Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2016; 7(12):1694-1700. doi:10.7150/jca.15086 This issue Cite
Research Paper
Lentivirus-mediated Knockdown of HDAC1 Uncovers Its Role in Esophageal Cancer Metastasis and Chemosensitivity
Min Song1,3, Gang He1, Yan Wang1, Xueli Pang2, Bo Zhang1 ![]()
1. Department of Medical Genetics, Third Military Medical University
2. Department of oncology, Southwest Hospital, Third Military Medical University
3. Department of neurology, the second Affiliated Hospital of Chongqing Medical University
Received 2016-1-25; Accepted 2016-6-7; Published 2016-7-26
Citation:
Song M, He G, Wang Y, Pang X, Zhang B. Lentivirus-mediated Knockdown of HDAC1 Uncovers Its Role in Esophageal Cancer Metastasis and Chemosensitivity. J Cancer 2016; 7(12):1694-1700. doi:10.7150/jca.15086. https://www.jcancer.org/v07p1694.htm
Other stylesAbstract
Histone deacetylationase 1 (HDAC1) is ubiquitously expressed in various cell lines and tissues and play an important role of regulation gene expression. Overexpression of HDAC1 has been observed in various types of cancers, which indicated that it might be a target for cancer therapy. To test HDAC1 inhibition for cancer treatment, the gene expression of HDAC1 was knockdown mediated by a lentivirus system. Our data showed the gene expression of HDAC1 could be efficiently knockdown by RNAi mediated by lentivirus in esophageal carcinoma EC109 cells. Knockdown of HDAC1 led to significant decrease of cell growth and altered cell cycle distribution. The result of transwell assay showed that the numbers of cells travelled through the micropore membrane was significantly decreased as HDAC1 expression was knockdown. Moreover, HDAC1 knockdown inhibited the migration of EC109 cells as determining by scratch test. Additionally, enhancement of cisplatin-stimulated apoptosis was detected by HDAC1 knockdown. Our data suggested inhibition of HDAC1 expression by lentivirus mediated shRNA might be further applied for esophageal cancer chemotherapy.
Keywords: histone deacetylationase, lentivirus, chemosensitivity, EC109 cells
Introduction
In eukaryocyte cells, histone acetylation is governed by histone acetyltransferases (HATs) and histone deacetylationase (HDACs). Under normal conditions, the balance between histone acetylation and deacetylation is well-controlled for cell proliferation and differentiation. However, the balance can be disrupted under pathological conditions, which leads to hyperacetylation of core histones and subsequently altered gene expression. Numerous studies have showed that aberrant histone modifications and the deregulation of gene expression are involved in the process of transformation and tumorigenesis [1-3].
There are 18 characterized members of HDACs in human, which can be grouped into four classes based on biological function and DNA sequence similarity [4]. Among these members, HDAC1 is shown to be ubiquitously expressed in various cell lines and tissues. As a component of the histone deacetylase complex, HDAC1 is believed to regulate most of the observed changes in histone acetylation. For example, HDAC1 and its homologous HDAC2 were found to be associated with Sin3A and Sin3B and several other proteins to form the Sin3 complex, which was thought to deacetylate histones near Sin3 regulated promoter regions leading to a repressed chromatin structure [5]. It also interacts with retinoblastoma tumor-suppressor protein (RB) and this complex is a key element in the control of cell proliferation and differentiation [6]. By using affinity purification and liquid mass spectrophotemetery, HDAC1 interacting partners were identified in HepG2 cells, which provided new insight into mechanism of HDAC1 regulation [7].
One of the most interesting findings about HDAC1 is that it was aberrantly expressed in various types of cancer cells, which suggested that the gene expression of HDAC1 might be associated with tumor progress [8]. For example, a significantly higher level of HDAC1 mRNA was expressed in human urinary bladder cancer specimens [9]. Moreover, high level of HDAC1 expression was associated with clinicopathological factors such as Gleason grade, advanced pathological tumor stage, positive nodal status, elevated preoperative PSA-level, early PSA recurrence and increased cell proliferation. Similar results were found in other cancer types [10-12]. All these data suggested that HDAC1 might be a target for cancer therapy.
