Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2016; 7(11):1497-1514. doi:10.7150/jca.14922 This issue Cite
Review
Cancer of the Pancreas: Molecular Pathways and Current Advancement in Treatment
Kishore Polireddy, Qi Chen ![]()
Department of Pharmacology, Toxicology and Therapeutics, University of Kansas Medical Center, 3901 Rainbow Blvd., Kansas City, KS, USA 66160.
Received 2016-1-8; Accepted 2016-4-26; Published 2016-7-7
Abstract
Pancreatic cancer is one of the most lethal cancers among all malignances, with a median overall survival of <1 year and a 5-year survival of ~5%. The dismal survival rate and prognosis are likely due to lack of early diagnosis, fulminant disease course, high metastasis rate, and disappointing treatment outcome. Pancreatic cancers harbor a variety of genetic alternations that render it difficult to treat even with targeted therapy. Recent studies revealed that pancreatic cancers are highly enriched with a cancer stem cell (CSC) population, which is resistant to chemotherapeutic drugs, and therefore escapes chemotherapy and promotes tumor recurrence. Cancer cell epithelial to mesenchymal transition (EMT) is highly associated with metastasis, generation of CSCs, and treatment resistance in pancreatic cancer. Reviewed here are the molecular biology of pancreatic cancer, the major signaling pathways regulating pancreatic cancer EMT and CSCs, and the advancement in current clinical and experimental treatments for pancreatic cancer.
Keywords: pancreatic cancer, cancer stem cell, epithelial to mesenchymal transition.
Pancreatic cancer: types and molecular biology
Pancreatic cancer is the 4th leading cause of cancer-related death in the United States, and is expected to become the 2nd by 2030. The American Cancer Society estimated that 53,070 (men=27,670 women=25,400) people will be diagnosed with pancreatic cancer in 2016 and that 41,780 (men=21,450, women=20,330) will die from the disease (1). Most of the pancreatic cancer cases (96%) are cancers of the exocrine pancreas. Most patients with localized disease have no recognizable symptoms, and currently there are no early detection tests for pancreatic cancer. As a result, >50% of the patients are diagnosed at a stage where metastases have developed, for whom the overall 5-year survival is only 2% (2).
A spectrum of distinct pancreatic malignancies have been identified that resemble the normal cellular counterparts in the pancreas, such as pancreatic ductal adenocarcinoma (PDAC), acinar cell carcinoma, pancreato blastoma, solid pseudopapillary neoplasm, serous cystadenoma, and pancreatic endocrine tumors. Among these, PDAC, whose name is derived from its histological resemblance to ductal cells, is the most common pancreatic neoplasm and accounts for >85% of pancreatic cancer cases (3).
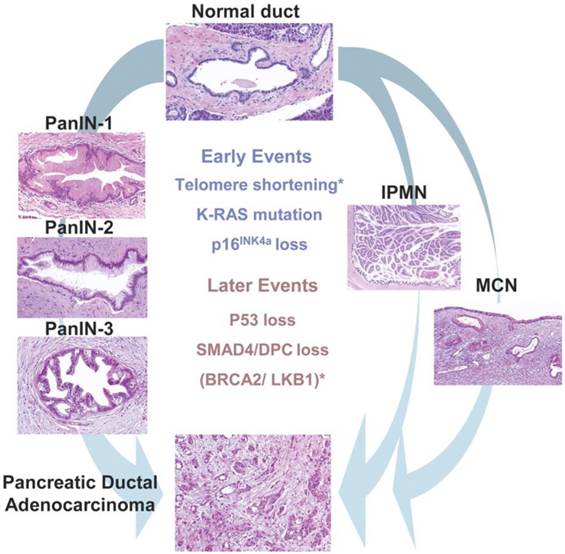
The majority of PDACs arises in the head region of the pancreas and exhibits a glandular pattern resembling ductal epithelial cells with varying degree of differentiation. Clinical and histological studies identified three different types of precursor lesions that lead to PDAC: pancreatic intra epithelial neoplasia (PanIN), mucinous cystic neoplasm (MCN), and intraductal papillary mucinous neoplasms (IPMN) (Fig. 1). Of these, PanIN is the most extensively studied. PanIN is graded into stages 1 to 3. PanIN stage 1 is characterized by columnar mucinous epithelium with slight nuclear atypia. PanIN stage 2 and 3 are characterized by more disorganized structural architecture and nuclear atypia. MCN and IPMN are less common precursor lesions. MCNs are large mucin producing columnar epithelial cystic lesions supported by ovarian type stroma usually found in the body and tail of the pancreas. IPMN arises in the main pancreatic duct or its major branches, and resembles PanIN at cellular levels but grows into large cystic structures (4).
Pancreatic precursor lesions and genetic events involved in pancreatic adenocarcinoma progression. PanIN, IPMN, MCN represents three known precursor lesions of PDAC. PanINs classified into three grades: PanIN-1, 2 and 3 which then eventually develop to PDAC. Development of PDAC from IPMN and MCN shown on right side of the picture. Early genetic alterations (K-Ras mutations, P16 loss) and late genetic alterations (P53 loss, SMAD4 loss,) that occur in adenocarcinomas also occur in PanIN and to lesser extent in IPMNs and MCNs represented in the middle of the picture. Asterisks indicate events (telomere shortening, BRCA2 mutations) that are not common to all precursor lesions. (Republished from Hezel et al. Gene Dev. 2006, 20(10):1218-49, with permission from the Cold Spring Harbor Laboratory Press).
The mutational landscape of pancreatic cancer has been explored using whole genome sequencing analysis (5), and revealed average of 119 somatic chromosomal structural variants per individual patient, which exceeded the previously reported average of 63 mutations by another comprehensive genetic analysis (6). Majority of these variants were intra-chromosomal deletions, duplications, tandem duplications, inversions, fold back inversions, amplified inversions, and intra chromosomal rearrangements. These mutations are involved in at least 12 different core signaling pathways that were altered in 67-100% of the tumors. The most commonly observed signature genetic lesions in pancreatic cancer are kirsten rat sarcoma viral oncogene homolog (K-Ras), P16/ cyclin-dependent kinase inhibitor 2A (CDKN2A), tumor protein P53 (P53), breast cancer 2 early onset (BRCA2) and SMAD family member 4 (SMAD4)/ deleted in pancreatic carcinoma 4 (DPC4) (6). In pancreatic cancer patients, K-Ras mutations are often considered as an initiating event occurring in adult cells, soon followed by mutation to P16 and later P53 and SMAD4 loss.
The K-Ras oncogene. K-Ras is activated by point mutations in >90% of the pancreatic cancer patients and is the earliest genetic alteration, being found in low-grade PanIN lesions (6, 7). Continuous K-Ras signaling is required for pancreatic cancer cells for sustained proliferation and survival (8). Ras proteins belong to the small G protein superfamily, and their activity is regulated by guanine nucleotides such as GTP and GDP. Ras downstream signaling pathways are activated if Ras binds to GTP, and are inactivated if Ras binds to GDP. Active and inactive states of Ras signaling are regulated by guanine nucleotide exchange factors (GEFs) and GTPase activating proteins (GAPs). GEFs aid in exchange of GDP for GTP. GAPs activate intrinsic GTPase activity of Ras protein to hydrolyze GTP into GDP (9). Any mutations that inactivate the GTPase constitutively activate Ras signaling and downstream effector pathways. For example, a single point mutation of codons G12 or G13 in K-Ras abolishes GAP induced GTP hydrolysis, thereby making K-Ras a constitutively active form. Therefore, a pancreatic specific mutation of codons G12D or G12V is sufficient to develop acinar to ductal metaplasia and PanIN, which then progress to PDAC. PDAC development can be accelerated in the K-Ras mutant mouse by introducing additional mutations in tumor suppressor genes such as p53, SMAD4, and P16/CDKN2A, all of which occur frequently in precursor lesions as they progress to invasive PDAC (10).
K-Ras signaling engages various downstream effectors. In PDAC, K-Ras predominantly signals through canonical Raf/mitogen activated protein kinase (MAPK)/ extracellular signal-regulated kinase (Erk), phosphoinositide 3-kinases (PI3Ks)/ 3-phosphoinositide dependent protein kinase-1 (PDK-1)/Akt, Ral guanine nucleotide exchange factors (RalGEFs), and phospholipase Cε (9).
Mutations or alternations in these downstream pathways complicate K-RAS driven PDAC. For example, expression of PIK3CA H1047R (codes for p110α H1047R), a constitutively active oncogenic class 1A PI3K in Ptf1a positive cells, induced acinar to ductal metaplasia and premalignant PanIN, recapitulating K-Ras G12D driven PDAC. Elimination of PDK-1, blocked K-RasG12D driven PDAC (11). RalGEF induces oncogenic activity by activating its substrate RalA (12). RalA is required for tumor initiation, whereas the other RalGEF substrate, RalB, is required for metastasis in Ras-driven pancreatic cancers (13).
Tumor suppressor genes (P16/CDKN2A, TP53, and SMAD4/DPC4). P16/CDKN2A is the most commonly inactivated tumor suppressor gene in pancreatic cancer. P16/CDKN2A inhibits CDK4/6 mediated phosphorylation of retinoblastoma (RB) protein, thereby blocking entry into the S phase of the cell cycle. Inactivation of P16/CDKN2A occurs by different mechanisms, including homozygous deletions, loss of heterozygosity and epigenetic silencing by promoter methylation (14). P16/CDKN2A cooperated with K-Ras in the development of PDAC (15). Mutations in K-Ras exert selective pressure for subsequent mutation in P16/CDKN2A, which cooperate to lead to the development of PDAC. In precursor lesions, oncogenic K-Ras expression co-exists with P16/CDKN2A and other markers of senescence, while in PDAC, expression of P16/CDKN2A and senescence markers are missing (16).
P53 is inactivated in 50-75% of PDAC cases and the inactivation occurs via intragenic mutations combined with loss of the second allele (17). P53 mutations observed in the late PanIN stage usually lead to loss of p53 function, and subsequently provides growth and survival advantage for the cells which harbor chromosomal aberrations (18).
SMAD4 is a key signal transducer of TGF-β signaling pathway. SMAD4 is inactivated in ~55% of pancreatic cancer cases either by homozygous deletions or by intergenic mutations and loss of the second allele (19). Loss of SMAD4 provides growth advantage for pancreatic cancer cells by abrogating the growth inhibitory signals mediated by TGF-β (20) in late PanIN stage (PanIN-3) (21). Patients undergoing surgical resection of their pancreatic adenocarcinoma survived longer if their cancer expressed SMAD4 (22).
Growth factor receptor signaling in PDAC. PDACs overexpress multiple mitogenic growth factors and their ligands. These include: the epidermal growth factor (EGF) and its receptor (EGFR), and multiple ligands that bind to EGFR; fibroblast growth factor (FGF) and its receptor (FGFR) and ligands; insulin-like growth factor (IGF) and its receptor (IGFR); platelet derived growth factor (PDGF); and vascular endothelial growth factor (VEGF) (23, 24).
EGFR is a transmembrane receptor tyrosine kinase, activated upon binding of its ligands, EGF and transforming growth factor-α (TGF-α). EGFR overexpression was detected in up to 90% of pancreatic tumors (25), and plays an important role in liver metastasis and recurrence of human pancreatic cancer (26). EGFR inhibitors decrease PDAC cell growth and tumorigenesis in vitro (27) and inhibited growth of orthotopic tumors when combined with chemotherapy (28). However, EGFR targeting agents in combination with gemcitabine did not provide many beneficial effects to pancreatic cancer patients in the clinic (25).
The IGFs and their receptors have been acknowledged as important players in a variety of cancers (29) by regulating cell survival, invasion, and angiogenesis (30). In PDAC patients, elevated expression of IGF-1 and its receptor IGF1R are associated with higher tumor grade and poor survival (31). In vitro experiments showed that exogenously added IGF-1 enhanced the growth of human pancreatic cancer cells and this effect was inhibited by IGF-1 neutralizing antibody (32). However, clinical trials performed with IGF1R blocking antibodies were largely disappointing (33). In 2012, Amgen announced termination of a large phase III clinical trial in patients with metastatic PDAC treated with the IGF1R blocking antibody ganitumab (AMG 479) and gemcitabine. The ganitumab and gemcitabine combination failed to improve overall survival compared to gemcitabine alone (http://www.amgen.com/media/media_pr_detail.jsp?releaseID=1723925).
FGFR is a transmembrane protein that triggers phosphorylation of an adaptor protein FGFR substrate 2 (FRS2), upon binding of FGF. Phosphorylated FRS2 then recruits and activates elements of Ras/MAPK and PI3K/Akt pathways. Overexpression of ligands (FGF1-7) and receptors (FGFR-1 and FGFR-2) contributed to mitogenesis and angiogenesis in a subset of pancreatic cancers (34). Inhibition of FGFR signaling using shRNA or dovitinib (a tyrosine-kinase inhibitor) achieved significant anti-cancer effects in preclinical pancreatic cancer models (35). Dovitinib is currently at clinical development for patients with metastatic pancreatic cancer, biliary cancers and pancreatic neuroendocrine tumors (https://clinicaltrials.gov identifier: NCT01497392, NCT01888965, NCT02108782).
