Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2016; 7(12):1668-1679. doi:10.7150/jca.15423 This issue Cite
Research Paper
An Integrated Analysis of the Genome-Wide Profiles of DNA Methylation and mRNA Expression Defining the Side Population of a Human Malignant Mesothelioma Cell Line
Myung-Chul Kim1,2, Na-Yon Kim1,2, Yu-Ri Seo1, Yongbaek Kim1,3 ![]()
1. Laboratory of Veterinary Clinical Pathology, College of Veterinary Medicine, Seoul National University, Seoul, The Republic of Korea.
2. BK21 PLUS Program for Creative Veterinary Science Research, College of Veterinary Medicine, Seoul National University, Seoul, The Republic of Korea.
3. Research Institute for Veterinary Science, College of Veterinary Medicine, Seoul National University, Seoul, The Republic of Korea.
Received 2016-3-2; Accepted 2016-5-18; Published 2016-7-26
Abstract
Intratumoral heterogeneity is a hallmark of all cancers and functions as the major barrier against effective cancer therapy. In contrast to genetic mutations, the role of epigenetic modifications in the generation and maintenance of heterogeneous cancer cells remains largely undetermined. This study was performed to evaluate the epigenetic mechanisms involved in the tumor cell heterogeneity using side population (SP) and non-SP cells isolated from a human malignant mesothelioma (HMM) cell line. The subpopulations of cancer cells were analyzed by methylated DNA immunoprecipitation combined with high-throughput sequencing (MeDIP-seq) and RNA-seq methodology. The RNA-seq data were analyzed with the MeDIP-seq data in an integrated way to identify the epigenetically modified genes that defined the SP. Concomitant changes in mRNA expression and DNA methylation were found in 122 genes, including 118 down-regulated genes with hypermethylation and 4 up-regulated genes with hypomethylation. Gene ontology revealed that a large portion of the genes belonged to the groups of biological processes such as stem cell maintenance, stem cell development, stem cell differentiation, and the negative regulation of the developmental process. Among these genes, BNC1, RPS6KA3, TWSG1 and DUSP15 contained aberrant methylation in the CpG islands of the promoter region, indicating that the genes regulated by DNA methylation characterized a distinct subpopulation of HMM cells. The present study provided valuable information to shed light on the epigenetic contributions to the generation and maintenance of tumor cell heterogeneity.
Keywords: mesothelioma, side population, epigenetics, MeDIP-seq, RNA-seq, DNA methylation
Introduction
Intratumoral heterogeneity refers to a mixture of phenotypically, functionally, and genetically different cancer cells with various cellular hierarchies or differentiation statuses within a tumor [1]. It is a common feature of almost all cancer types [1]. Tumor cell heterogeneity has been considered to be the major obstacle for effective cancer therapy, as heterogeneous cancer cells differ in their sensitivity to cancer therapy [2]. To date, studies have focused on genetic alterations as an underlying mechanism for the generation and maintenance of the tumor cell heterogeneity [3]. However, an increasing body of evidence supports the role of epigenetic modifications in the emergence of tumor heterogeneity. The formation of considerably heterogenetic spheres in vitro from a single cell, altered gene expression without genetic changes in many cancers, and the reversible changes in acquired drug resistance support epigenetic involvement in tumor heterogeneity [4, 5]. Nevertheless, limited information is available about the function of epigenetic modifications in intratumoral heterogeneity.
Epigenetic modifications are heritable and stable alterations of genes that do not change the DNA sequence and include DNA methylation, histone modification, and non-coding RNA interference [6]. DNA methylation has been extensively investigated regarding cancer development and progression [7]. While hypermethylation in the promoter of the cancer-related genes induces the silencing or down-regulation of tumor suppressor genes or DNA repair genes, global DNA hypomethylation drives oncogene activation and genomic instability [8]. Emerging evidence has suggested that aberrant DNA methylation may play a critical role in the generation of cancer cell heterogeneity [4]. Recent studies illustrated that epigenetic alterations, including aberrant DNA methylation, promote the heterogeneity of cancer stem cells (CSCs), preceding and/or predisposing to genetic changes [5, 9, 10].
Methylated DNA immunoprecipitation-based high throughput sequencing technology (MeDIP-seq) is the next generation sequencing (NGS) approach to analyze the DNA methylome [11]. MeDIP-seq is a useful tool for the rapid and efficient genome-wide assessment of DNA methylation in cancer epigenetics [11, 12]. For transcriptomic profiling, RNA-seq has been increasingly adopted to investigate the transcription profile in cells, tissues, and organisms because of its higher resolution relative to microarray-based methodology [13]. An integrated analysis of gene expression data with epigenetic profiling could be an effective approach to uncover the role of epigenetic modifications in cancer development and progression.
Human malignant mesothelioma (HMM) is an invariably fatal tumor arising from serosal surfaces of body cavities [14]. Many factors, including asbestos fibers and simian virus 40, are known to be closely associated with HMM tumorigenesis [15]. The annual incidence of HMM is relatively low, estimated to range from 0.6 to 30/1,000,000, but the global occurrence is expected to increase continuously in the coming decades [16]. HMM is extremely heterogeneous in its morphology and molecular phenotypes [17]. Using side population (SP) assays based on fluorescence-activated cell sorting (FACS), it has been shown that the SP fraction is enriched for more aggressive cells in HMM cell lines [18]. The prognosis of HMM is generally poor, with a median survival of 12 months from the diagnosis [19]. Despite progress in the understanding of its molecular carcinogenesis, the mechanisms underlying the resistance of HMMs to anticancer therapy remain unclear.
The present study was carried out to elucidate the molecular mechanism involved in the tumor cell heterogeneity using genome-wide analysis of SP and NSP cells isolated from a HMM cell line. Integrated analysis of the data from the MeDIP-seq and RNA-seq revealed that epigenetically regulated genes defined cancer cell subpopulations and their biological characteristics. To the best of our knowledge, this is the first genome-wide analysis of DNA methylation and transcriptomes in HMM cell subpopulations that sought to elucidate the molecular mechanisms underlying the development of intratumoral heterogeneity.