In our previous study, we found that inhibition of HDAC1 expression by shRNA enhanced radiosensitivity of EC109 cells as measured by immunofluorescence staining of γH2AX foci and single-cell electrophoresis [13]. Inhibition of HDAC1 expression was performed based on plasmid, which limited its application for gene deliver in vivo. To further test HDAC1 inhibition for cancer treatment, the gene expression of HDAC1 was knockdown mediated by a lentivirus system, which has been intensively applied for gene transferring. It has been reported that HDACis enhanced the chemosensitivity of a wide range of DNA-damaging drugs. Thus, we tested the sensitivity of DNA-damaging drugs as HDAC was knockdown. We found that inhibition of HDAC1 enhanced the chemosensitivity in esophageal cancer cells.
Materials and methods
Cell culture
The human EC109 cell line (esophageal squamous carcinoma) and 293T cell line were obtained from the Shanghai Cell Bank (http://www.ctcccas.ac.cn/xibao). Cells were cultivated in DMEM supplemented with fetal bovine serum (FBS,10%) in a humidified at mosphere containing 5% CO2 in air at 37℃.
Plasmid constructs
A shuttle vector named pGCSIL-GFP was applied for gene transferring in this work. This plasmid contains an expression cassette driven by the human U6 promotor that can produce dsRNA as a stem-loop structure in mammalian cells. Synthesized oligonucleotides (Table 1) were annealed and ligated to the AgeI/EcoRI sites of pGCSIL-GFP to produce pGCSIL-GFP-siHDAC1 or pGCSIL-GFP-siCon. The inserted sequences were subsequently confirmed by sequencing.
Table 1
sequences of shRNA against HDAC or negative control (NC)
| Types | Sequences |
|---|---|
| SiHDAC | aattcaaaaa GAAGTCCGAGGCATCTGGC tctcttgaa GCCAGATGCCTCGGACTTC |
| ccgg GAAGTCCGAGGCATCTGGC ttcaagaga GCCAGATGCCTCGGACTTC tttttg | |
| NC | aattcaaaaa TTCTCCGAACGTGTCACGT tctcttgaa ACGTGACACGTTCGGAGAA |
| ccgg TTCTCCGAACGTGTCACGT ttcaagaga ACGTGACACGTTCGGAGAA tttttg |
Production of lentivirus
The 293T packaging cell line was grown in DMEM supplemented with 10% FBS. Twenty-four hours before transfection, cells in monolayer culture during the logarithmic growth phase were trypsinized and plated in a six-well plate. Cells were transfected with shuttle vectors combined with helper plamids of pMD2.G and psPAX.2 by Lipofectamine 2000 according to the manufacturer's instructions (Invitrogen, USA). Four hour later, the medium containing the transfection mixture was replaced with fresh medium. Transfected cells were incubated for an additional period of 2 days and the supernatant were harvested for cell infection.
Cell infection
On the day before virus infection, EC109 cells were seeded in a six-well plate. On the following day, the cells were incubated with recombinant virus supernatant. Four hour later, the culture media was replaced with fresh medium. Cells were further screened in media containing puromycin (2μg/mL) for 7~10 days. Then individual cell clone with GFP-positive cells was picked up for further analysis.
Realtime RT-PCR
Total RNA was isolated from EC109 cells with Trizol reagent (Invitrogen). An amount of 1.0 µg total RNA was reverse transcribed to cDNA according to the manufacturer's directions (Roche (China) Ltd., Shanghai, China). Quantitative PCR amplification was performed as previously described 13 Primers were list in Table 2. The gene expression was normalized to the corresponding GAPDH level and fold changes was calculated by the 2-△△Ct method according to reference sample and amplification efficiency.
Table 2
sequence of real-time RT-PCR primers
| Gene Name | Sequence | Product (bp) |
|---|---|---|
| HDAC1 | F: TTCAAGCTCCACATCAGTCCTTC | 237 |
| R: CTCTTCCTCACAGGCAATTCGTT | ||
| GAPDH | F: GGGAAGGTGAAGGTCGGAGTC | 232 |
| R: CCTGGAAGATGGTGATGGGAT |
Western blotting
EC109 cells were harvested and homogenized in ice-cold RIPA buffer containing complete protease inhibitors cocktail (Roche). After centrifugation at 13,200 g for 10min, the protein concentration was determined using the Bradford dye-binding assay with bovine serum albumin as the standard. Western blotting was performed as previously described 13. The expression of HDAC1 protein was normalized by that of GAPDH. To validate the reproducibility, the tests were repeated at least 3 times.