VEGF is an angiogenic polypeptide. It promotes endothelial cell proliferation and survival by binding to its receptors VEGFR-1 and VEGFR-2 (36). Though PDAC is not a highly vascularized tumor, foci of endothelial cell proliferation are often observed in this malignancy. Patient tumor samples showed increased expression of VEGF mRNA and its expression correlated with high micro vessel density and disease progression (37, 38). TNP-40, an analog of fumagillin, which is an anti-angiogenic agent, decreased tumor growth and metastasis of pancreatic cancer cell lines in subcutaneous mouse model (39). Adenoviral vectors carrying the VEGFR tyrosine kinase inhibitor PTK 787 also inhibited the growth and metastasis of pancreatic cancer in preclinical models (40). Phase I/II studies of PTK787 plus gemcitabine (https://clinicaltrials.gov identifier: NCT00185588) in advanced pancreatic cancer showed promising results, and phase II studies are on-going (https://clinicaltrials.gov identifier: NCT00226005).
As our understanding has grown tremendously in oncogenic K-Ras, tumor suppressors, growth factors and their receptors in pancreatic cancer initiation and progression, approaches are being tested to target oncogenic K-Ras and growth factors. However, it remains challenging to improve treatment outcomes, because of the complicated genetic and molecular alternations. It also remains challenging for researchers to further understand the molecular genetics of tumor suppressor proteins in pancreatic cancer, to restore their normal function by gene therapy, or to develop small-molecule inhibitors that reactivate tumor suppressor function (41). Novel approaches to target tumor suppressor genes need to be discovered and tested with open mind.
Epithelial to mesenchymal transition (EMT) in pancreatic cancer
Most of the pancreatic cancer-related deaths are due to metastatic disease. Recent studies in animal models revealed that pancreatic cancer cells undergo dissemination from the primary tumor and get metastasized to liver even before frank malignancy was detected at the primary site of origin. The developmental program of epithelial to mesenchymal transition (EMT) is of virtual importance for this rapid tumor progression (42).
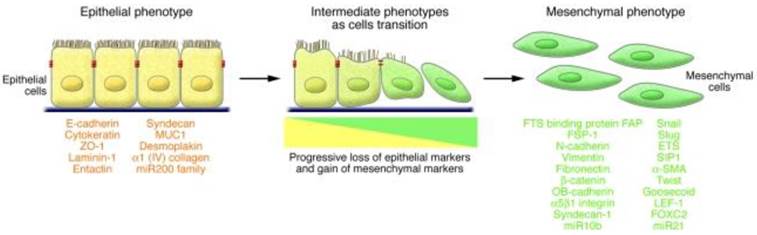
EMT is a multistage trans-differentiation process which allows highly polarized epithelial cells to undergo multiple biochemical changes to attain mesenchymal phenotype (Fig.2). Epithelial cells display apical-basolateral polarity and are organized in cell layers with strong cell-cell adhesion. Mesenchymal cells are spindle shaped, exhibit anterio-posterior polarity and strong migratory potential. During EMT progression, epithelial cells lose their epithelial markers (such as E-cadherin, occludin, claudin, and laminin 1) and gain mesenchymal markers (such as N-cadherin, vimentin, and fibronectin) (43). EMT is classified into three major types based on the context in which it occurs. Type 1 EMT is associated with implantation, embryo formation, and organ development. Mesenchymal cells generated by Type 1 EMT have potential to undergo mesenchymal to epithelial transition (MET) and generate secondary epithelia. Type 2 EMT is associated with inflammation processes and plays a major role in wound healing, tissue regeneration, and organ fibrosis (43).
Characteristics of EMT involve a functional transition of polarized epithelial cells into mesenchymal cells. Epithelial cells are highly polarized in nature with apical and basolateral polarity. E-cadherin, claudins, occludins, desmoplakin, type IV collagen, and laminin-1 are markers of epithelial cells. Mesenchymal cells are spindle shaped and are highly motile in nature. N-cadherin, α5 β1 integrin, αvb6 integrin, vimentin, type-1 collagen, laminin-5 and fibronectin are the markers of mesenchymal cells. During EMT progression, epithelial cells loose epithelial markers and gain mesenchymal markers. Cells that express markers of epithelial and mesenchymal cells represent intermediate phenotype. (Republished from Kalluri et al. J Clin Invest. 2009, 119(6): 1420-8, with permission from ASCI).
Type 3 EMT occurs in carcinoma cells and is considered important at several different stages (dissemination, invasion, intravasation and extravasation) during tumor metastasis (44). Metastatic tumor cells have the potential to migrate through the bloodstream and in some cases, form secondary tumors at other sites through MET (45). During type 3 EMT, some cells retain epithelial traits while acquiring mesenchymal features and other cells shed most epithelial features and become fully mesenchymal (45, 46).
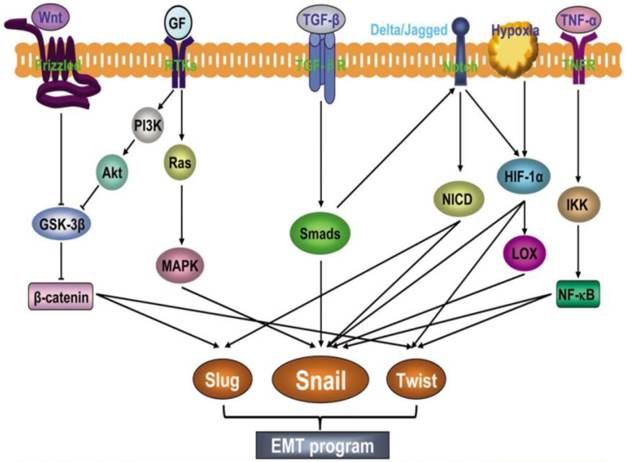
A complex network of signaling pathways governs EMT in tumor progression. Both soluble factors (TGF-β family members, FGF, HGF, Wnt, TNFα, Notch, HIF1-α), as well as non-soluble components of the extracellular matrix (collagen and hyaluronic acid) guide EMT during cancer progression (47). These signaling events primarily induce EMT by inducing transcription factors such as Snail family of zinc finger transcription factors (Snail 1 and 2), zinc-finger-enhancer binding protein (Zeb-1 and 2) and basic helix loop helix (bHLH) family members (E12, E-47, and Twist) (Fig.3). A common feature of these transcription factors is repression of the CDH1 gene that encodes E-cadherin (16). Reduced E-cadherin expression is a key initial step in the trans-differentiation of epithelial to a mesenchymal phenotype, invasion and metastasis.
EMT involves an intricate interplay of multiple signaling pathways. Activation of different signaling pathways such as Wnt, growth factors (GF), TGF-β, Notch, HIF1α and TNF-α induces expression of transcription factors such as Slug, Snail and Twist to promote EMT. (Republished from Wang et al. Chinese J of Can. 2011, 30(9):603-11. Open Access article).
TGF-β signaling pathway in EMT. TGF-β is one of the most important EMT-inducing factors in a diverse range of tumor cells, including pancreatic cancer cells (48). In canonical TGF-β signaling, binding of TGF-β to a TGF-β type II receptor leads to the trans-activation of type I receptor (TβR I). TβR I is a serine/threonine kinase that subsequently phosphorylates SMAD2 and 3, which then forms a complex with SMAD4 and translocates to the nucleus to regulate the transcription of target genes (49). Transcriptional activation of Twist, Snail, Slug, and Zeb-1 has been shown to be critical for TGF-β mediated EMT induction (50).
In pancreatic cancer with SMAD4 inactivation, TGF-β might induce EMT through the non-canonical pathway (i.e. SMAD-independent pathway) (49), which involves ERK/MAPK, PI3K, p38, JNK, RhoA, and other signaling pathways (51). In some pancreatic cancer cell lines such as Colo-357, depletion of SMAD4 using RNAi did not disrupt EMT responses in these cells (52). In other pancreatic cancer cell lines, TGF-β induced EMT was reversed by the MEK-1 inhibitor PD98059 (48).
Wnt/β-catenin signaling pathway. The canonical Wnt/β-catenin signaling pathway proceeds as the following: In the absence of Wnt ligand, β-catenin is kept low by a destruction complex consisting of axin, adenomatous polyposis coli, glycogen synthase kinase-3β (GSK-3β) and casein kinase (CK-1). CK-1 first phosphorylates β-catenin at Ser45, which primes β-catenin. Primed β-catenin is phosphorylated by GSK-3β, at Thr41, Ser33 and Ser37, ultimately leading to ubiquitination and proteosomal degradation by β-Trcp. This persistent removal of β-catenin prevents β-catenin nuclear accumulation, where Wnt target genes are repressed by DNA bound T cell factor (TCF)/lymphoid enhancer factor (LEF) and HDAC. Upon Wnt ligand binding to Frizzled and LRP5/6 receptors, complex is formed and leads to phosphorylation of LRP5/6. Phospho LRP5/6 binds axin leading to dissociation of the destruction complex and inactivation of GSK-3β, thereby stabilizing cytosolic β-catenin and promoting its nuclear localization. In the nucleus β-catenin forms a complex with TCF/LFE and activates expression of Wnt target genes that are important in the regulation of cell growth and proliferation (53).
The Wnt/β-catenin pathway is associated with EMT induction (54). GSK-3β phosphorylates Snail, and promotes Snail proteosomal degradation. Wnt suppresses the activity of GSK-3β and stabilizes the protein levels of Snail, and therefore induces EMT and stem-like properties in cancer cells (55). Oncogenic K-Ras activate the Wnt/β-catenin signaling pathway, which in turn up-regulates EMT stimulators (56). Inhibition of the Wnt/β-catenin signaling pathway blocks EMT, and restoration of the Wnt inhibitory factor 1 (WIF-1) expression resulted in increased expression of epithelial markers and decreased expression of mesenchymal markers, by decreased expression of Slug and Twist (57). Small hairpin RNA knockdown of β-catenin resulted in elevated expression of E-cadherin, decreased expression of the mesenchymal markers vimentin, N-cadherin, MMP-2, indicating the reversal of EMT (58).
Notch signaling pathway. The Notch-signaling pathway plays an important role in the development of organs, tissue proliferation, differentiation and apoptosis (59). To date, four Notch receptors (Notch1-4) and five Notch ligands (Delta-like 1, 3, 4 and Jagged-1 and 2) have been discovered. Notch signaling is activated when the Notch ligand binds to an adjacent receptor. Upon activation, Notch is cleaved through a cascade of proteolytic cleavages by the metalloproteases, tumor necrosis factor α-converting enzyme and γ-secretase. Gamma secretase complex releases an active fragment called Notch intracellular domain (NICD). NICD translocates to the nucleus and then binds to the transcription factor CSL (CBF1, Suppressor of Hairless, and Lag-1). The CSL-NICD complex recruits a co-activator complex containing p300 and other co-activators leading to the activation of Notch target genes that are important in the regulation of cell growth, proliferation and apoptosis (eg. Akt, Cyclin D1, c-Myc, COX-2, MMP-9, ERK, mTOR, NF-kB, p21, p27, p53, VEGF) (59, 60). Recently, the Notch signaling pathway has been found to directly upregulate Snail-1 and Slug thereby inducing EMT (61, 62). In pancreatic cancer cells, knockdown of Notch-2 or midkine (a downstream target of Notch-2) resulted in EMT inhibition (63).
Several other signaling pathways are also suggested in EMT induction in cancer cells, such as NF-κB (64), growth factor (65), and TNF-α (66). The end result of activation of all these signaling pathways is to induce the expression of EMT transcription factors (Snail, Slug, Zeb-1, Twist), which in turn decreases expression of epithelial markers and increases the expression of mesenchymal markers.
Transcription factors that play important roles in regulating EMT include the Snail family of zinc finger transcription factors, the bHLH transcription factors and the Zeb family transcription factors. Together with other co-repressors and co-activators, these master regulators decrease expression of epithelial markers and increase expression of mesenchymal markers during EMT. The mechanism of such dual regulation of target genes by these transcription factors is only partially understood (67, 68).
Snail transcription factors include Snail-1 (Snail), Snail-2 (Slug), and Snail-3 (Smuc), of which, Snail and Slug activate EMT during development and pathological conditions. All these transcription factors contain highly conserved carboxy terminal Cys2His2 (C2H2) type zinc finger motifs. The Snail1/GFI domain at the amino terminus is required for protein stability and transcriptional repression of target genes (69).
Snail and Slug are key mediators of EMT in pancreatic tumor progression. Moderate to strong Snail expression was found in 78% of pancreatic cancer cases while Slug was expressed in 50% of the cases and at a lower level than Snail (70). Snail expression levels in pancreatic cancers correlated with lymph node invasion and distant metastasis (71). Snail-transfected pancreatic cancer cell lines displayed highly metastatic and invasive abilities in an orthotopic mouse pancreatic cancer model. Snail enabled these pancreatic cancer cell lines to undergo EMT at the invasive front of the tumor (71, 72). Knocking out Snail enhanced sensitivity to gemcitabine, and resulted in increased overall survival in a genetically engineered mouse model of PDAC (73).
Snail acts as a repressor of genes involved in the maintenance of epithelial phenotype (E-cadherin, occludin, claudin, and cytokeratin-18) with the key target being E-cadherin. On the other hand, Snail activates the expression of mesenchymal genes such as vimentin, fibronectin, and N-cadherin. Snail also has been shown to regulate expression of genes involved in cell polarity (Crumbs3, Lgl2 and dlg3) and apoptosis (P53, BID and DFF40) (74). Among these, E-cadherin is the only direct target of Snail (70, 75).