Materials and methods
Cell line and culture
A HMM cell line, MS-1, was kindly provided by Dr. Jablons (University of California San Francisco, USA). The cell line was cultured in RPMI 1640 medium (Mediatech Inc., Manassas, VA, USA) supplemented with 10% fetal bovine serum (FBS; Mediatech Inc.), 10 mM HEPES (Sigma-Aldrich, St. Louis, MO, USA), 1.5 g/L sodium bicarbonate (Sigma-Aldrich), 1 mM sodium pyruvate (Sigma-Aldrich), and 100 U/100 μg/mL penicillin/streptomycin (Gibco-Life Technology, Gaithersburg, MD, USA) at 37◦C in a humidified atmosphere containing 5% CO2.
SP analysis
Cancer cell subpopulations were analyzed and isolated using an SP assay composed of Hoechst 33342 dye staining and subsequent flow cytometric analysis as described by Kai et al. [18]. Briefly, cultured cells were washed with phosphate-buffered saline (PBS), trypsinized and resuspended at 106 cells/mL in pre-warmed RPMI containing 2% FBS and 10 mM HEPES. The cells were incubated with Hoechst 33342 dye (5 μg/mL final concentration; Sigma-Aldrich, St. Louis, MO, USA) for 90 min at 37◦C with intermittent mixing. At the end of incubation, cells were spun down at 480 × g for 5 min and washed in cold PBS containing 2% FBS at 4◦C. Hoechst 33342 staining was detected using a flow cytometer sorter (Becton-Dickinson FACS Aria III, Becton-Dickinson, San Jose, CA, USA), exciting at 355 nm and detecting Hoechst Blue with a 450/50 broad pass filter and Hoechst Red with a 675/30 broad pass filter. At least 50,000 events within the live gate were examined to define an SP region, and an SP gate was identified using 50 μM verapamil hydrochloride, which blocks Hoechst 33342 dye efflux. SP and NSP cells were collected by a flow cytometry assisted cell sorter (FACS) based on the intensities of fluorescence and subjected to further analyses.
DNA preparation, library construction and MeDIP-seq
The DNA library was constructed as described [20]. Briefly, genomic DNAs were extracted from the sorted SP and NSP cells by an Exgene Genomic DNA Micro Kit (GeneAll Biotechnology Co. Ltd., Seoul, The Republic of Korea) according to the manufacturer's recommendations. The concentration and purity of the DNA were measured using Nanodrop (Nanodrop Technologies, Wilmington, DE, USA). The high quality of total DNA was subjected to MeDIP-seq analysis performed at Theragen Bio Institute (Suwon-city, Gyeonggi-do, The Republic of Korea). The qualified library was directly sequenced using an Illumina HiSeq 2000 sequencer (Illumina Inc, San Diego, CA, USA) according to the manufacturer's protocol.
Identification of differential DNA methylation regions (DMRs)
Detailed analysis of the MeDIP-seq data was performed as described [20]. Briefly, the high quality of clean reads was mapped to a human reference genome (hg19) using an alignment program, the Short Oligonucleotide Analysis Package software (SOAP, http://soap.genomics.org.cn). Whole genome peak scanning was performed using the Model-based Analysis of ChIP-seq (MACS: Software version-1.4.0). Dynamic Poisson distribution was used to calculate the p-value of a specific region based on the number of unique mapped reads. The region with a p-value of less than 10e-5 was defined as a peak. The distribution of peaks was analyzed in the upstream 2k, 5' UTR, exon, intron, 3' UTR, downstream 2k, and repeat element regions and in each class of repetitive elements. CpG islands and the gene body were divided into 40 equal regions. The upstream and downstream 2 kb regions were divided into 20 equal regions. For each region, the normalized number of reads was calculated. Peaks of MeDIP-seq data from SP and NSP cells were merged to find candidate DMRs. Then, numbers of the reads were assessed by chi-square statistics and false discovery rate statistics to obtain true DMRs. The p-value of the filtering standard was 0.05, and a 2-fold change in the difference of read numbers was used as a criterion for the DMRs. The methylation status of the NSP sample was utilized as a reference for the up- or down-regulation of DNA methylation in the SP fraction.
MeDIP-seq data validation by bisulfite sequencing
To verify differentially methylated genes between SP and NSP cells, 500 ng of genomic DNA from each SP and NSP sample was treated with sodium bisulfite using the EZ DNA Methylation-Gold kit (Zymo Research, Orange, CA, USA). Methylated and unmethylated primer sets were designed in MethyPrimer-Design (http://www.urogene.org/methprimer). Primer sequences for the genes selected for validation are documented in Table S3.
RNA isolation and RNA-seq
The RNA library was constructed as described [21]. Briefly, total RNAs from the sorted SP and NSP cells were isolated using a phenol-chloroform method with Trizol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. The quality and quantity of the total RNAs were determined using Nanodrop (Nanodrop Technologies). Total RNAs with high quality were subjected to NGS assay performed at the DNA Link Incorporation (Songpa-gu, Seoul, The Republic of Korea). Sequencing libraries of mRNAs were prepared using an Illumina TruSeq RNA Prep kit v2 (Illumina Inc.) according to the manufacturer's instructions. The quality of the amplified libraries was verified using an Agilent Technologies 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA). Cluster generation was carried out in the flow cells on the cBot automated cluster generation system (Illumina Inc.), and then the flow cells were loaded on a HiSeq 2000 sequencing system (Illumina Inc.) with 200 bps paired-end reads.
Identification of differentially expressed genes (DEGs)
Detailed analysis of the RNA-seq data was performed as described [21]. Briefly, the high quality of clean reads was mapped to the hg19 with TopHat (ver. 2.0.9). The Bam file was used as the output to store a list of read alignments and was added to the Cufflinks software package (ver. 2.0.2) to predict transcript structures and compare transcriptome profiles based on the RNA-Seq data [22]. To compare the expression level of a gene across samples, read counts obtained from RNA-seq were normalized as fragments per kilobase of transcript per million mapped fragments (FPKM) [23]. The FPKM was used to identify DEGs in SP and NSP subpopulations, and then the FPKM in each sample was compared and transformed to the Log2 ratio (log2(number of SP reads) - log2(number of NSP reads)). The gene expression of the NSP subpopulation was used as control data for the up- or down-regulation of gene expression in SP cells. Genes with a p-value of < 0.05 and a log2-transformed value smaller than -1 or greater than 1 were considered to be statistically significant DEGs.