Cell cycle and cell growth
For cell cycle, exponentially growing EC109 cells were cultivated and synchronized for 24 h in serum-free medium, and then changed with a complete medium. Then cells were washed with ice-cold PBS, fixed in 70% ethanol and labeled with propidium iodide (PI). Cell cycle distributions were analyzed on a FACSort (Becton Dickinson, San Jose, CA) with Cell Quest software (version 313) for the proportions of cells in G1, S and G2/M phases of the cell cycle. Cell growth was determined by CCK-8 kit (Beyotime Biotechnology Inc., Nantong, China) and the experiments were performed according to its instructions. The plates were read on a Dynatech MR600 microplate reader at 450 nm.
Cell migration assay and invasion assay
For migration assay, a scratch was made in a monolayer of cells using a pipet tip. Fresh media was added immediately to remove the floating cells and the scratch and surrounding cells were recorded immediately after scratching. Images were further captured at the end point from at least ten independent fields to determine the wound closure. Migration was calculated as a percentage of the area covered by the cells compared to the original wound area.
Invasion assay was performed in transwell plates (BD Biosciences) according to manufacturer's protocol. In brief, cells were seeded in duplicate in Matrigel-coated transwell inserts with fetal bovine serum as a chemoattractant in the bottom well. The lower side of the transwell membranes were fixed and stained with 0.05% crystal violet 48 hours after plating. After counting the cells, the invasion of normal cells was considered as 1 and the relative fold change was calculated accordingly.
Treatment of cisplatin and cell apoptosis determination
HDAC1 knockdown EC109 cells (2x102/well) and its control cell (NC) were seeded into 6-well plates and further treated with cisplatin (50μM) for 24h. This concentration was chosen according to other study [14]. Then cell viability was determined by clonogenic assay, normalized by untreated cells. For cell apoptosis, similar treated cells (1x105/well) were measured using the Annexin-V fluorescein isothiocyanate (FITC) Apoptosis Kit (Beyotime) according to the manufacturer's instructions as previously described [13].
Statistics
All data were expressed as mean ± standard deviation. One-way analysis of variance (ANOVA) and Student's t-test or Mann-Whitney test were used to analyze the significance between groups. A p value less than 0.05 was considered statistically significant.
Results
Construct of lentivirus-mediated shRNA expression vector against HDAC1
Lentirivus can integrate virus DNA into host genome, which has been intensively applied for gene transferring. The shuttle vector pGCSIL-GFP, which contains a U6 promoter, was used to express the shRNA as a stem-loop structure (Fig.1A). Synthesized oligonucleotides were annealed and ligated to the shuttle vector by digesting with AgeI and EcoRI. As expected, result of sequencing showed that synthesized oligonucleotide was inserted into the shuttle vector (Data not shown). Then the recombinant vector and the helper plasmids were transfected into 293T cells to product active lentivirus. GFP-positive cells were visible 24h after transfection in 80-90% of the cells, representing the fraction of the population that was successfully transfected (Fig.1B). The cells were further cultivated for 48h to product sufficient active lentivirus and the supernatant was collected for cell infection.
Knockdown of HDAC1 by lentivirus-mediated shRNA
To determine the efficiency of HDAC1 knockdown, EC109 cells were infected with recombinant lentivirus or contol virus, and were further screened in media containing puromycin (2μg/mL). Then individual cell clone with GFP-positive cells was picked up for further analysis of gene expression (Fig.2A). Result of realtime RT-PCR showed HDAC1 mRNA was efficiently knockdown in cell clone Si-HDAC1-2, while none was observed in the other cell clone (Fig.2B). To further confirm the knockdown of HDAC1, the protein level of HDAC1 was detected by Western blotting. As shown in Fig.2C, the protein level of HDAC1 was greatly decreased in the cell clone Si-HDAC1-2, which was coincident with RNA level. Thus, this cell clone was applied in the following experiments.