The mechanism of E-cadherin repression by Snail is a complex process and it involves several co-repressor proteins. Epigenetic modification of the chromatin structure at the promoter region of CDH1 gene is the major regulatory event. The C2H2 type zinc finger domain allows sequence-specific binding of Snail to the E-box in the proximal promoter region of the CDH1 gene, which encodes E-cadherin. Upon binding to the E-box region, Snail recruits co-repressor complexes such as histone deacetylases (HDAC)1/2/Sin3 (75) and polycomb repressive complex 2 (PRC-2) (76). HDAC1/2/Sin3 caused deacetylation of H3 and H4 at the CDH1 promoter resulting in decreased expression of E-cadherin (75). The PRC2 complex catalyzes histone 3 lysine 27 trimethylation, which is a hallmark of epigenetically silenced chromatin, resulting in decreased expression of E-cadherin (76).
Pharmacological agents that target Snail have been an attractive strategy to inhibit EMT and cancer cell metastasis. A cobalt (III) Schiff base complexe ([Co(acacen)(NH3)2]+) has been shown to inhibit C2H2 zinc finger transcription factors non-specifically. To enhance the specificity, an E-box DNA oligonucleotide specifically targeting Snail was attached to the acacen equatorial ligand (77). This Co(III)-E-box complex was a potent inhibitor of Snail- mediated transcriptional repression in breast cancer cells and in the neural crest of Xenopus (78). However, this complex has not been tested in pancreatic cancer.
Zeb transcription factors. The zinc-finger-enhancer binding protein (Zeb) family of transcription factors (Zeb 1 and Zeb2) are one of the best studied EMT inducing transcription factors (79). In PDAC patients, both pancreatic cancer cells and tumor associated stroma showed high level of Zeb-1 expression, and Zeb-1 expression was associated with poor prognosis. Inverse correlation between E-cadherin and Zeb-1 expression was observed in patient tumor samples, as well as in pancreatic cancer cell lines (80, 81). Silencing Zeb-1 has been shown to reduce cell migration, tumorigenicity and tumor dissemination (81-83). Zeb-1 has been shown to decrease transcription of key determinants of epithelial differentiation, cell adhesion and cell polarity genes (84). E-cadherin is a well-known direct target of Zeb-1. Zeb-1 decreases E-cadherin expression by recruiting HDAC-1/2 or Switch/sucrose non-fermentable chromatin remodeling protein BRG1 to the promoter region of CDH-1 gene (81, 85).
A negative feedback loop between Zeb-1 and the microRNA-200 family (miR-200c and miR141) has been shown in regulating EMT (86-88). Zeb-1 is a major target of miR-200c and miR-141. On the other hand, Zeb-1 strongly repress the expression of miR-200 family members (89). This suggests that Zeb-1 activates microRNA mediated feed forward loop that stabilizes EMT and promotes invasion of cancer cells. Pancreatic cancer patients with a high level of miR-200c expression had better survival rates than those with a low-level of miR-200c expression. In these patients, there also existed a strong correlation between the levels of miR-200c and E-cadherin expression (87, 90, 91).
Drugs that inhibit Zeb-1 function might have clinical relevance for pancreatic cancer patients. Unfortunately, effective small-molecule inhibitors of Zeb-1 are yet to be identified.
Basic helix loop helix transcription factors (bHLH). Among all the bHLH transcription factors studied, E12, E47, Twist 1 and Twist 2 have been shown to play important roles in EMT (92).
E-12 and E-47 act as a repressor of E-cadherin expression and trigger EMT. The mechanism of E-cadherin repression by E-12/47 is not well understood (93). Inhibition of differentiation 1 might be required for E-47 induced EMT (94, 95).
Twist 1 and Twist 2 are major regulators of EMT during development and pathogenesis (96). Twist expression is either absent or very weak in tissue samples from patients with PDAC. Pancreatic cancer cell lines also (MiaPaCa-2, PANC-1, Capan-1, HPAF-2 and AsPC-1) showed low expression of Twist. However, hypoxic conditions induced Twist expression in MiaPaCa-2, PANC-1, Capan-1 and HPAF-2 cell lines (70). As pancreatic tumors are often under hypoxic conditions (97), Twist may play a role in the invasive behavior of pancreatic tumors.
Twist has differential effects on expression of E-cadherin and N-cadherin. It decreases E-cadherin expression but induces N-cadherin expression. The mechanism of such dual regulation of target gene expression by Twist is not completely understood, but it is known that Twist recruits the Set-8 methyl transferase (a member of the SET domain-containing methyltransferase family) to the promoter region of E-cadherin and N-cadherin and enhances H4K20 monomethylation (98). Recently, it has been showed that Twist interacted with several components of the Mi2/nucleosome remodeling and deacetylase complex to repress the transcription of E-cadherin (99). Twist has also been shown to specifically interact with BRD4, a bromo domain and extra terminal domain protein, to regulate the expression of target genes. Inhibition of BRD4 and Twist interaction by small molecular inhibitors such as JQ1 and MS417 suppressed tumor growth in a few animal models (100).
Pancreatic cancer stem cell
Recent studies have shown that cancer stem cells (CSCs) are the predominant factors responsible for tumor recurrence (101). In 1997 Bonnet and colleagues discovered that a minor subpopulation of acute myeloid leukemia (AML) cells expressing markers of normal hematopoietic stem cells presented the potential to propagate AML in immune deficient mice (102). It was therefore hypothesized that malignancy originated from mutated stem cells that were transformed to generate daughter cancer cells. These tumor-generating subpopulations of cancer cells were later termed stem-like cancer cells, or cancer stem cells (CSCs). Although the biology of CSCs needs to be further understood, the existence of CSCs has been demonstrated in almost all kinds of hematological malignancies and solid tumors (103).
CSCs share a lot of characteristics with normal stem cells, such as asymmetric cell division, where each stem cell generates one daughter cell with stem cell fate (self-renewal) and another daughter cell destined to differentiate (progenitor cell). The self-renewal capacity helps to maintain the number of CSCs within the tumor and its descendent progeny constitute the bulk of the tumor (104). Many pathways (Wnt/β-catenin, Sonic hedgehog, and Notch) that are classically associated with cancer are known to regulate stem cell self-renewal (105).
Another important characteristic shared by normal and cancer stem cells is quiescence. Like the mammalian adult stem cells, CSCs are in a quiescent, non-dividing G0-state, which protect them from chemotherapeutic drugs that are developed to target actively dividing cells (104). Late relapse after initial treatment in malignancies is explained with the existence of dormant CSCs (106).
In addition, CSCs exhibit unique features such as metastasis ability. It was reported that the subset of CXCR4+ CD133+ pancreatic CSCs exhibited strong migratory potential and showed liver metastasis in an orthotropic model system. On the contrast, CXCR4+ CD133- cells representing the non-stem-like cancer cells failed to undergo liver metastasis (107).
The exact relationship between CSCs and metastasis is not clear yet. It might lie in the close association of CSCs with cancer cell EMT. EMT and CSCs share many signaling pathways, such as Wnt/β-catenin and Notch signaling pathway. Induction of EMT generated cells with stem-like properties, either by over expressing Twist or Snail (108), or by inhibiting the expression of E-cadherin using shRNA targeting CDH-1 (109). In a mouse model, it was shown that circulating pancreatic cancer cells maintained a mesenchymal phenotype and exhibited stem cell properties (42). The circulating cancer cells are considered equivalent to stem-like cancer cells. Overexpression of other EMT inducers, such as FoxM1 and Notch-1, also resulted in enrichment of pancreatic CSCs (110).
The mechanism by which CSCs become drug resistant is largely unknown, but it is very likely that ATP binding cassette (ABC) drug transporters are involved. Studies showed that CSCs gain drug resistance partly due to over expression of ABCG2 (BCRP) (111), which pumps cytotoxic drugs out of cancer cells (112). In another study, CD44+ pancreatic CSCs that were expanded during the acquisition of gemcitabine resistance, showed significantly elevated levels of ABCB1 (MDR1). The ABC transporter inhibitor, verapamil, re-sensitized the resistant cells to gemcitabine (113). It is also very likely that CSCs drug resistance is caused by both intrinsic and acquired properties such as quiescence, detoxifying enzymes, DNA repair ability and overexpression of anti-apoptotic proteins (114). Understanding the biology of drug resistance related to CSCs is necessary and would provide clues to design novel therapeutic options.
Several cellular markers or their combinations have been used to identify pancreatic CSCs, include aldehyde dehydrogenase (ALDH) (115, 116), CD133 (117) and the combination of CD133+ CXCR4+ (107). Another commonly used combination of markers are CD44+ CD24+ EpCAM+ , because CD44+ CD24+ EpCAM+ pancreatic cancer cells showed properties of self-renewal and the ability to produce differentiated progeny (118). Other markers such as c-Met (119), doublecortin-like kinase 1 (120), and CD44v6 (121) were also proposed as putative pancreatic CSC markers. Although none of the proposed markers definitively identify a pure population of pancreatic CSCs, these markers are used widely because they provide consistent data for a strong enrichment of pancreatic CSCs.
So far, the gold-standard assay to measure the self-renewal and lineage capacity of CSCs is serial transplantation in animal models. Cells are injected into the orthotopic site of NOD/SCID mice which are then assayed at different time points for tumor formation. Cells isolated from the tumor are xenografted into a second recipient animal to measure the self-renewal capacity. However, this serial transplantation model is costly and time consuming, and is therefore not feasible to be used in drug screening (122). An in vitro spheroid formation assay has been widely used to determine the self-renewal capacity of CSCs. The assay is based on the principle that only CSCs survive in suspension culture with non-stem-like cancer cells die by anoikis. The ability to form several generations of spheres in serial non-adherent passages is related to the self-renewal ability of CSCs. For example, the CD44+CD24+EpCAM+ pancreatic CSCs formed pancreatospheres in vitro, whereas CD44-CD24-EpCAM- cells did not. The CD44+CD24+EpCAM+ pancreatospheres can be passaged multiple times without loss of the tumor sphere-forming capability (123).
The understanding of the molecular mechanism by which CSCs are formed will benefit not only basic research but also clinical cancer therapy. Recent studies have demonstrated that the formation of CSCs can be triggered and maintained by the interplay of multiple cellular signaling pathways, including Notch, Hedgehog, and Wnt/β-catenin (124).
Notch-signaling pathway plays an important role in maintaining CSC population. Pancreatic CSCs expressed considerably higher levels of Notch-1 than the rest of the cancer cell population (125). Notch-1 over expression in pancreatic cancer cell lines induced EMT and increased the formation of pancreatospheres, which are indicative of CSCs (126). Activation of Notch pathway with an exogenous Notch peptide ligand increased the percentage of CSCs (127). However, further investigation is required to elucidate the molecular mechanism that explains how Notch signaling regulates pancreatic CSC self-renewal.
Hedgehog signaling pathway in maintaining CSC population. The Hedgehog (HH) pathway plays a critical role in the processes of embryonic development, it also plays an important role in maintenance of CSCs (128). The HH signaling pathway consists of HH ligands (Sonic HH, SHH; Indian HH, IHH; and Desert HH, DHH), Patched proteins (Patched-1 and 2), Smoothened (Smo), inhibitory complex (Fused, Suppressor of Fused) and the 5-zinc-finger transcription factors, Gli1, Gli2 and Gli3. In the absence of HH ligand, Patched-1 and 2 suppresses Smo. In the presence of HH ligand, inhibition of Smo by Patched is released, providing a signal for the dissociation of Gli transcription factor from the inhibitory complex. Dissociated Gli translocates to the nucleus and regulates the transcription of target genes such as Cyclin D1, N-Myc, p21, Wnt, Patched and Gli itself (129, 130).
Deregulation of HH signaling has been observed in pancreatic cancers (131). Transgenic mice overexpressing SHH in pancreatic epithelium developed PanIN lesions, suggesting that SHH might be an early mediator of pancreatic cancer tumorigenesis (131). Two independent studies showed that pancreatic CSCs had >40-fold higher SHH expression than the non-CSC population (118, 132). Therefore, inhibition of HH signaling might be an alternative therapeutic approach for pancreatic cancer patients.
Wnt/β-catenin signaling pathway in maintaining CSC population. Wnt/β-catenin signaling plays an important role in CSCs of a variety of cancers. For example, constitutively activated β-catenin signaling in stem cells is essential for intestinal neoplasia (133). Wnt-1 overexpression enhanced the sphere formation capacity of the gastric cancer AGS cell line (134). The canonical β-catenin signaling pathway is the most significantly deregulated signaling pathway in glioblastoma stem cells (135), it is also required for self-renewal of leukemia stem cells that were derived from either hematopoietic stem cells or from granulocyte-macrophage progenitors (136). The roles of Wnt/β-catenin signaling in pancreatic CSCs deserve further investigation.
Current and experimental treatment for pancreatic cancer
Current treatment for pancreatic cancer is far from satisfactory. As advancements in molecular and targeted therapies have greatly improved survival of patients with many types of cancers, the treatment outcome for pancreatic cancer has not changed much over the past 30 years. In the last a few decades of the 20th century, five fluoro uracil (5-FU) was the standard of care for pancreatic cancer patients (137). In 1997, a randomized phase 3 study demonstrated a survival benefit for gemcitabine (2′, 2′-difluoro 2′-deoxycytidine) over 5-FU (138). Since then, gemcitabine as mono-treatment has been the standard of care for pancreatic cancer patients. Gemcitabine is an analog of cytosine exhibited distinctive pharmacological properties (with multiple intracellular targets such as DNA polymerase, ribonucleotide reductase, cytidine triphosphate synthetase, deoxycytidylate deaminase) and a wide spectrum of anti-tumor activity (139). However, gemcitabine has little impact on median overall survival for patients with locally advanced or metastatic pancreatic cancer, who comprise the majority of cases (140, 141). Many studies used gemcitabine in combination with other chemotherapeutic drugs intending to improve the median survival in pancreatic cancer patients. However, the improvement has not been satisfactory. Gemcitabine in combination with either capacitance (prodrug of 5-FU, a pyrimidine analog) (142) or platinum drugs (cisplatin, carboplatin) (143, 144) did not result in significant improvement in overall survival. A combination of gemcitabine and nab-paclitaxel (145) improved overall survival, progression free survival and response rate compared to gemcitabine treatment alone. However, peripheral neuropathy and myelosuppression were significantly increased (146).