Validation of RNA-seq data by quantitative real-time RT-PCR
The total RNA 400 ng was used to synthesize cDNA using QuantiTect Reverse Transcription Kit (Qiagen, Valencia, CA, USA). Quantitative real-time PCR was performed using the Rotor-Gene SYBR Green RT-PCR Kit (Qiagen). The PCR conditions were as follows: 95◦C for 5 min followed by 45 cycles of 95◦C for 10 seconds and ending at 60◦C for 30 seconds. Expression levels of each target gene were normalized to the endogenous GAPDH level. The relative gene expression was analyzed as described [24]. Primer sequences for the target genes selected for validation are documented in Table S4.
Integrated analysis of DMGs and DEGs
MeDIP-seq and RNA-seq data were interpreted in an integrated way to identify epigenetically regulated genes that define SP subpopulations. DEGs that retain DMRs in the regulatory areas were selected as candidate genes. A fold change > 2 and p-value < 0.05 were used as filtering criteria. DEGs containing both differentially hypermethylated and hypomethylated genomic regions were excluded from the candidates. To investigate whether the epigenetically regulated DEGs have CpG islands, the genome browser embedded in the UCSC Genome Bioinformatics software (Santa Cruz, CA, USA; http://genome.ucsc.edu/cgi-bin/hgGateway) was performed using a WIG file of MeDIP-seq signals. All analyses were based on hg18.
Gene ontology (GO) analysis
To determine the key regulatory components and functional relationships of genes, gene products or gene-product groups from the high-throughput sequencing data, GO analysis was performed [25]. The web-based software toolkit for gene ontology enrichment analysis (GOEAST, http://omicslab.genetics.ac.cn/GOEAST) was used to visualize the biological process, molecular function, and cellular component terms for the target genes. Using the MeDIP-integrated RNA-seq data, GO enrichment revealed the biological implications of the list of genes showing epigenetically concomitant changes in aberrant DNA methylation and gene expression. GO terms with a fold change > 2 and p-value of less than 0.05 were considered functionally relevant.
Results
Isolation of DNA and RNA from the subpopulations of HMM cells
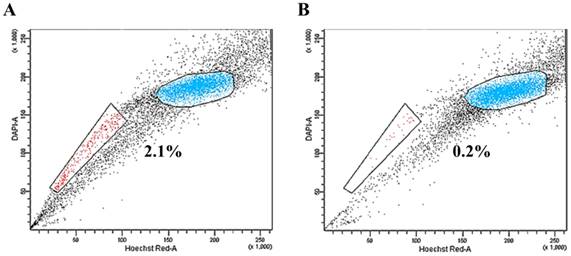
MS1 cells were subjected to SP analysis, which consisted of Hoechst 33342 dye staining and subsequent flow cytometry. A small subset of SP cells, ranging from 0.2% to 3.2% of the total cells, was identified in the MS1 cells as a distinct tail in the flow cytometry plot (Figure 1A). The SP fraction was decreased by treatment with 50 μM verapamil hydrochloride (Figure 1B). The quality of RNA samples from the sorted SP and NSP cells of MS1 cells was assessed by gel electrophoresis and absorbance at a A260/280 ratio by Agilent's 2100 Bioanalyzer and Nanodrop. RNAs with a A260/280 ratio greater than 1.8 and an RNA Integrity Number (RIN) value greater than 8 were subjected to the further analysis.
Identification of side population (SP) phenotypes in the MS1 cell line. (A) Side population assay revealed that the MS1 cell line contained a distinct region of SP cells indicated by a trapezoid on each panel. (B) Note the reduced fraction of SP cells with treatment with verapamil.
Global mapping of DNA methylation
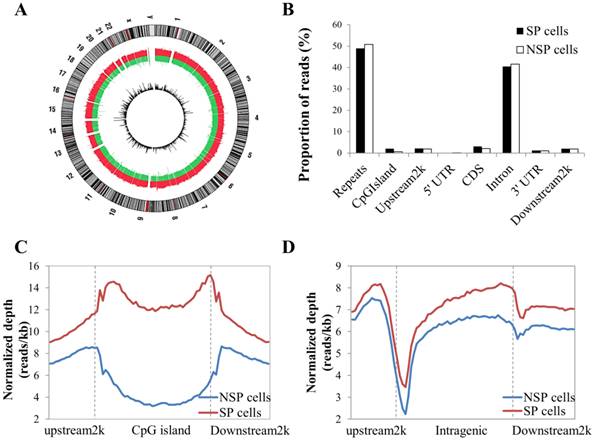
After data cleaning as described in the Materials and Methods section, a total of 48,979,592 reads were obtained by MeDIP-seq analysis of the SP and NSP cells. Approximately 92% of the total reads from each sample were aligned to the reference genome, and consequently, 68.26% and 71.90% of the reads from the SP and NSP samples were mapped to the human genome, respectively (Table S1). The MeDIP-seq reads were distributed across most human chromosomes with variable densities (Figure 2A). Most reads in both samples were concentrated in the repeat and intron regions, and a relatively small portion of reads was allocated to the other genomic elements (Figure 2B). The distribution of MeDIP-seq reads around CpG islands was further investigated because aberrant DNA methylation in CpG islands has been known to induce changes in gene expression more than other genomic regions. The levels of DNA methylation in the CpG islands and in the upstream 2 kb and downstream 2 kb regions of the CpG islands were significantly higher in SP cells than in NSP cells. The DNA methylation level of SP cells was progressively increased in the upstream 2k regions, markedly decreased at the beginning of the CpG islands, and dramatically increased at the end of the CpG islands (Figure 2C). On the other hand, the methylation level around the gene body was generally higher in SP cells than in NSP cells (Figure 2D).
Distribution of MeDIP-seq reads with the DNA methylation pattern in CpG islands and the coding gene region. (A) Circle plot showing chromosomal locations of Medip-seq reads obtained by NGS for SP and NSP cells. The outside circle depicts the chromosome ideogram in a clockwise rotation. The next track contains red and green colors, indicating genome-wide methylated regions in SP cells and NSP cells, respectively. The height of the histogram bins indicates the density of methylated regions. The innermost circle includes the differentially methylated regions indicated by log2 values between MeDIP-seq reads of SP cells. (B) Proportion of MeDIP-seq reads in SP and NSP cells. The x-axis indicates different genome regions. For each region, the normalized number of reads was calculated. The y-axis indicates the proportion of reads in a specific gene element. (C) The DNA methylation level of SP cells was particularly higher than that of NSP cells in CpG islands. (D) SP cells showed a generally increased level of DNA methylation in gene flanking and intragenic regions than that of NSP cells.