Figure 1
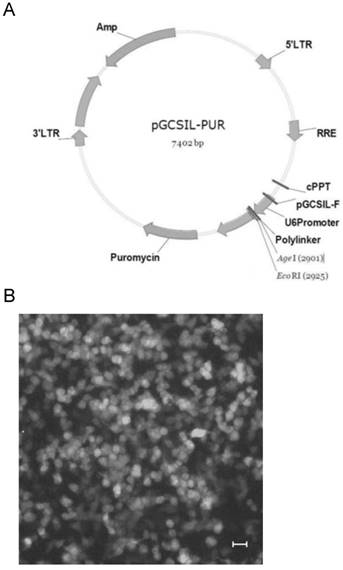
Construct of HDAC1 shRNA expression lentivirus. A: diagram of the shuttle vector pGCSIL-GFP. B: The lentivirus package 293T cells were transfected with recombinant vector and helper plasmids. GFP-positive cells were visible 24h after transfection in the package cells.
Figure 2
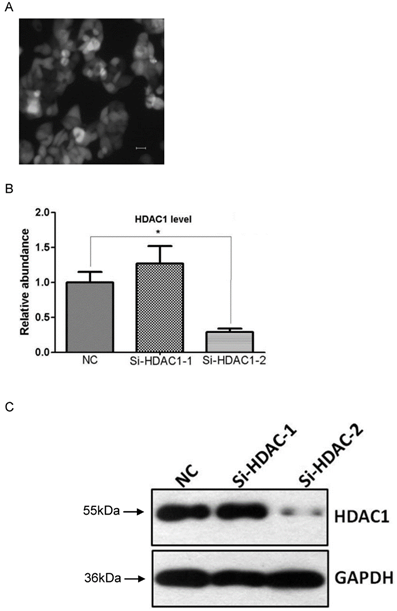
Knockdown of HDAC1 in EC109 cells. A: EC109 cells were infected with the lentivirus and GFP-positive cells were visible after screening with puromycin. B: The mRNA level of HDAC1 was determined by realtime RT-PCR. The gene expression was normalized to the corresponding GAPDH level and fold changes was calculated by the 2-△△Ct method according to reference sample and amplification efficiency. C: The protein level of HDAC1 was detected by Western blotting. Western blotting was repeated at least three times. *P< 0.05 by t-test.
Alteration of cell growth and cell cycle distribution by HDAC knockdown
As HDAC1 could be efficiently knockdown by lentivirus-mediated shRNA in EC109 cell, we further wonder its biological effects on cell growth. Cell line Si-HDAC1-2 and its control NC were seeded into 96-well plate and cell growth was determined by CCK-8. As shown in Fig.3A, cell growth of Si-HDAC1-2 significantly decreased, as compared with that of NC. Further analysis of cell cycle distribution showed that decreased S-phase and increased G2-phase population was observed as HDAC1 was knockdown (Fig.3B). These indicated that knockdown of HDAC1 affected the biological character of EC109 cells.
Inhibition of cell migration and invasion by HDAC1 knockdown
As metastasis is a strong independent prognostic factor for ESCC, we wonder whether HDAC1 knockdown would affect cell migration and invasion. Then transwell assay and scratch test were performed to evaluate its effects. The result of transwell assay showed that the numbers of cells travelled through the micropore membrane was significantly decreased as HDAC1 expression was knockdown (Fig.4A). Moreover, HDAC1 knockdown inhibited the migration of EC109 cells as determining by scratch test (Fig.4B). These data suggested that HDAC1 knockdown inhibited cancer metastasis.
Enhancement of cisplatin-stimulated apoptosis by HDAC knockdown
As acetylation of core histones is governed by opposing actions of a variety of HATs and HDACs, HDAC1 knockdown led to histone hyperacetylation, which was similar to that of HDAC inhibitors (HDACis) such as TSA and SAHA. As cisplatin is one of the most widely used DNA-damaging anticancer drugs, we examined the cytotoxic effect of this drug. As shown in Fig.5A, cell viability after exposure to cisplatin was much lower in HDAC1 knockdown group, compared with that of control. Then, we wonder the cell apoptosis level induced by cisplatin as HDAC was knockdown. The result of Annexin-V staining showed that the apoptotic cells were greatly increased after cisplatin treatment (Fig.5B). Moreover, the protein level of active caspase-3 was measured by western blotting. The apoptosis executer induced by cisplatin was also significantly increased in SiHDAC group, compared with that of NC group (Fig.5C). These data clearly indicated that HDAC1 knockdown enhanced the chemosensitivity of DNA-damaging drugs.
