang W, Chen CS, Reynolds K, He J. Global burden of obesity in 2005 and projections to 2030. Int J Obes (Lond). 2008;32:1431-7
2. Finkelstein EA, Khavjou OA, Thompson H. et al. Obesity and severe obesity forecasts through 2030. Am J Prev Med. 2012;42:563-70
3. Calle EE, Kaaks R. Overweight, obesity and cancer: epidemiological evidence and proposed mechanisms. Nat Rev Cancer. 2004;4:579-91
4. Flores MB, Rocha GZ, Damas-Souza DM. et al. Obesity-Induced Increase in Tumor Necrosis Factor-alpha Leads to Development of Colon Cancer in Mice. Gastroenterology. 2012;143:741-53 e4
5. Gravaghi C, Bo J, Laperle KM. et al. Obesity enhances gastrointestinal tumorigenesis in Apc-mutant mice. Int J Obes (Lond). 2008;32:1716-9
6. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87-108
7. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5-29
8. Ouchi N, Parker JL, Lugus JJ, Walsh K. Adipokines in inflammation and metabolic disease. Nat Rev Immunol. 2011;11:85-97
9. Karagiannides I, Pothoulakis C. Obesity, innate immunity and gut inflammation. Curr Opin Gastroenterol. 2007;23:661-6
10. Liu Z, Brooks RS, Ciappio ED. et al. Diet-induced obesity elevates colonic TNF-alpha in mice and is accompanied by an activation of Wnt signaling: a mechanism for obesity-associated colorectal cancer. J Nutr Biochem. 2012;23:1207-13
11. Jain SS, Bird RP. Elevated expression of tumor necrosis factor-alpha signaling molecules in colonic tumors of Zucker obese (fa/fa) rats. Int J Cancer. 2010;127:2042-50
12. Taketo MM. Shutting down Wnt signal-activated cancer. Nat Genet. 2004;36:320-2
13. Network CGA. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330-7
14. Schwabe RF, Jobin C. The microbiome and cancer. Nat Rev Cancer. 2013;13:800-12
15. Turnbaugh PJ, Backhed F, Fulton L, Gordon JI. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe. 2008;3:213-23
16. Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027-31
17. Kamada N, Seo SU, Chen GY, Nunez G. Role of the gut microbiota in immunity and inflammatory disease. Nat Rev Immunol. 2013;13:321-35
18. Liu Z, Ciappio ED, Crott JW. et al. Combined inadequacies of multiple B vitamins amplify colonic Wnt signaling and promote intestinal tumorigenesis in BAT-LacZxApc1638N mice. FASEB J. 2011;25:3136-45
19. Huffman DM, Augenlicht LH, Zhang X. et al. Abdominal obesity, independent from caloric intake, accounts for the development of intestinal tumors in Apc(1638N/+) female mice. Cancer Prev Res (Phila). 2013;6:177-87
20. Pfalzer AC, Nesbeth PD, Parnell LD. et al. Diet- and Genetically-Induced Obesity Differentially Affect the Fecal Microbiome and Metabolome in Apc1638N Mice. PLoS One. 2015;10:e0135758
21. Hummel KP, Dickie MM, Coleman DL. Diabetes, a new mutation in the mouse. Science. 1966;153:1127-8
22. Fodde R, Edelmann W, Yang K. et al. A targeted chain-termination mutation in the mouse Apc gene results in multiple intestinal tumors. Proc Natl Acad Sci U S A. 1994;91:8969-73
23. Borges-Canha M, Portela-Cidade JP, Dinis-Ribeiro M, Leite-Moreira AF, Pimentel-Nunes P. Role of colonic microbiota in colorectal carcinogenesis: A systematic review. Rev Esp Enferm Dig. 2015:107
24. Deng W, Wang Y, Liu Z, Cheng H, Xue Y. HemI: a toolkit for illustrating heatmaps. PLoS One. 2014;9:e111988
25. Villano JS, Rong F, Cooper TK. Bacterial infections in Myd88-deficient mice. Comp Med. 2014;64:110-4
26. Kawai T, Adachi O, Ogawa T, Takeda K, Akira S. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity. 1999;11:115-22
27. Bianchini F, Kaaks R, Vainio H. Overweight, obesity, and cancer risk. Lancet Oncol. 2002;3:565-74
28. Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med. 2003;348:1625-38
29. Murphy TK, Calle EE, Rodriguez C, Kahn HS, Thun MJ. Body mass index and colon cancer mortality in a large prospective study. Am J Epidemiol. 2000;152:847-54
30. Thun MJ, Calle EE, Namboodiri MM. et al. Risk factors for fatal colon cancer in a large prospective study. J Natl Cancer Inst. 1992;84:1491-500
31. Moghaddam AA, Woodward M, Huxley R. Obesity and risk of colorectal cancer: a meta-analysis of 31 studies with 70,000 events. Cancer Epidemiol Biomarkers Prev. 2007;16:2533-47
32. Everard A, Lazarevic V, Gaia N. et al. Microbiome of prebiotic-treated mice reveals novel targets involved in host response during obesity. ISME J. 2014;8:2116-30
33. Jones-Hall YL, Kozik A, Nakatsu C. Ablation of tumor necrosis factor is associated with decreased inflammation and alterations of the microbiota in a mouse model of inflammatory bowel disease. PLoS One. 2015;10:e0119441
34. Rossi G, Pengo G, Caldin M. et al. Comparison of microbiological, histological, and immunomodulatory parameters in response to treatment with either combination therapy with prednisone and metronidazole or probiotic VSL#3 strains in dogs with idiopathic inflammatory bowel disease. PLoS One. 2014;9:e94699