Surgical resection is the only curative therapy for pancreatic cancer patients with localized disease (147). However, only 15-20% of patients have resectable disease at the time of diagnosis. Patients who undergo surgery (e.g. Whipple procedure) have a perioperative mortality of 4-18% and an additional risk of post operational complications (3, 148, 149). Furthermore, the majority of the patients have locally invasive and micro metastasis at the time of surgery. Therefore, disease recurrence following operation is very high. Adjuvant therapy (5-FU or gemcitabine-based chemoradiation) is specified to decrease the risk of loco-regional and metastatic recurrence (150).
Recently, FOLFIRINOX (oxaliplatin, irinotecan, fluorouracil, and leucovorin), as the first gemcitabine free regimen, has been shown to be more efficient than gemcitabine. The median overall survival was 11.1 months in the FOLFIRNOX group as compared with 6.8 months in the gemcitabine group. Side effects of this new combination regimen were significant, including grade 3 and 4 neutropenia, suggesting a limited use for patients with good performance status (151).
Investigational chemotherapies for pancreatic cancer
Pancreatic cancer is a multifactorial complicated disease. Conventional chemotherapies or radiation therapies often yield disappointing results and are largely ineffective in depleting CSCs, which are responsible for disease recurrence. Therefore, new innovative treatment options need to be tested. As the understanding of pancreatic cancer biology and pathophysiology increases, some novel approaches for targeted therapies are currently under investigation. In the following paragraphs some of the investigational therapies that have potential clinical success for pancreatic cancer patients are summarized.
Targeting K-Ras
K-Ras is an attractive therapeutic target because it is mutated in the vast majority of PDAC cases. An ideal mechanism to prevent K-Ras signaling would be to directly block the GTP-binding site of K-Ras. However, there has not been an effective small-molecule inhibitor identified. Alternatively, other approaches have been investigated to 1) block membrane localization of K-Ras, 2) block Son of Sevenless (SOS)/K-Ras interactions, and 3) block K-Ras downstream effector targets such as PI3K, Raf, MEK 1/2 and Akt (152).
Blocking K-Ras membrane localization. Membrane localization of K-Ras brings it into contact with Ras activating protein to activate downstream signaling (153). Following translation, K-Ras undergoes a lipid modification called farnesylation and/or geranylgeranylation (together referred as prenylation). Phosphodiesterase delta (PDEδ) is a prenyl binding protein and interacts with prenylated K-Ras to aid K-Ras to translocate to the membrane (154, 155). It was hypothesized that inhibition of farnesylation and or geranylgeranylation would prevent K-Ras membrane localization, thus inhibiting its signaling pathway. A number of inhibitors for farnesyl transferase (FTIs) and geranylgeranylation transferase (GGTIs) were synthesized and tested for their anti-tumor activity (156-160). At least six FTIs have been tested in clinical trials, but unfortunately have predominantly proven unsuccessful for K-Ras driven tumors (161, 162). A geranylgeranylation transferase inhibitor, GGTI-2418 is currently in phase I clinical evaluation (http://www.prnewswire.com/news-releases/first-patient-dosed-in-phase-i-clinical-trial-of-tigris-pharmaceuticals-ggti-2418-61683932.html). The combination of FTI and GGTI induced greater apoptotic response in cancer cells than a single agent alone, but high toxicities limited their clinical use (163). Deltarasin, inhibited PDEδ and K-Ras interactions and K-Ras membrane localization, suppressed in vitro and in vivo proliferation of PDAC cells (164). Salirasib (S-trans, trans-farnesylthiosalycilic acid) dislodged K-Ras from the membrane thereby promoting K-Ras degradation. In a patient derived xenograft model, salirasib showed a wide range of activity and showed a heightened tumor response when combined with gemcitabine (165). Salirasib is under clinical investigation (165).
Blocking SOS/K-Ras interactions. At the membrane, K-Ras is activated by SOS (guanine nucleotide exchange factor), which aids in K-Ras binding to GTP. Blocking these interactions potentially inhibits K-Ras signaling pathway (166). Using an NMR based fragment-screening approach, a small molecule, 4, 6-dichloro-2-methyl-3-aminoethyl-indole was identified that blocked the interaction between K-Ras and SOS and inhibited the nucleotide exchange (167). Several other compounds were also discovered which bound to GDP-bound K-RasG12D and thus inhibited SOS catalyzed nucleotide exchange (168). Anti-cancer activities of these compounds need to be investigated in preclinical and clinical studies.
Blocking K-Ras downstream effector targets. Effector pathways are activated downstream of K-Ras in a context- and tissue-specific manner. MAPK and PI3K-Akt signaling pathways are the two most commonly activated signaling pathways in pancreatic cancer. Efforts have been made to develop pharmacological agents targeting MAPK and PI3K-Akt signaling pathways. The MAPK pathway consists of a kinase cascade, where K-Ras activates Raf kinases, which in turn activate MEK1/2 (166). In preclinical models, MEK inhibitor PD325901 reduced tumor burden and prolonged survival time and showed a synergistic effect with Akt inhibitor GSK690693 (169). In a patient derived xenograft model, another MEK inhibitor, trametinib (GSK1120212) reduced tumor mass, and ddding an EGFR/HER2 inhibitor lapatinib, significantly enhanced trametinib effects (170). PI3K and Akt inhibitors were also tested in preclinical models and showed potent anti-cancer activity (11, 169). These results suggest that blocking K-Ras downstream targets is a potential therapeutic option for pancreatic cancer patients who harbor K-Ras mutations.
High-throughput screening assays have been performed using cells harboring K-Ras mutation and its isogenic cells without K-Ras mutation, in order to discover compounds that specifically inhibit K-Ras mutant cells. Several such compounds were identified, including sulfinyl cytidine and its derivative triphenyltetrazolium (171), oncrasin-1 (172), tolperisone and its derivative lanperisone (173), and SLI501 (174). These compounds are under preclinical and clinical testing.
Histone deacetylase (HDAC) inhibitors
A growing body of literature suggests that deregulation of histone deacetylases (HDACs) could contribute to pancreatic cancer development and progression (175-178). HDACs play critical roles in the epigenetic regulation of gene expression by catalyzing the removal of acetyl groups from histones, causing compaction of the DNA/histone complex. This compaction blocks gene transcription and inhibits differentiation (179, 180). In eukaryotic cells, 18 different HDACs have been identified and are classified into 4 groups based on their homology to yeast proteins. Class I includes HDAC 1, 2, 3, and 8 and are exclusively located in the nucleus. Class IIa includes HDAC 4, 5, 7 and 9 and are localized to the cytoplasm. HDAC 6 and 10 belong to class IIb and HDAC 11 is classified in class IV. HDAC 11 is localized in both cytoplasm and nucleus. Classes I, II, and IV HDACs are Zn-dependent enzymes. Class III HDACs include sirtuins, which are NAD+ dependent enzymes and have homology to yeast Sir2 (180-182). In pancreatic cancer, overexpression of HDAC 1, (177), HDAC 2 (175), HDAC7 (176), and HDAC 3 (178) were observed. As described before, HDACs also involve in Snail mediated EMT process. Thus, targeting HDACs by inhibitors could be a promising strategy for pancreatic cancer treatment.
To date, more than 15 HDAC inhibitors have been tested in preclinical and early clinical studies. Based on the chemical structure, HDAC inhibitors are classified in into 4 different classes: hydroxamates (e.g. suberanilohydroxamic acid (SAHA) and panobinostat), cyclic peptides (e.g. romidepsin), aliphatic acids (e.g. sodium butyrate, valproic acid and phenyl butyrate) and benzamides (e.g. entinostat or known as MS-275 and mocetinostat) (180). The Food and Drug Administration approved SAHA for the treatment of cutaneous T-Cell Lymphoma (CTCL). In animal models, SAHA in combination with bortezomib (proteasome inhibitor) or Zebularine (nucleoside analog of cytidine) reduced pancreatic tumor weight with minimal noted toxicity (183, 184). SAHA also reduced migration, colony formation and sphere formation ability of pancreatic CSCs. Specifically, SAHA inhibited the expression of Zeb-1, Snail, and Slug, suggesting that SAHA inhibited EMT in pancreatic CSCs. Furthermore, SAHA inhibited the Notch signaling pathway by upregulating miR-134 (185). Other HDAC inhibitors like MS-275, TSA and FK228 showed potent anti-tumor effects both in vitro and in vivo (186). However, all these HDAC inhibitors so far showed limited to no efficacy in clinical trials in solid malignancies including PDAC, either alone or in combination with other chemotherapeutic drugs (187-190).
The lack of effects might partially come from the non-specificity of current HDAC inhibitors. Majority of the HDAC inhibitors in clinical trials inhibit all HDAC isoforms nonspecifically (pan inhibitors). Such nonspecific inhibition leads to many toxic side effects that limited the dose that can apply (191). Selective HDAC inhibitors, which affect a single HDAC isoform, would be ideal chemical tools to elucidate the functions of each individual HDAC isoform. Also, development of class-selective HDAC inhibitors might provide more effective HDAC inhibitors than the pan HDAC inhibitors.
Targeting pancreatic CSCs
Reactivation of developmental signaling pathways (Notch, SHH) is involved in the formation of CSCs (192). Inhibitors of these signaling pathways could be valuable tools to target CSCs.
Hedgehog (HH) inhibitors. Several small-molecule HH inhibitors are under preclinical and clinical development (193). For example, the HH inhibitor cyclopamine inhibited pancreatic cancer cell EMT, CSCs, and reduced metastasis in an orthotopic xenograft mouse model (194). Cyclopamine down regulated the expression of CSC markers CD44 and CD133 in gemcitabine resistant pancreatic cancer cells and restored gemcitabine sensitivity (195). In combination with the mTOR inhibitor, rapamycin, cyclopamine reduced the number of pancreatic CSCs to undetectable levels in vitro and in vivo (196). IPI-269609 and GDC-0449, two other small-molecule inhibitors of the HH signaling pathway, also effectively depleted pancreatic CSCs (197, 198). A recent study found that the anti-malarial agent chloroquine (CQ) significantly decreased pancreatic CSCs by inhibiting the HH signaling pathway (199). CQ in combination with gemcitabine improved the overall survival of mice bearing PDAC patient derived xenografts.
However, these preclinical results were not recapitulated in clinical trials. A single arm pilot study in 25 metastatic PDAC patients was conducted to study the effect of GDC-0449 plus gemcitabine. The treatment resulted in inhibition of the HH signaling pathway, without significant changes in CSCs, fibrosis, progression free survival and overall survival. However, grade ≥3 toxicities in 56% of patients were observed. This pilot study concluded that GDC-0449 and gemcitabine were not superior to gemcitabine alone in the treatment of metastatic pancreatic cancer (200). Similarly, a double-blind, randomized phase II study, IPI-926 plus gemcitabine or gemcitabine plus placebo, showed no improvement in overall survival (http://www.businesswire.com/news/home/20120127005146/en/Infinity-Reports-Update-Phase-2-Study-Saridegib#.VXGcwcvwvcs). A phase II GDC-0449 plus gemcitabine or gemcitabine plus placebo study also showed no statistical improvement in progression free survival and overall survival (https://clinicaltrials.gov identifier: NCT01064622).
Reasons for the failure of HH inhibition in PDAC patients were explored using a genetically engineered mouse model (GEMM) (201). Unexpectedly, in this GEMM, loss or pharmacological inhibition of SHH were associated with decreased survival, higher frequency of gross metastasis. SHH deficient tumors had reduced stromal content, and increased blood vessel density (201). Therefore, SHH inhibitors warned for further clinical use in PDAC patients.
Notch inhibitors. Gamma secretase activates the Notch signaling pathway. Hence γ-secretase inhibitors (GSI) are attractive inhibitors for CSCs. The GSI MRK-003 effectively inhibited intratumoral Notch signaling and prolonged survival of tumor bearing mice when combined with gemcitabine (202). Pretreatment of PDAC cells with MRK-003 in cell culture significantly inhibited pancreatic CSCs and subsequent tumor formation in immunocompromised mice (203). PF-03084014, a selective GSI, was also shown to induce pancreatic tumor regression by depleting CSCs (204). Inhibition of Notch singling pathway using a GSI RO4929097 or Hes1 ShRNA reduced both in vitro sphere formation and in vivo tumor growth of orthtopic pancreatic tumors (127). However, a Phase II study was completed recently using RO4929097 in previously treated metastatic pancreatic cancer patients and showed no potential advantage over gemcitabine (https://clinicaltrials.gov identifier: NCT01232829). PF-03084014 recently entered into a phase I/II study in combination with gemcitabine and Nab-paclitaxel in patients with previously untreated metastatic PDAC (https://clinicaltrials.gov identifier: NCT02109445). A phase III study of gemcitabine plus Nab-paclitaxel combination showed slight improvement in survival (8.5 months, statistically significant) compared to gemcitabine (6.7 months) (146). Adding PF-03084014 to this combination is in hope to further improve the survival.