Figure 3
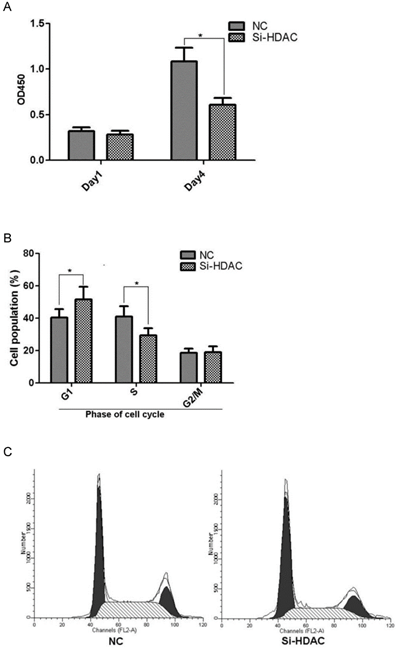
Knockdown of HDAC1 inhibited cell growth and cell cycle distribution. A: HDAC1 knockdown EC109 cells and its control cells (NC) were seeded into 96-well plate and cell growth was determined by CCK-8. B: Cells cycle distribution was performed as described in Material and Methods. Experiments were repeated at least three times. *P< 0.05 by t-test. C: Typical cell distribution detected by FACS.
Figure 4
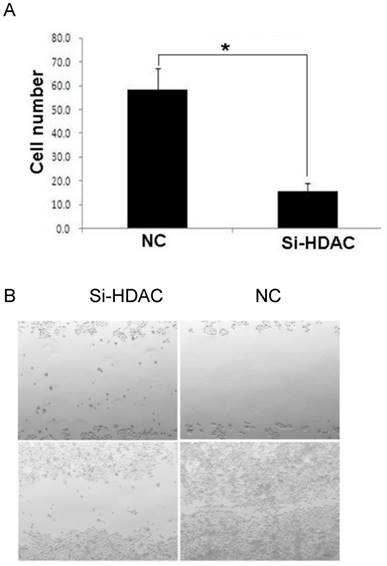
Knockdown of HDAC1 inhibited cell migration and invasion. A: HDAC1 knockdown EC109 cells and its control cells (NC) were plated in Matrigel-coated transwell. The lower side of the transwell membranes were fixed, stained and counted. B: Cell migration of EC109 cells was determined by scratch test. *P< 0.05 by t-test.
Figure 5
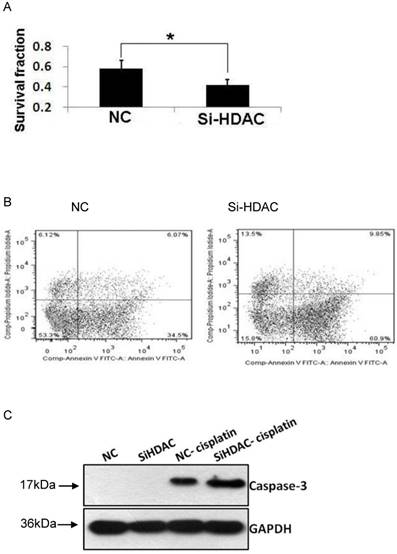
Knockdown of HDAC1 enhanced cisplatin-stimulated apoptosis. A: HDAC1 knockdown EC109 cells and its control cells (NC) were seeded into 96-well plate and further treated with cisplatin for 24h. Cell viability was detected by clonogenic assay as described in Material and Methods. B: Similarly treated cells were harvested for Annexin-V/FITC staining and analyzed by FACS. *P< 0.05 by t-test. C: The protein level of active caspase-3 was detected by Western blotting. Western blotting was repeated at least three times.
Discussion
Many studies have shown that the biochemical function of HDAC1 is to regulate acetylation status of histone and non-histone proteins. As histone acetylation is associated with gene expression, the function of HDAC1 in the regulation of gene transcription has been widely studied [15-17]. However, its physiological significance has not been fully understood. In this study, the gene expression of HDAC1 was efficiently inhibited by lentivirus mediated RNAi technology and its effects on cell biological function have been detected. The most interesting finding of this work is that HDAC knockdown enhanced the cell death induced by cisplatin.