35. Sokol H, Pigneur B, Watterlot L. et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci U S A. 2008;105:16731-6
36. Remely M, Tesar I, Hippe B, Gnauer S, Rust P, Haslberger AG. Gut microbiota composition correlates with changes in body fat content due to weight loss. Benef Microbes. 2015;6:431-9
37. Stenman LK, Burcelin R, Lahtinen S. Establishing a causal link between gut microbes, body weight gain and glucose metabolism in humans - towards treatment with probiotics. Benef Microbes. 2015:1-12
38. Fava F, Gitau R, Griffin BA, Gibson GR, Tuohy KM, Lovegrove JA. The type and quantity of dietary fat and carbohydrate alter faecal microbiome and short-chain fatty acid excretion in a metabolic syndrome 'at-risk' population. Int J Obes (Lond). 2013;37:216-23
39. Everard A, Belzer C, Geurts L. et al. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc Natl Acad Sci U S A. 2013;110:9066-71
40. Wang T, Cai G, Qiu Y. et al. Structural segregation of gut microbiota between colorectal cancer patients and healthy volunteers. ISME J. 2012;6:320-9
41. Singh J, Rivenson A, Tomita M, Shimamura S, Ishibashi N, Reddy BS. Bifidobacterium longum, a lactic acid-producing intestinal bacterium inhibits colon cancer and modulates the intermediate biomarkers of colon carcinogenesis. Carcinogenesis. 1997;18:833-41
42. Naugler WE, Sakurai T, Kim S. et al. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science. 2007;317:121-4
43. Uronis JM, Muhlbauer M, Herfarth HH, Rubinas TC, Jones GS, Jobin C. Modulation of the intestinal microbiota alters colitis-associated colorectal cancer susceptibility. PLoS One. 2009;4:e6026
44. Ochi A, Nguyen AH, Bedrosian AS. et al. MyD88 inhibition amplifies dendritic cell capacity to promote pancreatic carcinogenesis via Th2 cells. J Exp Med. 2012;209:1671-87
45. Salcedo R, Worschech A, Cardone M. et al. MyD88-mediated signaling prevents development of adenocarcinomas of the colon: role of interleukin 18. J Exp Med. 2010;207:1625-36
46. Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006;127:469-80
47. Waller-Evans H, Hue C, Fearnside J. et al. Nutrigenomics of high fat diet induced obesity in mice suggests relationships between susceptibility to fatty liver disease and the proteasome. PLoS One. 2013;8:e82825
48. Mao J, Hu X, Xiao Y. et al. Overnutrition stimulates intestinal epithelium proliferation through beta-catenin signaling in obese mice. Diabetes. 2013;62:3736-46
49. Sun P, Zhou K, Wang S. et al. Involvement of MAPK/NF-kappaB signaling in the activation of the cholinergic anti-inflammatory pathway in experimental colitis by chronic vagus nerve stimulation. PLoS One. 2013;8:e69424
50. Karin M. The regulation of AP-1 activity by mitogen-activated protein kinases. J Biol Chem. 1995;270:16483-6
Author contact
![]() Corresponding author: Zhenhua Liu, Ph.D. Nutrition and Cancer Prevention Laboratory, School of Public Health and Health Sciences, University of Massachusetts, Amherst, 100 Holdsworth Way, Amherst, MA, 01003 Email: zliuumass.edu; Tel: 1-413-545-1075; Fax: 1-413-545-1074.
Corresponding author: Zhenhua Liu, Ph.D. Nutrition and Cancer Prevention Laboratory, School of Public Health and Health Sciences, University of Massachusetts, Amherst, 100 Holdsworth Way, Amherst, MA, 01003 Email: zliuumass.edu; Tel: 1-413-545-1075; Fax: 1-413-545-1074.
Citation styles
APA
Liu, W., Crott, J.W., Lyu, L., Pfalzer, A.C., Li, J., Choi, S.W., Yang, Y., Mason, J.B., Liu, Z. (2016). Diet- and Genetically-induced Obesity Produces Alterations in the Microbiome, Inflammation and Wnt Pathway in the Intestine of Apc+/1638N Mice: Comparisons and Contrasts. Journal of Cancer, 7(13), 1780-1790. https://doi.org/10.7150/jca.15792.
ACS
Liu, W.; Crott, J.W.; Lyu, L.; Pfalzer, A.C.; Li, J.; Choi, S.W.; Yang, Y.; Mason, J.B.; Liu, Z. Diet- and Genetically-induced Obesity Produces Alterations in the Microbiome, Inflammation and Wnt Pathway in the Intestine of Apc+/1638N Mice: Comparisons and Contrasts. J. Cancer 2016, 7 (13), 1780-1790. DOI: 10.7150/jca.15792.
NLM
Liu W, Crott JW, Lyu L, Pfalzer AC, Li J, Choi SW, Yang Y, Mason JB, Liu Z. Diet- and Genetically-induced Obesity Produces Alterations in the Microbiome, Inflammation and Wnt Pathway in the Intestine of Apc+/1638N Mice: Comparisons and Contrasts. J Cancer 2016; 7(13):1780-1790. doi:10.7150/jca.15792. https://www.jcancer.org/v07p1780.htm
CSE
Liu W, Crott JW, Lyu L, Pfalzer AC, Li J, Choi SW, Yang Y, Mason JB, Liu Z. 2016. Diet- and Genetically-induced Obesity Produces Alterations in the Microbiome, Inflammation and Wnt Pathway in the Intestine of Apc+/1638N Mice: Comparisons and Contrasts. J Cancer. 7(13):1780-1790.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.