MACS, a peak scanning software, revealed 194,781 peaks and 177,650 peaks in SP and NSP cells, respectively, accounting for approximately 6% of the peak coverage rate in the whole genome (Table S2). By merging the peaks of methylation, 18,154 differentially hypermethylated peaks and 8,577 differentially hypomethylated peaks were identified with various genomic contexts in SP cells compared to NSP cells. Distribution of the peaks was analyzed in upstream 2k, 5' UTR, exon, intron, 3' UTR and downstream 2k regions. A large proportion of peaks were found in the intron region, followed by CDS, upstream 2k, downstream 2k, 3' UTR, and 5' UTR in SP and NSP cells (Figure S1). To evaluate the general methylation level of each gene element, the coverage of peaks that equaled the length of the peak occupying region divided by the total length of the corresponding element was measured. In contrast to the distribution of peak reads, the peak coverage of introns was the lowest among other genomic elements (Figure S1).
Differentially methylated genes (DMGs) and the validation of MeDIP-seq data using bisulfite sequencing
The distribution of differentially methylated genes between SP and NSP cells in different genomic contexts was determined (Table S5). A total of 6,400 genes were differentially hypermethylated, and 3,483 genes were differentially hypomethylated in SP cells compared to NSP cells, containing a total of 2,161 DMGs of simultaneous hypermethylated and hypomethylated regions. The sequencing data of the methylome in the present study are available in the National Center for Biotechnology Information Sequence Read Archive (http://www.ncbi.nlm.nih.gov/Traces/sra/) under accession numbers SRR2062225 and SRR2062224.
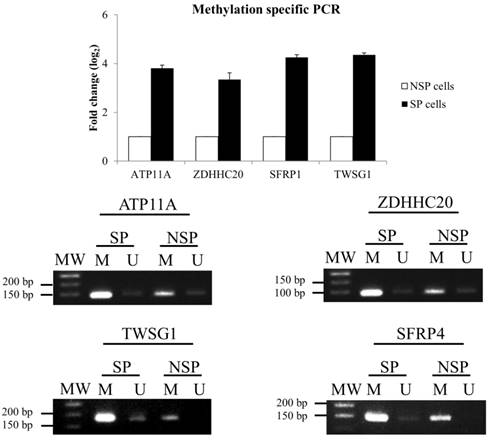
To validate the MeDIP-seq data, methylation-specific PCR (MSP) was performed for 4 selected DMGs: ATPase, Class VI, Type 11 (ATP11A), zinc finger, DHHC-type containing 20 (ZDHHC20), twisted gastrulation BMP signaling modulator 1 (TWSG1), and secreted frizzled-related protein 1 (SFRP1) (Figure 3). MSP results confirmed the data from the MeDIP-seq analysis.
Validation of MeDIP-seq data using methylation-specific PCR (MSP). (A) Relative gene expression level of DMGs. The DMGs in NSP cells were used as a reference for the up- or down-regulation of DNA methylation in the SP fraction. (B) DNA methylation patterns of four selected DMGs.
Mapping of the RNA-seq library sequencing data
After removing poor quality sequences during the quality control steps, approximately 85 million clean reads were generated from SP and NSP samples and subsequently mapped to the hg18 using a TopHat aligner. A total of 73,888,650 and 72,799,414 RNA-seq reads were mapped to the hg18, with 96.32% and 96.10% of the mapping rate in SP and NSP samples, respectively (Table S6). The quality score across all bases represented high accuracy based on the base-calling process using Illumina pipeline software.
Identification of DEGs and the validation of RNA-seq data using quantitative real-time RT-PCR
Differentially expressed genes (DEGs) were analyzed from the Bam file, a standard file of the resulting read alignments. The Cuffdiff program was employed to find potential genes that showed significant differences in expression levels. Among the total genes and transcripts, Cuffdiff filtered a small portion of inadequate reads, including too complex or shallowly sequenced alignments and too short or too numerous fragments in the locus. By comparing the RNA-seq data of SP and NSP, the differential expression of 1,130 genes from a total of 17,122 mRNAs was identified. Among these genes, 795 DEGs were significantly up-regulated and 335 DEGs were down-regulated in the SP cells compared to the NSP cells. The sequencing data of the transcriptome in the present study are available in the National Center for Biotechnology Information Sequence Read Archive (http://www.ncbi.nlm.nih.gov/Traces/sra/) under accession number SRR2062223 and SRR2064655.
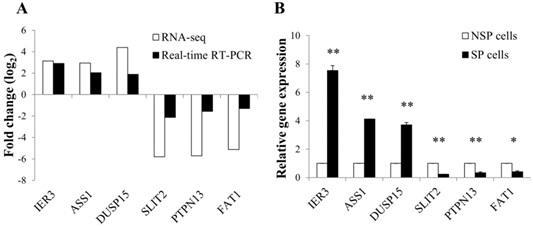
To validate the expression level of differentially regulated genes in RNA-seq data, real-time RT-PCR was performed for 6 selected DEGs: immediate early response 3 (IER3), argininosuccinate synthase 1 (ASS1), dual specificity phosphatase 15 (DUSP15), slit homolog 2-(Drosophila) (SLIT2), protein tyrosine phosphatase, non-receptor type 13 (PTPN13), and FAT atypical cadherin 1 (FAT1). The RT-PCR results were generally concordant with RNA-seq data, although there was a small difference in the degree of fold changes (Figure 4A and 4B).
Validation of RNA-seq data using quantitative real-time PCR. (A) Correlation in the expression of differentially regulated genes between RNA-seq and real-time RT-PCR. Log2 values were calculated by comparing read counts in SP cells to NSP cells. RT-PCR results showed general consistency with RNA-seq data. (B) Relative mRNA level of differentially up or down-regulated genes found by RNA-seq. GAPDH was used as the housekeeping gene. Relative gene expression was generated according to the 2-Δ ΔCt method. *p-value <0.05, **p-value <0.01; Student's t-test.