HDAC1 is shown to be ubiquitously expressed in various cell lines and tissues. Recent studies indicated that HDAC1 were associated with DNA damage response, cell cycle control and tumorigenesis [18-21]. In the field of cancer research, many studies showed HDAC1 was over-expressed in various types of cancer cells, which suggested that the gene expression of HDAC1 might be associated with tumor progress. To counteract the excess activity of deacetylation, scientists have developed various types of inhibitors of HDACs, which have been currently investigated for the antitumor activities [22]. The most potential inhibitors are hydroxamic acid (SAHA) and Romidepsin (FK 228), which have been approved for cancer therapy. Although these inhibitors have been shown perfect antitumor effects, it is uncertain which member is responsible for its activity as such small molecule inhibitors have little selectivity for the different subtypes of HDAC family [23]. In our previous work, we konockdown the HDAC1 expression using RNAi strategy mediated by plasmid. The mRNA level of HDAC1 was significantly inhibited, with a decrease of more than 50% [13]. To further analyze the gene function of HDAC1 for cancer therapy, lentivirus was applied for gene transfer. We found that the gene expression was efficiently inhibited in current work. As lentivirus can infect cancer cell in vivo, it can be further applied for animal models. More importantly, lentivirus can lead to integration of virus DNA into host genome. In this study, we picked up GFP-positive cell clones for further study. However, the gene expression of HDAC1 remained unchanged both at transcriptional and translational level in one cell clone, which might be related to the integration site of virus DNA. This might be the shortness of lentivirus system.
Overexpression of HDAC1 was linked to a higher proliferation rate of cancer cells. For example, Burdelski C et al. found high HDAC1 expression was associated with increased cell proliferation of prostate cancer. For molecular mechanism, HDAC1 was shown to repression of the cell cycle inhibitors p21 and p27 at transcriptional level [24]. Consequently, inhibition of HDAC1 could lead to growth arrest and activation of cell cycle inhibitor such as p21 and p27 [25,26]. In this work, we found a significant decrease of cell growth as HDAC1 was knockdown by shRNA. Cell cycle analysis revealed an altered distribution associated with HDAC1 knockdown (Fig.3). This observation was consistent with our previous data and other studies. In line with the altered cell growth, cancer metastasis of EC109 cells was also significantly decreased as HDAC1 expression was knockdown, as determined by cell migration and invasion assay.
As histone hyperacetylation induced by HDAC inhibition can lead to complicated changes in chromatin structure, the changes may expose sections of DNA that are normally protected in tightly packed chromatin. Numerous studies have examined the effects of HDACi in combination therapy with other agents such as DNA-damaging drugs and and EFGR inhibitor [27,28]. Inhibition of HDACs activity contributes to the enhancement of chemosensitivity. For examples, Ozaki K et al. found that TSA and FK228 potentiated induction of apoptosis by cisplatin [29]. Moreover, enhanced radiosensitivity was also observed as combined HDACi with radiation. We observed decreased cell viability and increased cell apoptosis induced by cisplatin as HDAC1 was knockdown (Fig.5), which was inconsistent with other studies [30]. However, there are many limitations of the study. Most importantly, in vivo study was needed to further confirm the gene functions of HDAC1. Also, to unambiguously confirm the gene functions, rescue experiments in which forced gene expression of HDAC1 in lentivirus infected cells are also needed.