Other potential approaches to target pancreatic CSCs. Targeting the cell-surface antigens that are characteristic to CSCs is an attractive strategy to eliminate pancreatic CSCs. Specific monoclonal antibody targeting CD44, eliminated leukemic stem cells by inducing terminal differentiation (205). Inhibition of c-Met, a marker of highly tumorigenic CSCs has been found to inhibit tumor growth and metastasis (119). A bispecific antibody that recognizes both epithelial surface antigen (EpCAM) and CD3 has been shown to eliminate pancreatic CSCs (206). Monoclonal antibodies targeting CD44, EpCAM and c-Met needs to be tested in clinical trials.
A monoclonal antibody NPC-1C (NEO-102) has been recently developed targeting MUC5ac which is a member of the mucins family and is a highly expressed prognostic biomarker in pancreatic cancer (207). MUC5ac is upregulated by the GLI1, a downstream transcription factor in the Hedgehog signaling pathway. MUC5ac upregulation leads to disruption in the function of E-cadherin and β-catenin, and thus contributes to pancreatic carcinogenesis (208). NPC-1C is a chimeric immunoglobulin that was derived from a Tumor Associated Antigen (TAA) based vaccine that was previously tested in a Phase I/II clinical trial in colon cancer (209). In a phase I study, NPC-1C showed activity in patients with refractory pancreatic cancer and significantly prolonged overall survival in two patients who failed standard therapy (210). An improved formulation of NPC-1C, NEO-102, is been tested in phase II studies in refractory pancreatic cancer, either given alone (https://clinicaltrials.gov identifier: NCT01040000), or in combination with gemcitabine and nab-paclitaxel in refractory pancreatic cancer (211, 212) (https://clinicaltrials.gov identifier: NCT01834235).
The anti-cancer agents from diet or natural plants (e.g. genistein, curcumin, resveratrol) have been found to inhibit pancreatic CSCs self-renewal through modulation of important signaling pathways (213). Genistein, one of the isoflavones found in soybeans inhibited the pancreatospheres in vitro by down regulating the Notch signaling pathway (126, 214). Genistein also inhibited EMT, which is highly associated with CSCs (110). Curcumin and its synthetic analog, 3, 4-difluoro-benzo-curcumin, inhibited the sphere formation ability of pancreatic cancer cell lines and attenuated the pancreatic CSC markers CD44 and EpCAM (215). Curcumin was reported to decrease the CSC population by targeting the histone methyl transferase, enhancer of zeste homolog 2 miRNA regulatory circuit (216). Resveratrol inhibited the self-renewal capacity of pancreatic CSCs derived from K-Ras transgenic mice and from human primary tumors. Moreover, resveratrol decreased the expression of pluripotency maintaining factors such a Sox-2, c-Myc, Nanog and Oct-4 in pancreatic CSCs and induced CSCs apoptosis through activation of caspase-3/7 (217).
Clinical studies have been carried out on some of these dietary compounds, however, so far showed little benefit over gemcitabine treatment. A phase II trials of curcumin in patients with advanced pancreatic cancer showed good tolerance and biological activity in 2 out of 21 patients. One patient had stable disease for >18 months. Another patient had marked tumor regression (73%) (218). Two other studies investigated curcumin in combination with gemcitabine in patients with advanced pancreatic cancer. The combination did not provide survival advantage over gemcitabine (median survival 5.3 and 6 months in the two trials, respectively) (219, 220). A phase II study adding genistein to gemcitabine and erlotinib combination in advanced pancreatic cancer patients, also resulted in no significant improvement (221, 222). As for resveratrol, studies so far indicated it is a promising cancer preventing agent instead of chemotherapeutic agent (223).
Taken together, many efforts have been made targeting various molecular pathways using either pharmacological agents or nutraceuticals to inhibit pancreatic cancer growth, metastasis and CSCs. Despite many preclinical advancements, it remains a huge challenge to find a clinically effective therapeutic agent.
Discussion
Though the oncology community has made significant progress in understanding pancreatic cancer biology, there are no effective therapies available to date, because of its multifactorial nature. At the cellular level, neoplastic cells harbor activating K-Ras mutation at high frequencies in pancreatic cancer patients. Pharmacological agents targeting K-Ras for pancreatic cancer is currently an active area of research (224). However, it is possible that blocking K-Ras function may not be sufficient to treat pancreatic cancer, because these cancer cells may adapt to K-Ras inhibition either by overexpressing or mutating genes in downstream pathways of K-Ras. Several other growth factor signaling pathways also impact disease progression and pathogenesis, which might drive growth and cell survival in the absence of K-Ras signaling (225). Layered on top of oncogenic mutation is a host of tumor suppressor genes inactivation (226). Developing effective anticancer regimens against changes of tumor suppressor pathways has emerged to be a promising area of research (41). Novel approaches need to be investigated with an open mind, as new knowledge and concepts in pancreatic cancer biology develop.
CSCs is a relatively new concept in cancer biology. A growing body of literature suggests that pancreatic tumors are enriched with CSCs (227). CSCs are frequently associated with metastatic foci and chemo-resistance and are increasingly linked to an EMT phenotype (108, 228). The resistance to chemotherapy of this subpopulation is responsible for tumor recurrence. EMT in cancer cells is triggered by several different signaling pathways, which then activate a set of transcription factors to induce EMT (67, 75). Therefore, inhibitors of EMT signaling pathways or the transcription factors hold the promise to inhibit pancreatic cancer metastasis and CSCs. Efforts have been directed in looking for agents that selectively targeting CSCs.
Immunotherapies have recently shown significant results in cancer treatment. Successful examples include anti-PD1/PD-L1 antibodies in treating advanced melanoma (229), and CAR-T cell therapy in acute lymphoblastic leukemia (ALL) (230). In pancreatic cancer, immunotherapy is a largely unexplored landscape; however, an area holds great potential (231). Some promising therapies such as the NPC-1C antibody are under clinical testing. Efforts should be directed in identification and selection of antigens specific to pancreatic cancer, and to attenuation of the immunosuppressive microenvironment of pancreatic cancer. Appropriate selection of target antigens and combination treatment are critical in enhancing efficacy and lowering treatment related toxicities.
As our understanding for pancreatic cancer biology and cancer stem cells keep growing, more clinically relevant models will be established that serve as better tools for development of drugs against pancreatic cancer. Novel regimens that are promising in preclinical tests should be investigated with an open mind. These agents , or in combination with standard chemotherapy, would eliminate both the bulk of cancer cell population, and the stem-like cancer cells, therefore achieve long-term tumor regression and eventually cure of the disease.
Competing Interests
The authors have declared that no competing interest exists.
References
1. American Cancer Society. Cancer Facts and Figures 2016. 2016.
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5-29
3. Hezel AF, Kimmelman AC, Stanger BZ, Bardeesy N, Depinho RA. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2006;20:1218-1249
4. Distler M, Aust D, Weitz J, Pilarsky C, Grutzmann R. Precursor lesions for sporadic pancreatic cancer: PanIN, IPMN, and MCN. Biomed Res Int. 2014;2014:474905
5. Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P, Johns AL. et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518:495-501
6. Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, Mankoo P. et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801-1806
7. Kanda M, Matthaei H, Wu J, Hong SM, Yu J, Borges M, Hruban RH. et al. Presence of somatic mutations in most early-stage pancreatic intraepithelial neoplasia. Gastroenterology. 2012;142:730-733 e739
8. Collins MA, Bednar F, Zhang Y, Brisset JC, Galban S, Galban CJ, Rakshit S. et al. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J Clin Invest. 2012;122:639-653
9. Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003;3:11-22
10. Herreros-Villanueva M, Hijona E, Cosme A, Bujanda L. Mouse models of pancreatic cancer. World J Gastroenterol. 2012;18:1286-1294
11. Eser S, Reiff N, Messer M, Seidler B, Gottschalk K, Dobler M, Hieber M. et al. Selective requirement of PI3K/PDK1 signaling for Kras oncogene-driven pancreatic cell plasticity and cancer. Cancer Cell. 2013;23:406-420
12. Lim KH, Baines AT, Fiordalisi JJ, Shipitsin M, Feig LA, Cox AD, Der CJ. et al. Activation of RalA is critical for Ras-induced tumorigenesis of human cells. Cancer Cell. 2005;7:533-545
13. Lim KH, O'Hayer K, Adam SJ, Kendall SD, Campbell PM, Der CJ, Counter CM. Divergent roles for RalA and RalB in malignant growth of human pancreatic carcinoma cells. Curr Biol. 2006;16:2385-2394
14. Maitra A, Kern SE, Hruban RH. Molecular pathogenesis of pancreatic cancer. Best Pract Res Clin Gastroenterol. 2006;20:211-226
15. Aguirre AJ, Bardeesy N, Sinha M, Lopez L, Tuveson DA, Horner J, Redston MS. et al. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev. 2003;17:3112-3126
16. Collado M, Gil J, Efeyan A, Guerra C, Schuhmacher AJ, Barradas M, Benguria A. et al. Tumour biology: senescence in premalignant tumours. Nature. 2005;436:642
17. Redston MS, Caldas C, Seymour AB, Hruban RH, da Costa L, Yeo CJ, Kern SE. p53 mutations in pancreatic carcinoma and evidence of common involvement of homocopolymer tracts in DNA microdeletions. Cancer Res. 1994;54:3025-3033
18. Boschman CR, Stryker S, Reddy JK, Rao MS. Expression of p53 protein in precursor lesions and adenocarcinoma of human pancreas. Am J Pathol. 1994;145:1291-1295
19. Hahn SA, Schutte M, Hoque AT, Moskaluk CA, da Costa LT, Rozenblum E, Weinstein CL. et al. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science. 1996;271:350-353
20. Siegel PM, Massague J. Cytostatic and apoptotic actions of TGF-beta in homeostasis and cancer. Nat Rev Cancer. 2003;3:807-821
21. Wilentz RE, Iacobuzio-Donahue CA, Argani P, McCarthy DM, Parsons JL, Yeo CJ, Kern SE. et al. Loss of expression of Dpc4 in pancreatic intraepithelial neoplasia: evidence that DPC4 inactivation occurs late in neoplastic progression. Cancer Res. 2000;60:2002-2006
22. Tascilar M, Skinner HG, Rosty C, Sohn T, Wilentz RE, Offerhaus GJ, Adsay V. et al. The SMAD4 protein and prognosis of pancreatic ductal adenocarcinoma. Clin Cancer Res. 2001;7:4115-4121
23. Korc M. Role of growth factors in pancreatic cancer. Surg Oncol Clin N Am. 1998;7:25-41
24. Balaz P, Friess H, Buchler MW. Growth factors in pancreatic health and disease. Pancreatology. 2001;1:343-355
25. Troiani T, Martinelli E, Capasso A, Morgillo F, Orditura M, De Vita F, Ciardiello F. Targeting EGFR in pancreatic cancer treatment. Curr Drug Targets. 2012;13:802-810
26. Tobita K, Kijima H, Dowaki S, Kashiwagi H, Ohtani Y, Oida Y, Yamazaki H. et al. Epidermal growth factor receptor expression in human pancreatic cancer: Significance for liver metastasis. Int J Mol Med. 2003;11:305-309
27. Li J, Kleeff J, Giese N, Buchler MW, Korc M, Friess H. Gefitinib ('Iressa', ZD1839), a selective epidermal growth factor receptor tyrosine kinase inhibitor, inhibits pancreatic cancer cell growth, invasion, and colony formation. Int J Oncol. 2004;25:203-210
28. Bruns CJ, Solorzano CC, Harbison MT, Ozawa S, Tsan R, Fan D, Abbruzzese J. et al. Blockade of the epidermal growth factor receptor signaling by a novel tyrosine kinase inhibitor leads to apoptosis of endothelial cells and therapy of human pancreatic carcinoma. Cancer Res. 2000;60:2926-2935
29. Moschos SJ, Mantzoros CS. The role of the IGF system in cancer: from basic to clinical studies and clinical applications. Oncology. 2002;63:317-332
30. Baserga R, Hongo A, Rubini M, Prisco M, Valentinis B. The IGF-I receptor in cell growth, transformation and apoptosis. Biochim Biophys Acta. 1997;1332:F105-126
31. Valsecchi ME, McDonald M, Brody JR, Hyslop T, Freydin B, Yeo CJ, Solomides C. et al. Epidermal growth factor receptor and insulinlike growth factor 1 receptor expression predict poor survival in pancreatic ductal adenocarcinoma. Cancer. 2012;118:3484-3493