Integrated analysis of DMGs and DEGs
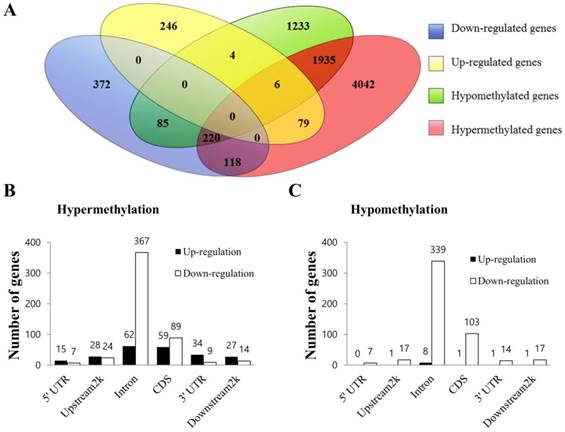
Data from MeDIP-seq and RNA-seq were analyzed in an integrated manner to identify a subset of genes that were regulated by DNA methylation. The comprehensive distribution pattern of the genes with both differential methylation and expression levels were illustrated (Figure 5A). After merging overlapping DMGs containing DMRs with different gene elements into the unitary DMG, a total of 7722 DMGs, including 2,161 simultaneously hypermethylated and hypomethylated DMGs, were identified. Among the many unique DMGs, 512 DMGs with 89 up-regulated and 423 down-regulated genes were found to be significantly regulated in SP cells compared to NSP cells. Their bidirectional gene expression patterns were located in the various genomic regions (Figure 5B and 5C). The DMGs showing the opposite expression relative to their corresponding methylation status or simultaneously containing at least two regions of differential hypermethylation and hypomethylation were excluded from the subsequent integrated analysis. Consequently, a total of 122 DEGs were considered as potential candidate genes regulated by aberrant DNA methylation, including 118 hypermethylated genes showing simultaneous down-expression and 4 hypomethylated genes showing concurrent up-expression (Table S7). Among those genes, DMGs containing methylated sequences in the upstream 2k region were further investigated using the UCSC genome browser to determine whether differentially methylated sequences included CpG islands. This analysis revealed that a total of 10 DMGs exhibited altered methylation in CpG islands (Table 1 and Figure S2). In particular, TWSG1, BNC1, RPS6KA3 and DUSP15 exhibited differential methylation in the CpG islands of the promoter region.
Integrated analysis of MeDIP-seq and RNA-seq data. (A) Venn diagram of DEGs and DMGs. Each group was divided into up-regulated and down-regulated subgroups. The number of genes is given in the middle of each figure section. (B) Bidirectional expression patterns of differentially hypermethylated genes with various genomic elements. (C) Differentially hypomethylated genes with various genomic elements. The number of genes is given at the top of each graph bar.
Differentially methylated and regulated genes with methylation profiles in CpG islands.
| Gene ID | Chromosome | Genomic context | Methylated region | Fold change (MeDIP-seq) | p-value | Fold change (RNA-seq) | p-value |
|---|---|---|---|---|---|---|---|
| ATP11A | chr13 | Intron | 112398122-112400894 | 2.590 | 3.40E-34 | -9.298 | 0.0105 |
| Intron | 112503894-112504883 | 3.263 | 8.69E-30 | ||||
| NDST1 | chr5 | Intron | 149895060-149895861 | 13.727 | 1.63E-164 | -5.134 | 0.04435 |
| CDS | 149895060-149895861 | 13.727 | 1.63E-164 | ||||
| CDS | 149886968-149888651 | 2.462 | 9.89E-49 | ||||
| MTMR1 | chrX | Intron | 149681001-149682087 | 4.667 | 2.55E-47 | -6.905 | 0.02025 |
| Intron | 149661398-149662354 | 2.778 | 1.21E-19 | ||||
| CDS | 149681001-149682087 | 4.667 | 2.55E-47 | ||||
| 3'-UTR | 149681001-149682087 | 4.667 | 2.55E-47 | ||||
| BNC1 | chr15 | Upstream 2k | 81744675-81745352 | 2.929 | 1.16E-17 | -6.420 | 0.03335 |
| EGR3 | chr8 | CDS | 22602725-22605431 | 4.278 | 3.35E-48 | -9.350 | 0.03775 |
| TWSG1 | chr18 | Upstream 2k | 9323013-9324201 | 3.867 | 1.62E-33 | -6.409 | 0.02105 |
| Intron | 9352211-9352680 | 2.739 | 5.38E-24 | ||||
| ZDHHC20 | chr13 | Intron | 20849079-20850389 | 9.308 | 1.26E-117 | -5.174 | 0.03855 |
| Intron | 20876367-20877118 | 2.238 | 9.14E-13 | ||||
| RPS6KA3 | chrX | Upstream 2k | 20195426-20197844 | 2.115 | 4.33E-13 | -7.811 | 0.0116 |
| ZZEF1 | chr17 | Intron | 3906929-3908389 | 2.139 | 5.17E-35 | -4.842 | 0.04695 |
| Downstream 2k | 3852723-3855666 | 2.055 | 9.06E-24 | ||||
| CDS | 3906929-3908389 | 2.139 | 5.17E-35 | ||||
| DUSP15 | chr20 | Upstream2k | 29922261-29922719 | 0.426 | 3.524E-26 | 21.024 | 0.0438 |
The methylation and expression statuses of the NSP samples were utilized as references for those in the SP samples.
GO analysis of the epigenetically regulated DEGs
To determine biologically functional relationships among epigenetically regulated DEGs, GO analysis was performed. A total of 122 DEGs, including 118 down-regulated genes with hypermethylation and 4 up-regulated genes with hypomethylation, were subjected to GO analysis. In total, 102 GO terms were enriched in the integrated epigenome data, including 7 enriched GO terms under the cellular component category, 46 enriched GO terms under the molecular function category, and 49 enriched GO terms under the biological process category. Statistically significant profiles of GO terms with a p-value less than 0.05 in the 3 categories were determined (Table S8). It was noteworthy that the 3 most significantly enriched GO terms in the biological process were stem cell maintenance, stem cell development, and stem cell differentiation. GO terms could not be acquired for hypomethylated DEGs due to the insufficient number of the submitted genes.