In summary, the gene expression of HDAC1 was efficiently inhibited by lentivirus mediated RNAi technology. Knockdown of HDAC1 affected the biological character of EC109 cells including cell growth, cell cycle distribution. It also inhibited the cell migration and invasion, as well as chemosensitivity. Our data helps to understand the role of HDAC1 for esophageal cancer therapy.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (No. 81272497 and 81000147).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Wagner T, Brand P, Heinzel T, Krämer OH. Histone deacetylase 2 controls p53 and is a critical factor in tumorigenesis. Biochim Biophys Acta. 2014;1846(2):524-38
2. Li Z, Zhu WG. Targeting histone deacetylases for cancer therapy: from molecular mechanisms to clinical implications. Int J Biol Sci. 2014;10(7):757-70
3. Seto E, Yoshida M. Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harb Perspect Biol. 2014;6(4):a018713
4. Højfeldt JW, Agger K, Helin K. Histone lysine demethylases as targets for anticancer therapy. Nat Rev Drug Discov. 2013;12(12):917-30
5. Yang L, Mei Q, Zielinska-Kwiatkowska A, Matsui Y, Blackburn ML, Benedetti D. et al. An ERG (ets-related gene)-associated histone methyltransferase interacts with histone deacetylases 1/2 and transcription co-repressors mSin3A/B. Biochem J. 2003;369(Pt 3):651-7
6. Jeon HS, Choi YY, Fukuoka J, Fujii M, Lyakh LA, Song SH. et al. High expression of SNIP1 correlates with poor prognosis in non-small cell lung cancer and SNIP1 interferes with the recruitment of HDAC1 to RB in vitro. Lung Cancer. 2013;82(1):24-30
7. Farooq M, Hozzein WN, Elsayed EA, Taha NA, Wadaan MA. Identification of histone deacetylase 1 protein complexes in liver cancer cells. Asian Pac J Cancer Prev. 2013;14(2):915-21
8. Willis-Martinez D, Richards HW, Timchenko NA, Medrano EE. Role of HDAC1 in senescence, aging, and cancer. Exp Gerontol. 2010;45(4):279-85
9. Ozawa A, Tanji N, Kikugawa T, Sasaki T, Yanagihara Y, Miura N. et al. Inhibition of bladder tumour growth by histone deacetylase inhibitor. BJU Int. 2010;105(8):1181-6
10. Zhang Z, Yamashita H, Toyama T, Sugiura H, Ando Y, Mita K. et al. Quantitation of HDAC1 mRNA expression in invasive carcinoma of the breast. Breast Cancer Res Treat. 2005;94(1):11-6
11. Weichert W, Röske A, Gekeler V, Beckers T, Ebert MP, Pross M. et al. Association of patterns of class I histone deacetylase expression with patient prognosis in gastric cancer: a retrospective analysis. Lancet Oncol. 2008;9(2):139-48
12. Halkidou K, Gaughan L, Cook S, Leung HY, Neal DE, Robson CN. Upregulation and nuclear recruitment of HDAC1 in hormone refractory prostate cancer. Prostate. 2004;59(2):177-89
13. Zhang B, Wang Y, Pang X. Enhanced radiosensitivity of EC109 cells by inhibition of HDAC1 expression. Med Oncol. 2012;29(1):340-8
14. Flores-Pérez A, Rafaelli LE, Ramírez-Torres N, Aréchaga-Ocampo E, Frías S, Sánchez S. et al. RAD50 targeting impairs DNA damage response and sensitizes human breast cancer cells to cisplatin therapy. Cancer Biol Ther. 2014;15(6):777-88
15. Segré CV, Chiocca S. Regulating the regulators: the post-translational code of class I HDAC1 and HDAC2. J Biomed Biotechnol. 2011;2011:690848
16. Brunmeir R, Lagger S, Seiser C. Histone deacetylase HDAC1/HDAC2-controlled embryonic development and cell differentiation. Int J Dev Biol. 2009;53(2-3):275-89
17. Reichert N, Choukrallah MA, Matthias P. Multiple roles of class I HDACs in proliferation, differentiation, and development. Cell Mol Life Sci. 2012;69(13):2173-87
18. Robert T, Vanoli F, Chiolo I, Shubassi G, Bernstein KA, Rothstein R. et al. HDACs link the DNA damage response, processing of double-strand breaks and autophagy. Nature. 2011;471(7336):74-9
19. Di Micco R, Sulli G, Dobreva M, Liontos M, Botrugno OA, Gargiulo G. et al. Interplay between oncogene-induced DNA damage response and heterochromatin in senescence and cancer. Nat Cell Biol. 2011Mar;13(3):292-302