32. Bergmann U, Funatomi H, Yokoyama M, Beger HG, Korc M. Insulin-like growth factor I overexpression in human pancreatic cancer: evidence for autocrine and paracrine roles. Cancer Res. 1995;55:2007-2011
33. Rieder S, Michalski CW, Friess H, Kleeff J. Insulin-like growth factor signaling as a therapeutic target in pancreatic cancer. Anticancer Agents Med Chem. 2011;11:427-433
34. Nowak NJ, Gaile D, Conroy JM, McQuaid D, Cowell J, Carter R, Goggins MG. et al. Genome-wide aberrations in pancreatic adenocarcinoma. Cancer Genet Cytogenet. 2005;161:36-50
35. Zhang H, Hylander BL, LeVea C, Repasky EA, Straubinger RM, Adjei AA, Ma WW. Enhanced FGFR signalling predisposes pancreatic cancer to the effect of a potent FGFR inhibitor in preclinical models. Br J Cancer. 2014;110:320-329
36. Olsson AK, Dimberg A, Kreuger J, Claesson-Welsh L. VEGF receptor signalling - in control of vascular function. Nat Rev Mol Cell Biol. 2006;7:359-371
37. Tsuzuki Y, Mouta Carreira C, Bockhorn M, Xu L, Jain RK, Fukumura D. Pancreas microenvironment promotes VEGF expression and tumor growth: novel window models for pancreatic tumor angiogenesis and microcirculation. Lab Invest. 2001;81:1439-1451
38. Seo Y, Baba H, Fukuda T, Takashima M, Sugimachi K. High expression of vascular endothelial growth factor is associated with liver metastasis and a poor prognosis for patients with ductal pancreatic adenocarcinoma. Cancer. 2000;88:2239-2245
39. Hotz HG, Reber HA, Hotz B, Sanghavi PC, Yu T, Foitzik T, Buhr HJ. et al. Angiogenesis inhibitor TNP-470 reduces human pancreatic cancer growth. J Gastrointest Surg. 2001;5:131-138
40. Solorzano CC, Baker CH, Bruns CJ, Killion JJ, Ellis LM, Wood J, Fidler IJ. Inhibition of growth and metastasis of human pancreatic cancer growing in nude mice by PTK 787/ZK222584, an inhibitor of the vascular endothelial growth factor receptor tyrosine kinases. Cancer Biother Radiopharm. 2001;16:359-370
41. Guo XE, Ngo B, Modrek AS, Lee WH. Targeting tumor suppressor networks for cancer therapeutics. Curr Drug Targets. 2014;15:2-16
42. Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, McAllister F, Reichert M. et al. EMT and dissemination precede pancreatic tumor formation. Cell. 2012;148:349-361
43. Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420-1428
44. Huber MA, Kraut N, Beug H. Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr Opin Cell Biol. 2005;17:548-558
45. Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2:442-454
46. Zeisberg M, Neilson EG. Biomarkers for epithelial-mesenchymal transitions. J Clin Invest. 2009;119:1429-1437
47. Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. 2006;7:131-142
48. Ellenrieder V, Hendler SF, Boeck W, Seufferlein T, Menke A, Ruhland C, Adler G. et al. Transforming growth factor beta1 treatment leads to an epithelial-mesenchymal transdifferentiation of pancreatic cancer cells requiring extracellular signal-regulated kinase 2 activation. Cancer Res. 2001;61:4222-4228
49. Moustakas A, Heldin CH. Signaling networks guiding epithelial-mesenchymal transitions during embryogenesis and cancer progression. Cancer Sci. 2007;98:1512-1520
50. Heldin CH, Vanlandewijck M, Moustakas A. Regulation of EMT by TGFbeta in cancer. FEBS Lett. 2012;586:1959-1970
51. Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577-584
52. Levy L, Hill CS. Smad4 dependency defines two classes of transforming growth factor {beta} (TGF-{beta}) target genes and distinguishes TGF-{beta}-induced epithelial-mesenchymal transition from its antiproliferative and migratory responses. Mol Cell Biol. 2005;25:8108-8125
53. Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006;127:469-480
54. Gavert N, Ben-Ze'ev A. Epithelial-mesenchymal transition and the invasive potential of tumors. Trends Mol Med. 2008;14:199-209
55. Zhou BP, Deng J, Xia W, Xu J, Li YM, Gunduz M, Hung MC. Dual regulation of Snail by GSK-3beta-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat Cell Biol. 2004;6:931-940
56. Xu W, Wang Z, Zhang W, Qian K, Li H, Kong D, Li Y. et al. Mutated K-ras activates CDK8 to stimulate the epithelial-to-mesenchymal transition in pancreatic cancer in part via the Wnt/beta-catenin signaling pathway. Cancer Lett. 2015;356:613-627
57. Yee DS, Tang Y, Li X, Liu Z, Guo Y, Ghaffar S, McQueen P. et al. The Wnt inhibitory factor 1 restoration in prostate cancer cells was associated with reduced tumor growth, decreased capacity of cell migration and invasion and a reversal of epithelial to mesenchymal transition. Mol Cancer. 2010;9:162
58. Zhao JH, Luo Y, Jiang YG, He DL, Wu CT. Knockdown of beta-Catenin through shRNA cause a reversal of EMT and metastatic phenotypes induced by HIF-1alpha. Cancer Invest. 2011;29:377-382
59. Guruharsha KG, Kankel MW, Artavanis-Tsakonas S. The Notch signalling system: recent insights into the complexity of a conserved pathway. Nat Rev Genet. 2012;13:654-666
60. Wang Z, Zhang Y, Li Y, Banerjee S, Liao J, Sarkar FH. Down-regulation of Notch-1 contributes to cell growth inhibition and apoptosis in pancreatic cancer cells. Mol Cancer Ther. 2006;5:483-493
61. Wang Z, Li Y, Kong D, Sarkar FH. The role of Notch signaling pathway in epithelial-mesenchymal transition (EMT) during development and tumor aggressiveness. Curr Drug Targets. 2010;11:745-751
62. Xie M, Zhang L, He CS, Xu F, Liu JL, Hu ZH, Zhao LP. et al. Activation of Notch-1 enhances epithelial-mesenchymal transition in gefitinib-acquired resistant lung cancer cells. J Cell Biochem. 2012;113:1501-1513
63. Gungor C, Zander H, Effenberger KE, Vashist YK, Kalinina T, Izbicki JR, Yekebas E. et al. Notch signaling activated by replication stress-induced expression of midkine drives epithelial-mesenchymal transition and chemoresistance in pancreatic cancer. Cancer Res. 2011;71:5009-5019
64. Chua HL, Bhat-Nakshatri P, Clare SE, Morimiya A, Badve S, Nakshatri H. NF-kappaB represses E-cadherin expression and enhances epithelial to mesenchymal transition of mammary epithelial cells: potential involvement of ZEB-1 and ZEB-2. Oncogene. 2007;26:711-724
65. Agbunag C, Bar-Sagi D. Oncogenic K-ras drives cell cycle progression and phenotypic conversion of primary pancreatic duct epithelial cells. Cancer Res. 2004;64:5659-5663
66. Ho MY, Tang SJ, Chuang MJ, Cha TL, Li JY, Sun GH, Sun KH. TNF-alpha induces epithelial-mesenchymal transition of renal cell carcinoma cells via a GSK3beta-dependent mechanism. Mol Cancer Res. 2012;10:1109-1119
67. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15:178-196
68. Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer. 2007;7:415-428
69. Nieto MA. The snail superfamily of zinc-finger transcription factors. Nat Rev Mol Cell Biol. 2002;3:155-166
70. Hotz B, Arndt M, Dullat S, Bhargava S, Buhr HJ, Hotz HG. Epithelial to mesenchymal transition: expression of the regulators snail, slug, and twist in pancreatic cancer. Clin Cancer Res. 2007;13:4769-4776
71. Yin T, Wang C, Liu T, Zhao G, Zha Y, Yang M. Expression of snail in pancreatic cancer promotes metastasis and chemoresistance. J Surg Res. 2007;141:196-203
72. Nishioka R, Itoh S, Gui T, Gai Z, Oikawa K, Kawai M, Tani M. et al. SNAIL induces epithelial-to-mesenchymal transition in a human pancreatic cancer cell line (BxPC3) and promotes distant metastasis and invasiveness in vivo. Exp Mol Pathol. 2010;89:149-157
73. Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H, Wu CC. et al. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature. 2015;527:525-530
74. Wu Y, Zhou BP. Snail: More than EMT. Cell Adh Migr. 2010;4:199-203
75. Peinado H, Ballestar E, Esteller M, Cano A. Snail mediates E-cadherin repression by the recruitment of the Sin3A/histone deacetylase 1 (HDAC1)/HDAC2 complex. Mol Cell Biol. 2004;24:306-319
76. Herranz N, Pasini D, Diaz VM, Franci C, Gutierrez A, Dave N, Escriva M. et al. Polycomb complex 2 is required for E-cadherin repression by the Snail1 transcription factor. Mol Cell Biol. 2008;28:4772-4781
77. Heffern MC, Kurutz JW, Meade TJ. Spectroscopic elucidation of the inhibitory mechanism of Cys2His2 zinc finger transcription factors by cobalt(III) Schiff base complexes. Chemistry. 2013;19:17043-17053
78. Harney AS, Meade TJ, LaBonne C. Targeted inactivation of Snail family EMT regulatory factors by a Co(III)-Ebox conjugate. PLoS One. 2012;7:e32318
79. Vandewalle C, Van Roy F, Berx G. The role of the ZEB family of transcription factors in development and disease. Cell Mol Life Sci. 2009;66:773-787
80. Bronsert P, Kohler I, Timme S, Kiefer S, Werner M, Schilling O, Vashist Y. et al. Prognostic significance of Zinc finger E-box binding homeobox 1 (ZEB1) expression in cancer cells and cancer-associated fibroblasts in pancreatic head cancer. Surgery. 2014;156:97-108
81. Aghdassi A, Sendler M, Guenther A, Mayerle J, Behn CO, Heidecke CD, Friess H. et al. Recruitment of histone deacetylases HDAC1 and HDAC2 by the transcriptional repressor ZEB1 downregulates E-cadherin expression in pancreatic cancer. Gut. 2012;61:439-448
82. Arumugam T, Ramachandran V, Fournier KF, Wang H, Marquis L, Abbruzzese JL, Gallick GE. et al. Epithelial to mesenchymal transition contributes to drug resistance in pancreatic cancer. Cancer Res. 2009;69:5820-5828
83. Wellner U, Schubert J, Burk UC, Schmalhofer O, Zhu F, Sonntag A, Waldvogel B. et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat Cell Biol. 2009;11:1487-1495
84. Dong P, Kaneuchi M, Watari H, Sudo S, Sakuragi N. MicroRNA-106b modulates epithelial-mesenchymal transition by targeting TWIST1 in invasive endometrial cancer cell lines. Mol Carcinog. 2014;53:349-359
85. Sanchez-Tillo E, Lazaro A, Torrent R, Cuatrecasas M, Vaquero EC, Castells A, Engel P. et al. ZEB1 represses E-cadherin and induces an EMT by recruiting the SWI/SNF chromatin-remodeling protein BRG1. Oncogene. 2010;29:3490-3500
86. Bracken CP, Gregory PA, Kolesnikoff N, Bert AG, Wang J, Shannon MF, Goodall GJ. A double-negative feedback loop between ZEB1-SIP1 and the microRNA-200 family regulates epithelial-mesenchymal transition. Cancer Res. 2008;68:7846-7854
87. Burk U, Schubert J, Wellner U, Schmalhofer O, Vincan E, Spaderna S, Brabletz T. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 2008;9:582-589
88. Korpal M, Lee ES, Hu G, Kang Y. The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J Biol Chem. 2008;283:14910-14914
89. Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, Vadas MA. et al. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol. 2008;10:593-601
90. Diaz-Lopez A, Moreno-Bueno G, Cano A. Role of microRNA in epithelial to mesenchymal transition and metastasis and clinical perspectives. Cancer Manag Res. 2014;6:205-216
91. Yu J, Ohuchida K, Mizumoto K, Sato N, Kayashima T, Fujita H, Nakata K. et al. MicroRNA, hsa-miR-200c, is an independent prognostic factor in pancreatic cancer and its upregulation inhibits pancreatic cancer invasion but increases cell proliferation. Mol Cancer. 2010;9:169
92. Teng Y, Li X. The roles of HLH transcription factors in epithelial mesenchymal transition and multiple molecular mechanisms. Clin Exp Metastasis. 2014;31:367-377
93. Perez-Moreno MA, Locascio A, Rodrigo I, Dhondt G, Portillo F, Nieto MA, Cano A. A new role for E12/E47 in the repression of E-cadherin expression and epithelial-mesenchymal transitions. J Biol Chem. 2001;276:27424-27431
94. Cubillo E, Diaz-Lopez A, Cuevas EP, Moreno-Bueno G, Peinado H, Montes A, Santos V. et al. E47 and Id1 interplay in epithelial-mesenchymal transition. PLoS One. 2013;8:e59948
95. Kondo M, Cubillo E, Tobiume K, Shirakihara T, Fukuda N, Suzuki H, Shimizu K. et al. A role for Id in the regulation of TGF-beta-induced epithelial-mesenchymal transdifferentiation. Cell Death Differ. 2004;11:1092-1101
96. Ansieau S, Morel AP, Hinkal G, Bastid J, Puisieux A. TWISTing an embryonic transcription factor into an oncoprotein. Oncogene. 2010;29:3173-3184
97. Buchler P, Reber HA, Lavey RS, Tomlinson J, Buchler MW, Friess H, Hines OJ. Tumor hypoxia correlates with metastatic tumor growth of pancreatic cancer in an orthotopic murine model. J Surg Res. 2004;120:295-303
98. Yang F, Sun L, Li Q, Han X, Lei L, Zhang H, Shang Y. SET8 promotes epithelial-mesenchymal transition and confers TWIST dual transcriptional activities. EMBO J. 2012;31:110-123
99. Fu J, Qin L, He T, Qin J, Hong J, Wong J, Liao L. et al. The TWIST/Mi2/NuRD protein complex and its essential role in cancer metastasis. Cell Res. 2011;21:275-289
100. Shi J, Wang Y, Zeng L, Wu Y, Deng J, Zhang Q, Lin Y. et al. Disrupting the interaction of BRD4 with diacetylated Twist suppresses tumorigenesis in basal-like breast cancer. Cancer Cell. 2014;25:210-225