Discussion
In the present study, integrated analysis of MeDIP-seq and RNA-seq data from distinct cancer cell subpopulations in a HMM cell line revealed a set of 122 differentially regulated genes, including 118 down-regulated genes with hypermethylation and 4 up-regulated genes with hypomethylation. The results indicated that genes defining the subpopulations of HMM cells could be regulated epigenetically. Although the use of additional HMM cell lines or human disease samples would solidify the conclusion, the data obtained from the present study clarify clearly suggests that DNA methylation may play a pivotal role in the genesis of intratumoral heterogeneity, possibly by regulating the more aggressive properties of CSCs.
Clonal evolution from a single cancer cell through genetic alterations has been conceptually believed to be a driving force for intratumoral heterogeneity, providing new properties favorable for survival and expansion [3]. However, emerging evidence suggests that tumor heterogeneity arises from epigenetic modifications of tumor cells independent of genetic mutations [4, 5, 26]. In a support of this notion, Feinberg and colleagues [5] demonstrated that epigenetic alterations in cancer progenitor cells drive intratumoral heterogeneity during the exacerbation of tumor progression. In solid tumors, CSCs exhibited more heterogeneous patterns of DNA methylation than their differentiated progeny and contributed to the generation of population heterogeneity [27]. It is evident that the dysregulation of DNA methylation as an early molecular event promotes the expression of malignant phenotypes in cancers [28].
HMM was selected as a paradigmatic model for the present study about intratumoral heterogeneity because HMM is extremely heterogeneous with regard to tumor cell morphology and molecular phenotypes [17] and contains distinct cancer cell subpopulations [18, 29]. HMM harbors fewer mutations than other cancers [30]. This feature is quite advantageous for the study of the potential involvement of non-genetic mechanisms relative to genetic alterations in the tumor heterogeneity of HMM.
Altered gene expression has been extensively investigated in HMM, and epigenetic deregulation leading to changes in the gene expression has been a constant finding in HMMs [31]. Altered DNA methylation, which is one of the most extensively studied epigenetic modifications, has been reported to play a key role in HMM tumorigenesis and the acquisition of malignant potentials during mesothelial transformation [28, 30]. Methylated CpG islands have been shown to affect a wide range of oncogenic processes in HMM, including uncontrolled cell proliferation, differentiation, apoptosis, and invasion. For example, asbestos fibers increase the prevalence of aberrant promoter methylation in the cell cycle control genes APC and RASSF1 [32, 33]. The survival of HMM cells has been attributed to promoter methylation and the silencing of genes, including SFRP4, SFRP5, FHIT, and SLC6A20 [30]. In cancers such as HMM, tumor heterogeneity is mainly controlled by the differentiation status of the tumor cells, which depends on the balance between self-renewal and the differentiation of tumorigenic CSCs into their progeny cells [9, 17]. Additionally, it is evident that DNA methylation is required for the maintenance of CSCs [34]. Thus, it is conceivable that deregulated DNA methylation not only confers CSCs better properties among heterogeneous populations but also facilitates the genesis of intratumoral heterogeneity. In the present study, epigenetically regulated genes are mainly attributed to the biological processes of not only stem cell maintenance, development, and differentiation (NOTCH2, STAG2, YAP1, and ZCCHC11) but also the negative regulation of differentiation and developmental processes (ROCK1, ROCK2, GTF2I, ITGB3, MED1, NOTCH2, PKP2, STAG2, THBS1, TWSG1, YAP1, ZCCHC11). The GO terms enriched for the candidate genes indicate that putative CSCs regulate stem cell properties epigenetically by modulating the DNA methylation status and, at least in part, by regulating the potency of differentiation. Of the aberrantly methylated genes, YAP1 (yes-associated protein 1) was shown to play a pivotal role in the differentiation of stem cells. It is highly expressed in mammalian undifferentiated pluripotent cells and known to expand stem cell populations upon up-regulation, whereas the inactivation of YAP1 generates more differentiated progeny [35]. YAP1 is also involved in the tumorigenesis of mesothelioma by improving the proliferative capacity of the cancer cells [36]. Hypermethylation in the 3' UTR region and the down-regulation of YAP1 observed in the present study may indicate that YAP1 promotes the differentiation of CSCs, recapitulating the heterogeneity of HMM cells. Methylation in the terminal exon of YAP1 is related to RNA stability and subcellular localization as well as translation regulation [37], which warrants further studies.
Aberrant methylation of CpG islands is closely associated with the disturbances of gene expression that result in the transformation of normal mesothelium [30]. Thus, attention has focused on the aberrant methylation of CpG islands in the promoter. The present study revealed 4 genes with differential methylation in the CpG islands of promoters in SP cells compared to NSP cells. These genes include hypermethylated basonuclin 1 (BNC1), ribosomal protein S6 kinase (90 kDa), polypeptide 3 (RPS6KA3), twisted gastrulation bone morphogenetic protein (BMP) signaling modulator 1 (TWSG1), and hypomethylated dual specificity phosphatases 15 (DUSP15).
BNC1 is a transcription factor that participates in the mitosis of actively dividing keratinocytes and germ cells of the testis and ovary [38]. Previous studies have revealed that it is commonly hypermethylated and silenced in many human malignancies, including solid tumors and hematopoietic neoplasms [39, 40], implicating its potential role as a tumor suppressor. Similarly, promoter hypermethylation of RPS6KA3 may cause a decrease in cell division. Hsu et al. [41] reported that RPS6KA3 is an important gene defining CSCs in triple-negative breast cancer, the most heterogeneous subtype of breast cancers. A breast cancer methylome showed that the expression of RPS6KA3 was regulated via the hypermethylation of CpG islands [42]. These previous studies indicate that the hypermethylation of BNC1 and RPS6KA3 could be potentially associated with the quiescent status of CSCs and the heterogeneity of cancer cells. As an inherent feature of CSCs, slowly growing or quiescent stem cell populations in cancer are primarily attributable to the resistance to chemotherapy [43]. Thus, the perturbed hypermethylation and consequent inactivation of BNC1 and RPS6KA3 may be a key strategy for putative CSCs to repopulate tumor cell progenies and promote the heterogeneity of HMM; however, the suggested biological functions of these genes require further study. On the other hand, TWSG1, a secreted cysteine-rich protein that regulates the extracellular cell-to-cell BMP signaling involved in normal embryogenesis [44], is required for myoepithelial differentiation during the postnatal development of the mammary gland, and its dysregulation induces apoptotic defects, leading to ductal proliferation [45]. In human blood leukocytes, exposure to environmental contaminants increases the systemic DNA methylation level in 92 genes, including TWSG1 [46]. When cancer cells are exposed to hostile microenvironments, DUSP15 is related to aberrant methylation [4747, 48]. Although the precise roles of DUSP15 and TWSG1 in the response of CSCs to cellular and environmental stresses remains to be determined, the aberrant methylation of CpG islands in their promoter region may promote tumor cell viability or differentiation when exposed to unfavorable conditions, ultimately contributing to tumor heterogeneity in HMM. Further studies may be necessary to investigate the functional correlation of aberrant methylation in the promoter regulatory regions and biological properties of a number of epigenetically regulated genes.