20. Santoro F, Botrugno OA, Dal Zuffo R, Pallavicini I, Matthews GM, Cluse L. et al. A dual role for Hdac1: oncosuppressor in tumorigenesis, oncogene in tumor maintenance. Blood. 2013;121(17):3459-68
21. Heideman MR, Wilting RH, Yanover E, Velds A, de Jong J, Kerkhoven RM. et al. Dosage-dependent tumor suppression by histone deacetylases 1 and 2 through regulation of c-Myc collaborating genes and p53 function. Blood. 2013;121(11):2038-50
22. Carafa V, Nebbioso A, Altucci L. Histone deacetylase inhibitors: recent insights from basic to clinical knowledge & patenting of anti-cancer actions. Recent Pat Anticancer Drug Discov. 2011;6(1):131-45
23. Furumai R, Komatsu Y, Nishino N, Khochbin S, Yoshida M, Horinouchi S. Potent histone deacetylase inhibitors built from trichostatin A and cyclic tetrapeptide antibiotics including trapoxin. Proc Natl Acad Sci USA. 2001;98(1):87-92
24. Lagger G, O'Carroll D, Rembold M, Khier H, Tischler J, Weitzer G. et al. Essential function of histone deacetylase 1 in proliferation control and CDK inhibitor repression. EMBO J. 2002;21(11):2672-81
25. Sachweh MC, Drummond CJ, Higgins M, Campbell J, Laín S. Incompatible effects of p53 and HDAC inhibition on p21 expression and cell cycle progression. Cell Death Dis. 2013;4:e533
26. Wu X, Yang N, Zhou WH, Xu J, Chen JJ, Zheng FM. et al. Up-regulation of P21 inhibits TRAIL-mediated extrinsic apoptosis, contributing resistance to SAHA in acute myeloid leukemia cells. Cell Physiol Biochem. 2014;34(2):506-18
27. Liffers K, Kolbe K, Westphal M, Lamszus K, Schulte A. Histone Deacetylase Inhibitors Resensitize EGFR/EGFRvIII-Overexpressing, Erlotinib-Resistant Glioblastoma Cells to Tyrosine Kinase Inhibition. Target Oncol. 2016;11(1):29-40
28. Raha P, Thomas S, Thurn KT, Park J, Munster PN. Combined histone deacetylase inhibition and tamoxifen induces apoptosis in tamoxifen-resistant breast cancer models, by reversing Bcl-2 overexpression. Breast Cancer Res. 2015;17:26
29. Ozaki K, Kishikawa F, Tanaka M, Sakamoto T, Tanimura S, Kohno M. Histone deacetylase inhibitors enhance the chemosensitivity of tumor cells with cross-resistance to a wide range of DNA-damaging drugs. Cancer Sci. 2008;99(2):376-84
30. Cacan E, Ali MW, Boyd NH, Hooks SB, Greer SF. Inhibition of HDAC1 and DNMT1 modulate RGS10 expression and decrease ovarian cancer chemoresistance. PLoS One. 2014;9(1):e87455
Author contact
![]() Corresponding author: Dr. Bo Zhang, Department of Medical Genetics, Third Military Medical University, Chongqing 400038, China. FAX: 86-23-65318610, TEL: 86-23-68753670, e-mail: bo_zhangcom; Co-corresponding author: Dr. Xueli Pang, 1303062457com
Corresponding author: Dr. Bo Zhang, Department of Medical Genetics, Third Military Medical University, Chongqing 400038, China. FAX: 86-23-65318610, TEL: 86-23-68753670, e-mail: bo_zhangcom; Co-corresponding author: Dr. Xueli Pang, 1303062457com
Citation styles
APA
Song, M., He, G., Wang, Y., Pang, X., Zhang, B. (2016). Lentivirus-mediated Knockdown of HDAC1 Uncovers Its Role in Esophageal Cancer Metastasis and Chemosensitivity. Journal of Cancer, 7(12), 1694-1700. https://doi.org/10.7150/jca.15086.
ACS
Song, M.; He, G.; Wang, Y.; Pang, X.; Zhang, B. Lentivirus-mediated Knockdown of HDAC1 Uncovers Its Role in Esophageal Cancer Metastasis and Chemosensitivity. J. Cancer 2016, 7 (12), 1694-1700. DOI: 10.7150/jca.15086.
NLM
Song M, He G, Wang Y, Pang X, Zhang B. Lentivirus-mediated Knockdown of HDAC1 Uncovers Its Role in Esophageal Cancer Metastasis and Chemosensitivity. J Cancer 2016; 7(12):1694-1700. doi:10.7150/jca.15086. https://www.jcancer.org/v07p1694.htm
CSE
Song M, He G, Wang Y, Pang X, Zhang B. 2016. Lentivirus-mediated Knockdown of HDAC1 Uncovers Its Role in Esophageal Cancer Metastasis and Chemosensitivity. J Cancer. 7(12):1694-1700.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.