101. Kreso A, Dick JE. Evolution of the cancer stem cell model. Cell Stem Cell. 2014;14:275-291
102. Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730-737
103. Beck B, Blanpain C. Unravelling cancer stem cell potential. Nat Rev Cancer. 2013;13:727-738
104. Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105-111
105. Karamboulas C, Ailles L. Developmental signaling pathways in cancer stem cells of solid tumors. Biochim Biophys Acta. 2013;1830:2481-2495
106. Dick JE. Stem cell concepts renew cancer research. Blood. 2008;112:4793-4807
107. Hermann PC, Huber SL, Herrler T, Aicher A, Ellwart JW, Guba M, Bruns CJ. et al. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell. 2007;1:313-323
108. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M. et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704-715
109. Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA, Lander ES. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell. 2009;138:645-659
110. Bao B, Wang Z, Ali S, Kong D, Banerjee S, Ahmad A, Li Y. et al. Over-expression of FoxM1 leads to epithelial-mesenchymal transition and cancer stem cell phenotype in pancreatic cancer cells. J Cell Biochem. 2011;112:2296-2306
111. Wang YH, Li F, Luo B, Wang XH, Sun HC, Liu S, Cui YQ. et al. A side population of cells from a human pancreatic carcinoma cell line harbors cancer stem cell characteristics. Neoplasma. 2009;56:371-378
112. Ni Z, Bikadi Z, Rosenberg MF, Mao Q. Structure and function of the human breast cancer resistance protein (BCRP/ABCG2). Curr Drug Metab. 2010;11:603-617
113. Hong SP, Wen J, Bang S, Park S, Song SY. CD44-positive cells are responsible for gemcitabine resistance in pancreatic cancer cells. Int J Cancer. 2009;125:2323-2331
114. Vinogradov S, Wei X. Cancer stem cells and drug resistance: the potential of nanomedicine. Nanomedicine (Lond). 2012;7:597-615
115. Ma S, Chan KW, Lee TK, Tang KH, Wo JY, Zheng BJ, Guan XY. Aldehyde dehydrogenase discriminates the CD133 liver cancer stem cell populations. Mol Cancer Res. 2008;6:1146-1153
116. Li T, Su Y, Mei Y, Leng Q, Leng B, Liu Z, Stass SA. et al. ALDH1A1 is a marker for malignant prostate stem cells and predictor of prostate cancer patients' outcome. Lab Invest. 2010;90:234-244
117. Grosse-Gehling P, Fargeas CA, Dittfeld C, Garbe Y, Alison MR, Corbeil D, Kunz-Schughart LA. CD133 as a biomarker for putative cancer stem cells in solid tumours: limitations, problems and challenges. J Pathol. 2013;229:355-378
118. Li C, Heidt DG, Dalerba P, Burant CF, Zhang L, Adsay V, Wicha M. et al. Identification of pancreatic cancer stem cells. Cancer Res. 2007;67:1030-1037
119. Li C, Wu JJ, Hynes M, Dosch J, Sarkar B, Welling TH, Pasca di Magliano M. et al. c-Met is a marker of pancreatic cancer stem cells and therapeutic target. Gastroenterology. 2011;141:2218-2227 e2215
120. Sureban SM, May R, Lightfoot SA, Hoskins AB, Lerner M, Brackett DJ, Postier RG. et al. DCAMKL-1 regulates epithelial-mesenchymal transition in human pancreatic cells through a miR-200a-dependent mechanism. Cancer Res. 2011;71:2328-2338
121. Gaviraghi M, Tunici P, Valensin S, Rossi M, Giordano C, Magnoni L, Dandrea M. et al. Pancreatic cancer spheres are more than just aggregates of stem marker-positive cells. Biosci Rep. 2011;31:45-55
122. Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CH, Jones DL, Visvader J. et al. Cancer stem cells-perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006;66:9339-9344
123. Simeone DM. Pancreatic cancer stem cells: implications for the treatment of pancreatic cancer. Clin Cancer Res. 2008;14:5646-5648
124. Beachy PA, Karhadkar SS, Berman DM. Tissue repair and stem cell renewal in carcinogenesis. Nature. 2004;432:324-331
125. Wang J, Sullenger BA, Rich JN. Notch signaling in cancer stem cells. Adv Exp Med Biol. 2012;727:174-185
126. Bao B, Wang Z, Ali S, Kong D, Li Y, Ahmad A, Banerjee S. et al. Notch-1 induces epithelial-mesenchymal transition consistent with cancer stem cell phenotype in pancreatic cancer cells. Cancer Lett. 2011;307:26-36
127. Abel EV, Kim EJ, Wu J, Hynes M, Bednar F, Proctor E, Wang L. et al. The Notch pathway is important in maintaining the cancer stem cell population in pancreatic cancer. PLoS One. 2014;9:e91983
128. Ruiz i Altaba A, Sanchez P, Dahmane N. Gli and hedgehog in cancer: tumours, embryos and stem cells. Nat Rev Cancer. 2002;2:361-372
129. Morton JP, Mongeau ME, Klimstra DS, Morris JP, Lee YC, Kawaguchi Y, Wright CV. et al. Sonic hedgehog acts at multiple stages during pancreatic tumorigenesis. Proc Natl Acad Sci U S A. 2007;104:5103-5108
130. Morton JP, Lewis BC. Shh signaling and pancreatic cancer: implications for therapy? Cell Cycle. 2007;6:1553-1557
131. Thayer SP, di Magliano MP, Heiser PW, Nielsen CM, Roberts DJ, Lauwers GY, Qi YP. et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature. 2003;425:851-856
132. Lee CJ, Dosch J, Simeone DM. Pancreatic cancer stem cells. J Clin Oncol. 2008;26:2806-2812
133. Fuchs E. The tortoise and the hair: slow-cycling cells in the stem cell race. Cell. 2009;137:811-819
134. Mao J, Fan S, Ma W, Fan P, Wang B, Zhang J, Wang H. et al. Roles of Wnt/beta-catenin signaling in the gastric cancer stem cells proliferation and salinomycin treatment. Cell Death Dis. 2014;5:e1039
135. Sandberg CJ, Altschuler G, Jeong J, Stromme KK, Stangeland B, Murrell W, Grasmo-Wendler UH. et al. Comparison of glioma stem cells to neural stem cells from the adult human brain identifies dysregulated Wnt- signaling and a fingerprint associated with clinical outcome. Exp Cell Res. 2013;319:2230-2243
136. Wang Y, Krivtsov AV, Sinha AU, North TE, Goessling W, Feng Z, Zon LI. et al. The Wnt/beta-catenin pathway is required for the development of leukemia stem cells in AML. Science. 2010;327:1650-1653
137. Fung MC, Takayama S, Ishiguro H, Sakata T, Adachi S, Morizane T. [Chemotherapy for advanced or metastatic pancreatic cancer: analysis of 43 randomized trials in 3 decades (1974-2002)]. Gan To Kagaku Ryoho. 2003;30:1101-1111
138. Burris HA 3rd, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR, Cripps MC. et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol. 1997;15:2403-2413
139. Mini E, Nobili S, Caciagli B, Landini I, Mazzei T. Cellular pharmacology of gemcitabine. Ann Oncol. 2006;17(Suppl 5):v7-12
140. Oettle H, Neuhaus P. Adjuvant therapy in pancreatic cancer: a critical appraisal. Drugs. 2007;67:2293-2310
141. Renouf D, Moore M. Evolution of systemic therapy for advanced pancreatic cancer. Expert Rev Anticancer Ther. 2010;10:529-540
142. Cunningham D, Chau I, Stocken DD, Valle JW, Smith D, Steward W, Harper PG. et al. Phase III randomized comparison of gemcitabine versus gemcitabine plus capecitabine in patients with advanced pancreatic cancer. J Clin Oncol. 2009;27:5513-5518
143. Louvet C, Labianca R, Hammel P, Lledo G, Zampino MG, Andre T, Zaniboni A. et al. Gemcitabine in combination with oxaliplatin compared with gemcitabine alone in locally advanced or metastatic pancreatic cancer: results of a GERCOR and GISCAD phase III trial. J Clin Oncol. 2005;23:3509-3516
144. Heinemann V, Quietzsch D, Gieseler F, Gonnermann M, Schonekas H, Rost A, Neuhaus H. et al. Randomized phase III trial of gemcitabine plus cisplatin compared with gemcitabine alone in advanced pancreatic cancer. J Clin Oncol. 2006;24:3946-3952
145. Von Hoff DD, Ramanathan RK, Borad MJ, Laheru DA, Smith LS, Wood TE, Korn RL. et al. Gemcitabine plus nab-paclitaxel is an active regimen in patients with advanced pancreatic cancer: a phase I/II trial. J Clin Oncol. 2011;29:4548-4554
146. Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, Seay T. et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. 2013;369:1691-1703
147. Vauthey JN, Dixon E. AHPBA/SSO/SSAT Consensus Conference on Resectable and Borderline Resectable Pancreatic Cancer: rationale and overview of the conference. Ann Surg Oncol. 2009;16:1725-1726
148. Freelove R, Walling AD. Pancreatic cancer: diagnosis and management. Am Fam Physician. 2006;73:485-492
149. Li D, Xie K, Wolff R, Abbruzzese JL. Pancreatic cancer. Lancet. 2004;363:1049-1057
150. Herman JM, Swartz MJ, Hsu CC, Winter J, Pawlik TM, Sugar E, Robinson R. et al. Analysis of fluorouracil-based adjuvant chemotherapy and radiation after pancreaticoduodenectomy for ductal adenocarcinoma of the pancreas: results of a large, prospectively collected database at the Johns Hopkins Hospital. J Clin Oncol. 2008;26:3503-3510
151. Conroy T, Desseigne F, Ychou M, Bouche O, Guimbaud R, Becouarn Y, Adenis A. et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364:1817-1825
152. Bryant KL, Mancias JD, Kimmelman AC, Der CJ. KRAS: feeding pancreatic cancer proliferation. Trends Biochem Sci. 2014;39:91-100
153. Schmick M, Vartak N, Papke B, Kovacevic M, Truxius DC, Rossmannek L, Bastiaens PI. KRas localizes to the plasma membrane by spatial cycles of solubilization, trapping and vesicular transport. Cell. 2014;157:459-471
154. Berndt N, Hamilton AD, Sebti SM. Targeting protein prenylation for cancer therapy. Nat Rev Cancer. 2011;11:775-791
155. Chandra A, Grecco HE, Pisupati V, Perera D, Cassidy L, Skoulidis F, Ismail SA. et al. The GDI-like solubilizing factor PDEdelta sustains the spatial organization and signalling of Ras family proteins. Nat Cell Biol. 2012;14:148-158
156. Sebti SM, Hamilton AD. Farnesyltransferase and geranylgeranyltransferase I inhibitors and cancer therapy: lessons from mechanism and bench-to-bedside translational studies. Oncogene. 2000;19:6584-6593
157. Sjogren AK, Andersson KM, Liu M, Cutts BA, Karlsson C, Wahlstrom AM, Dalin M. et al. GGTase-I deficiency reduces tumor formation and improves survival in mice with K-RAS-induced lung cancer. J Clin Invest. 2007;117:1294-1304
158. Lu J, Chan L, Fiji HD, Dahl R, Kwon O, Tamanoi F. In vivo antitumor effect of a novel inhibitor of protein geranylgeranyltransferase-I. Mol Cancer Ther. 2009;8:1218-1226
159. Sane KM, Mynderse M, Lalonde DT, Dean IS, Wojtkowiak JW, Fouad F, Borch RF. et al. A novel geranylgeranyl transferase inhibitor in combination with lovastatin inhibits proliferation and induces autophagy in STS-26T MPNST cells. J Pharmacol Exp Ther. 2010;333:23-33
160. Appels NM, Beijnen JH, Schellens JH. Development of farnesyl transferase inhibitors: a review. Oncologist. 2005;10:565-578
161. Van Cutsem E, van de Velde H, Karasek P, Oettle H, Vervenne WL, Szawlowski A, Schoffski P. et al. Phase III trial of gemcitabine plus tipifarnib compared with gemcitabine plus placebo in advanced pancreatic cancer. J Clin Oncol. 2004;22:1430-1438
162. Rao S, Cunningham D, de Gramont A, Scheithauer W, Smakal M, Humblet Y, Kourteva G. et al. Phase III double-blind placebo-controlled study of farnesyl transferase inhibitor R115777 in patients with refractory advanced colorectal cancer. J Clin Oncol. 2004;22:3950-3957
163. Lobell RB, Omer CA, Abrams MT, Bhimnathwala HG, Brucker MJ, Buser CA, Davide JP. et al. Evaluation of farnesyl:protein transferase and geranylgeranyl:protein transferase inhibitor combinations in preclinical models. Cancer Res. 2001;61:8758-8768
164. Zimmermann G, Papke B, Ismail S, Vartak N, Chandra A, Hoffmann M, Hahn SA. et al. Small molecule inhibition of the KRAS-PDEdelta interaction impairs oncogenic KRAS signalling. Nature. 2013;497:638-642
165. Laheru D, Shah P, Rajeshkumar NV, McAllister F, Taylor G, Goldsweig H, Le DT. et al. Integrated preclinical and clinical development of S-trans, trans-Farnesylthiosalicylic Acid (FTS, Salirasib) in pancreatic cancer. Invest New Drugs. 2012;30:2391-2399
166. Eser S, Schnieke A, Schneider G, Saur D. Oncogenic KRAS signalling in pancreatic cancer. Br J Cancer. 2014;111:817-822
167. Maurer T, Garrenton LS, Oh A, Pitts K, Anderson DJ, Skelton NJ, Fauber BP. et al. Small-molecule ligands bind to a distinct pocket in Ras and inhibit SOS-mediated nucleotide exchange activity. Proc Natl Acad Sci U S A. 2012;109:5299-5304
168. Wang Y, Kaiser CE, Frett B, Li HY. Targeting mutant KRAS for anticancer therapeutics: a review of novel small molecule modulators. J Med Chem. 2013;56:5219-5230
169. Collisson EA, Trejo CL, Silva JM, Gu S, Korkola JE, Heiser LM, Charles RP. et al. A central role for RAF->MEK->ERK signaling in the genesis of pancreatic ductal adenocarcinoma. Cancer Discov. 2012;2:685-693
170. Walters DM, Lindberg JM, Adair SJ, Newhook TE, Cowan CR, Stokes JB, Borgman CA. et al. Inhibition of the Growth of Patient-Derived Pancreatic Cancer Xenografts with the MEK Inhibitor Trametinib Is Augmented by Combined Treatment with the Epidermal Growth Factor Receptor/HER2 Inhibitor Lapatinib. Neoplasia;15:IN8-IN10.