Intratumoral heterogeneity has been studied with different perspectives, including genetic alterations, environmental differences, stochastic processes, cell and tissue plasticity, and the presence or absence of a cellular hierarchy [2, 9, 10]. Previous investigations about epigenetic mechanisms in HMM were limited by the lack of a genome-wide assay in DNA methylation profiles and focused on methylation profile in the CpG islands of selected genes. To the best of our knowledge, for the first time, the present study demonstrated the epigenetic heterogeneity of cancer cells using an integrated analysis of the DNA methylome and transcriptome. In the future, these findings may promote the development of novel diagnostic and therapeutic measures for cancers, including HMMs.
Supplementary Material
Additional File 1Table S1.
Table S2.
Table S3.
Table S4.
Table S5.
Table S6.
Table S7.
Table S8.
Figures S1-S2.
Acknowledgements
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (Grant #: NRF-2013R1A2A2A01068237) and the BK21 program and the Research Institute of Veterinary Sciences, College of Veterinary Medicine, Seoul National University. The authors are grateful to the DNA Link Incorporation (Songpa-gu, Seoul, The Republic of Korea) for their excellent technical assistance.
Abbreviations
HMM: Human malignant mesotheliom; SP: side population; NSP: non-side population; DMGs: Differentially methylated genes; DEGs: Differentially expressed genes; GO: Gene ontology; NGS: Next generation sequencing; FPKM: Fragments per kilobase of transcript per million mapped fragments; MTI: microRNA-target interactions; RIN: RNA Integrity Number; FC: Fold change; MACS: Model-based Analysis for ChIP-Seq; MeDIP-seq: methylated DNA immunoprecipitation combined with high-throughput sequencing; RNA-seq: RNA-sequencing; YAP1: yes-associated protein 1; BNC1: Basonuclin 1; RPS6KA3: Ribosomal Protein S6 Kinase, 90kDa, Polypeptide 3; TWSG1: twisted gastrulation BMP signaling modulator 1; DUSP15: dual specificity phosphatase-like 15.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Marusyk A, Polyak K. Tumor heterogeneity: causes and consequences. Biochimica et Biophysica Acta (BBA)-Reviews on Cancer. 2010;1805:105-17
2. Saunders NA, Simpson F, Thompson EW, Hill MM, Endo-Munoz L, Leggatt G. et al. Role of intratumoural heterogeneity in cancer drug resistance: molecular and clinical perspectives. EMBO molecular medicine. 2012;4:675-84
3. Heng HH, Bremer SW, Stevens JB, Ye KJ, Liu G, Ye CJ. Genetic and epigenetic heterogeneity in cancer: A genome-centric perspective. Journal of cellular physiology. 2009;220:538-47
4. Wilting RH, Dannenberg J-H. Epigenetic mechanisms in tumorigenesis, tumor cell heterogeneity and drug resistance. Drug Resistance Updates. 2012;15:21-38
5. Feinberg AP, Ohlsson R, Henikoff S. The epigenetic progenitor origin of human cancer. Nature reviews genetics. 2006;7:21-33
6. Esteller M. Epigenetics in cancer. New England Journal of Medicine. 2008;358:1148-59
7. Esteller M. Aberrant DNA methylation as a cancer-inducing mechanism. Annu Rev Pharmacol Toxicol. 2005;45:629-56
8. Laird PW, Jaenisch R. The role of DNA methylation in cancer genetics and epigenetics. Annual review of genetics. 1996;30:441-64
9. Magee JA, Piskounova E, Morrison SJ. Cancer stem cells: impact, heterogeneity, and uncertainty. Cancer cell. 2012;21:283-96
10. Alexander P. Cancer stem cells in tumor heterogeneity. Advances in cancer research. 2011;112:255-82
11. Laird PW. Principles and challenges of genome-wide DNA methylation analysis. Nature Reviews Genetics. 2010;11:191-203
12. Jacinto FV, Ballestar E, Esteller M. Methyl-DNA immunoprecipitation (MeDIP): hunting down the DNA methylome. Biotechniques. 2008;44:35
13. Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nature Reviews Genetics. 2009;10:57-63
14. Robinson BW, Lake RA. Advances in malignant mesothelioma. New England Journal of Medicine. 2005;353:1591-603
15. Robinson BW, Musk AW, Lake RA. Malignant mesothelioma. The Lancet. 2005;366:397-408
16. Bianchi C, Bianchi T. Malignant mesothelioma: global incidence and relationship with asbestos. Industrial health. 2007;45:379-87
17. Sun X, Wei L, Lidén J, Hui G, Dahlman-Wright K, Hjerpe A. et al. Molecular characterization of tumour heterogeneity and malignant mesothelioma cell differentiation by gene profiling. The Journal of pathology. 2005;207:91-101
18. Kai K, D'Costa S, Yoon B-I, Brody AR, Sills RC, Kim Y. Characterization of side population cells in human malignant mesothelioma cell lines. Lung Cancer. 2010;70:146-51
19. Jänne PA. Chemotherapy for malignant pleural mesothelioma. Clinical lung cancer. 2003;5:98-106
20. Su J, Wang Y, Xing X, Liu J, Zhang Y. Genome-wide analysis of DNA methylation in bovine placentas. BMC genomics. 2014;15:12
21. Yang HJ, Kim N, Seong KM, Youn H, Youn B. Investigation of radiation-induced transcriptome profile of radioresistant non-small cell lung cancer A549 cells using RNA-seq. PloS one. 2013;8:e59319
22. Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature protocols. 2012;7:562-78
23. Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nature methods. 2008;5:621-8
24. Livak KJ, Schmittgen TD. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2(-Delta Delta C(T)) Method. methods. 2001;25:402-8
25. Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM. et al. Gene Ontology: tool for the unification of biology. Nature genetics. 2000;25:25-9
26. Marquardt J, Factor V, Thorgeirsson S. Epigenetic regulation of cancer stem cells in liver cancer: current concepts and clinical implications. Journal of hepatology. 2010;53:568-77
27. Siegmund KD, Marjoram P, Woo Y-J, Tavaré S, Shibata D. Inferring clonal expansion and cancer stem cell dynamics from DNA methylation patterns in colorectal cancers. Proceedings of the National Academy of Sciences. 2009;106:4828-33
28. Bagwe A, Kay P, Spagnolo D. Evidence that DNA methylation imbalance is not involved in the development of malignant mesothelioma. Anticancer research. 1996;17:3341-3
29. Hadnagy A, Gaboury L, Beaulieu R, Balicki D. SP analysis may be used to identify cancer stem cell populations. Experimental cell research. 2006;312:3701-10
30. Zhang X, Tang N, Rishi AK, Pass HI, Wali A. Methylation Profile Landscape in Mesothelioma: Possible Implications in Early Detection, Disease Progression, and Therapeutic Options. Cancer Epigenetics: Springer. 2015:235-47
31. Vandermeers F, Sriramareddy SN, Costa C, Hubaux R, Cosse J-P, Willems L. The role of epigenetics in malignant pleural mesothelioma. Lung Cancer. 2013;81:311-8
32. Christensen BC, Godleski JJ, Marsit CJ, Houseman E, Lopez-Fagundo CY, Longacker JL. et al. Asbestos exposure predicts cell cycle control gene promoter methylation in pleural mesothelioma. Carcinogenesis. 2008;29:1555-9
33. Christensen BC, Houseman EA, Poage GM, Godleski JJ, Bueno R, Sugarbaker DJ. et al. Integrated profiling reveals a global correlation between epigenetic and genetic alterations in mesothelioma. Cancer research. 2010;70:5686-94
34. Pathania R, Ramachandran S, Elangovan S, Padia R, Yang P, Cinghu S. et al. DNMT1 is essential for mammary and cancer stem cell maintenance and tumorigenesis. Nature communications. 2015:6
35. Camargo FD, Gokhale S, Johnnidis JB, Fu D, Bell GW, Jaenisch R. et al. YAP1 increases organ size and expands undifferentiated progenitor cells. Current biology. 2007;17:2054-60
36. Mizuno T, Murakami H, Fujii M, Ishiguro F, Tanaka I, Kondo Y. et al. YAP induces malignant mesothelioma cell proliferation by upregulating transcription of cell cycle-promoting genes. Oncogene. 2012;31:5117-22
37. Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell. 2012;149:1635-46
38. Tseng H, Biegel JA, Brown RS. Basonuclin is associated with the ribosomal RNA genes on human keratinocyte mitotic chromosomes. Journal of cell science. 1999;112:3039-47
39. Tong W-G, Wierda WG, Lin E, Kuang S-Q, Bekele BN, Estrov Z. et al. Genome-wide DNA methylation profiling of chronic lymphocytic leukemia allows identification of epigenetically repressed molecular pathways with clinical impact. Epigenetics. 2010;5:499-508
40. Shames DS, Girard L, Gao B, Sato M, Lewis CM, Shivapurkar N. et al. A genome-wide screen for promoter methylation in lung cancer identifies novel methylation markers for multiple malignancies. PLoS medicine. 2006;3:e486
41. Hsu Y-H, Yao J, Chan L-C, Wu T-J, Hsu JL, Fang Y-F. et al. Definition of PKC-α, CDK6, and MET as Therapeutic Targets in Triple-Negative Breast Cancer. Cancer research. 2014;74:4822-35
42. Gu F, Doderer MS, Huang Y-W, Roa JC, Goodfellow PJ, Kizer EL. et al. CMS: a web-based system for visualization and analysis of genome-wide methylation data of human cancers. PloS one. 2013;8:e60980
43. Moore N, Lyle S. Quiescent, slow-cycling stem cell populations in cancer: a review of the evidence and discussion of significance. Journal of oncology. 2010;2011:1-11
44. Petryk A, Anderson RM, Jarcho MP, Leaf I, Carlson CS, Klingensmith J. et al. The mammalian twisted gastrulation gene functions in foregut and craniofacial development. Developmental biology. 2004;267:374-86
45. Forsman CL, Ng BC, Heinze RK, Kuo C, Sergi C, Gopalakrishnan R. et al. BMP-binding protein twisted gastrulation is required in mammary gland epithelium for normal ductal elongation and myoepithelial compartmentalization. Developmental biology. 2013;373:95-106
46. Sanders A, Smeester L, Rojas D, DeBussycher T, Wu M, Wright F. et al. Cadmium exposure and the epigenome: Exposure-associated patterns of DNA methylation in leukocytes from mother-baby pairs. Epigenetics. 2014;9:212-21
47. Ouyang B, Baxter CS, Lam H-M, Yeramaneni S, Levin L, Haynes E. et al. Hypomethylation of dual specificity phosphatase 22 promoter correlates with duration of service in firefighters and is inducible by low-dose benzo [a] pyrene. Journal of Occupational and Environmental Medicine. 2012;54:774
48. Cain EL, Beeser A. Emerging Roles of Atypical Dual Specificity Phosphatases in Cancer. Oncogene and Cancer-From Bench to Clinic. 2013:93
Author contact
![]() Corresponding author: Dr. Yongbaek Kim, Laboratory of Clinical Pathology, College of Veterinary Medicine, Seoul National University, 1 Gwanak-ro, Gwanak-gu, Seoul 151-742, The Republic of Korea. Tel: + 82 2 880 1273, Fax: +82 2 873 1213, Email: yongbaekac.kr
Corresponding author: Dr. Yongbaek Kim, Laboratory of Clinical Pathology, College of Veterinary Medicine, Seoul National University, 1 Gwanak-ro, Gwanak-gu, Seoul 151-742, The Republic of Korea. Tel: + 82 2 880 1273, Fax: +82 2 873 1213, Email: yongbaekac.kr