171. Torrance CJ, Agrawal V, Vogelstein B, Kinzler KW. Use of isogenic human cancer cells for high-throughput screening and drug discovery. Nat Biotechnol. 2001;19:940-945
172. Guo W, Wu S, Liu J, Fang B. Identification of a small molecule with synthetic lethality for K-ras and protein kinase C iota. Cancer Res. 2008;68:7403-7408
173. Shaw AT, Winslow MM, Magendantz M, Ouyang C, Dowdle J, Subramanian A, Lewis TA. et al. Selective killing of K-ras mutant cancer cells by small molecule inducers of oxidative stress. Proc Natl Acad Sci U S A. 2011;108:8773-8778
174. Ji Z, Mei FC, Lory PL, Gilbertson SR, Chen Y, Cheng X. Chemical genetic screening of KRAS-based synthetic lethal inhibitors for pancreatic cancer. Front Biosci (Landmark Ed). 2009;14:2904-2910
175. Fritsche P, Seidler B, Schuler S, Schnieke A, Gottlicher M, Schmid RM, Saur D. et al. HDAC2 mediates therapeutic resistance of pancreatic cancer cells via the BH3-only protein NOXA. Gut. 2009;58:1399-1409
176. Ouaissi M, Sielezneff I, Silvestre R, Sastre B, Bernard JP, Lafontaine JS, Payan MJ. et al. High histone deacetylase 7 (HDAC7) expression is significantly associated with adenocarcinomas of the pancreas. Ann Surg Oncol. 2008;15:2318-2328
177. Miyake K, Yoshizumi T, Imura S, Sugimoto K, Batmunkh E, Kanemura H, Morine Y. et al. Expression of hypoxia-inducible factor-1alpha, histone deacetylase 1, and metastasis-associated protein 1 in pancreatic carcinoma: correlation with poor prognosis with possible regulation. Pancreas. 2008;36:e1-9
178. Lehmann A, Denkert C, Budczies J, Buckendahl AC, Darb-Esfahani S, Noske A, Muller BM. et al. High class I HDAC activity and expression are associated with RelA/p65 activation in pancreatic cancer in vitro and in vivo. BMC Cancer. 2009;9:395
179. Johnstone RW. Histone-deacetylase inhibitors: novel drugs for the treatment of cancer. Nat Rev Drug Discov. 2002;1:287-299
180. Tan J, Cang S, Ma Y, Petrillo RL, Liu D. Novel histone deacetylase inhibitors in clinical trials as anti-cancer agents. J Hematol Oncol. 2010;3:5
181. Dokmanovic M, Clarke C, Marks PA. Histone deacetylase inhibitors: overview and perspectives. Molecular Cancer Research. 2007;5:981-989
182. Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5:769-784
183. Neureiter D, Zopf S, Leu T, Dietze O, Hauser-Kronberger C, Hahn EG, Herold C. et al. Apoptosis, proliferation and differentiation patterns are influenced by Zebularine and SAHA in pancreatic cancer models. Scand J Gastroenterol. 2007;42:103-116
184. Nawrocki ST, Carew JS, Pino MS, Highshaw RA, Andtbacka RH, Dunner K Jr, Pal A. et al. Aggresome disruption: a novel strategy to enhance bortezomib-induced apoptosis in pancreatic cancer cells. Cancer Res. 2006;66:3773-3781
185. Nalls D, Tang SN, Rodova M, Srivastava RK, Shankar S. Targeting epigenetic regulation of miR-34a for treatment of pancreatic cancer by inhibition of pancreatic cancer stem cells. PLoS One. 2011;6:e24099
186. Koutsounas I, Giaginis C, Patsouris E, Theocharis S. Current evidence for histone deacetylase inhibitors in pancreatic cancer. World J Gastroenterol. 2013;19:813-828
187. Tinari N, De Tursi M, Grassadonia A, Zilli M, Stuppia L, Iacobelli S, Natoli C. An epigenetic approach to pancreatic cancer treatment: the prospective role of histone deacetylase inhibitors. Curr Cancer Drug Targets. 2012;12:439-452
188. Kim HJ, Bae SC. Histone deacetylase inhibitors: molecular mechanisms of action and clinical trials as anti-cancer drugs. Am J Transl Res. 2011;3:166-179
189. Zafar SF, Nagaraju GP, El-Rayes B. Developing histone deacetylase inhibitors in the therapeutic armamentarium of pancreatic adenocarcinoma. Expert Opin Ther Targets. 2012;16:707-718
190. Richards DA, Boehm KA, Waterhouse DM, Wagener DJ, Krishnamurthi SS, Rosemurgy A, Grove W. et al. Gemcitabine plus CI-994 offers no advantage over gemcitabine alone in the treatment of patients with advanced pancreatic cancer: results of a phase II randomized, double-blind, placebo-controlled, multicenter study. Ann Oncol. 2006;17:1096-1102
191. Gojo I, Jiemjit A, Trepel JB, Sparreboom A, Figg WD, Rollins S, Tidwell ML. et al. Phase 1 and pharmacologic study of MS-275, a histone deacetylase inhibitor, in adults with refractory and relapsed acute leukemias. Blood. 2007;109:2781-2790
192. Hindriksen S, Bijlsma MF. Cancer Stem Cells, EMT, and Developmental Pathway Activation in Pancreatic Tumors. Cancers (Basel). 2012;4:989-1035
193. Tang SC, Chen YC. Novel therapeutic targets for pancreatic cancer. World J Gastroenterol. 2014;20:10825-10844
194. Feldmann G, Dhara S, Fendrich V, Bedja D, Beaty R, Mullendore M, Karikari C. et al. Blockade of hedgehog signaling inhibits pancreatic cancer invasion and metastases: a new paradigm for combination therapy in solid cancers. Cancer Res. 2007;67:2187-2196
195. Yao J, An Y, Wie JS, Ji ZL, Lu ZP, Wu JL, Jiang KR. et al. Cyclopamine reverts acquired chemoresistance and down-regulates cancer stem cell markers in pancreatic cancer cell lines. Swiss Med Wkly. 2011;141:w13208
196. Mueller MT, Hermann PC, Witthauer J, Rubio-Viqueira B, Leicht SF, Huber S, Ellwart JW. et al. Combined targeted treatment to eliminate tumorigenic cancer stem cells in human pancreatic cancer. Gastroenterology. 2009;137:1102-1113
197. Feldmann G, Fendrich V, McGovern K, Bedja D, Bisht S, Alvarez H, Koorstra JB. et al. An orally bioavailable small-molecule inhibitor of Hedgehog signaling inhibits tumor initiation and metastasis in pancreatic cancer. Mol Cancer Ther. 2008;7:2725-2735
198. Singh BN, Fu J, Srivastava RK, Shankar S. Hedgehog signaling antagonist GDC-0449 (Vismodegib) inhibits pancreatic cancer stem cell characteristics: molecular mechanisms. PLoS One. 2011;6:e27306
199. Balic A, Sorensen MD, Trabulo SM, Sainz B Jr, Cioffi M, Vieira CR, Miranda-Lorenzo I. et al. Chloroquine targets pancreatic cancer stem cells via inhibition of CXCR4 and hedgehog signaling. Mol Cancer Ther. 2014;13:1758-1771
200. Kim EJ, Sahai V, Abel EV, Griffith KA, Greenson JK, Takebe N, Khan GN. et al. Pilot clinical trial of hedgehog pathway inhibitor GDC-0449 (vismodegib) in combination with gemcitabine in patients with metastatic pancreatic adenocarcinoma. Clin Cancer Res. 2014;20:5937-5945
201. Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, Dekleva EN. et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell. 2014;25:735-747
202. Cook N, Frese KK, Bapiro TE, Jacobetz MA, Gopinathan A, Miller JL, Rao SS. et al. Gamma secretase inhibition promotes hypoxic necrosis in mouse pancreatic ductal adenocarcinoma. J Exp Med. 2012;209:437-444
203. Mizuma M, Rasheed ZA, Yabuuchi S, Omura N, Campbell NR, de Wilde RF, De Oliveira E. et al. The gamma secretase inhibitor MRK-003 attenuates pancreatic cancer growth in preclinical models. Mol Cancer Ther. 2012;11:1999-2009
204. Yabuuchi S, Pai SG, Campbell NR, de Wilde RF, De Oliveira E, Korangath P, Streppel MM. et al. Notch signaling pathway targeted therapy suppresses tumor progression and metastatic spread in pancreatic cancer. Cancer Lett. 2013;335:41-51
205. Jin L, Hope KJ, Zhai Q, Smadja-Joffe F, Dick JE. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat Med. 2006;12:1167-1174
206. Cioffi M, Dorado J, Baeuerle PA, Heeschen C. EpCAM/CD3-Bispecific T-cell engaging antibody MT110 eliminates primary human pancreatic cancer stem cells. Clin Cancer Res. 2012;18:465-474
207. Takikita M, Altekruse S, Lynch CF, Goodman MT, Hernandez BY, Green M, Cozen W. et al. Associations between selected biomarkers and prognosis in a population-based pancreatic cancer tissue microarray. Cancer Res. 2009;69:2950-2955
208. Inaguma S, Kasai K, Ikeda H. GLI1 facilitates the migration and invasion of pancreatic cancer cells through MUC5AC-mediated attenuation of E-cadherin. Oncogene. 2011;30:714-723
209. Arlen M, Wang X, Luka J, Gupta R, Saric O, Arlen PM. The use of specific monoclonal antibodies to target immunogenic tumor membrane proteins in patients with recurrent pancreatic and colon cancer. Curr Drug Deliv. 2012;9:52-56
210. Morse M, Diaz L, Azad N, Laheru D, Haley S, Sleer L, Arlen M. A Phase I/II safety study of NPC-1C: a novel, therapeutic antibody to treat pancreas and colorectal cancers. J Clin Oncol. 2012;30:Abstr 233
211. Patel S, Morse M, Beg M, Azad N, Beaston M, Mavroukakis S, Arlen PM. A phase Ib/IIa study of NEO-102: A therapeutic antibody to treat pancreatic ancd colorectal cancers. J Clin Oncol. 2014;32:Abst 243
212. Beg M, Morse M, Patel S, Mavroukakis S, Beatson M, Arlen PM, Azad N. A phase I/II multicenter study of the chimeric monoclonal antibody NEO102 (NPC-1C) in adults with refractory pancreatic (PC) and colorectal cancer (CC). J Clin Oncol. 2015;32:Abstr 240
213. Li Y, Go VL, Sarkar FH. The Role of Nutraceuticals in Pancreatic Cancer Prevention and Therapy: Targeting Cellular Signaling, MicroRNAs, and Epigenome. Pancreas. 2015;44:1-10
214. Mateos-Aparicio I, Redondo Cuenca A, Villanueva-Suarez MJ, Zapata-Revilla MA. Soybean, a promising health source. Nutr Hosp. 2008;23:305-312
215. Bao B, Ali S, Kong D, Sarkar SH, Wang Z, Banerjee S, Aboukameel A. et al. Anti-tumor activity of a novel compound-CDF is mediated by regulating miR-21, miR-200, and PTEN in pancreatic cancer. PLoS One. 2011;6:e17850
216. Bao B, Ali S, Banerjee S, Wang Z, Logna F, Azmi AS, Kong D. et al. Curcumin analogue CDF inhibits pancreatic tumor growth by switching on suppressor microRNAs and attenuating EZH2 expression. Cancer Res. 2012;72:335-345
217. Shankar S, Nall D, Tang SN, Meeker D, Passarini J, Sharma J, Srivastava RK. Resveratrol inhibits pancreatic cancer stem cell characteristics in human and KrasG12D transgenic mice by inhibiting pluripotency maintaining factors and epithelial-mesenchymal transition. PLoS One. 2011;6:e16530
218. Dhillon N, Aggarwal BB, Newman RA, Wolff RA, Kunnumakkara AB, Abbruzzese JL, Ng CS. et al. Phase II trial of curcumin in patients with advanced pancreatic cancer. Clin Cancer Res. 2008;14:4491-4499
219. Kanai M, Yoshimura K, Asada M, Imaizumi A, Suzuki C, Matsumoto S, Nishimura T. et al. A phase I/II study of gemcitabine-based chemotherapy plus curcumin for patients with gemcitabine-resistant pancreatic cancer. Cancer Chemother Pharmacol. 2011;68:157-164
220. Epelbaum R, Schaffer M, Vizel B, Badmaev V, Bar-Sela G. Curcumin and gemcitabine in patients with advanced pancreatic cancer. Nutr Cancer. 2010;62:1137-1141
221. El-Rayes BF, Philip PA, Sarkar FH, Shields AF, Ferris AM, Hess K, Kaseb AO. et al. A phase II study of isoflavones, erlotinib, and gemcitabine in advanced pancreatic cancer. Invest New Drugs. 2011;29:694-699
222. Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, Au HJ. et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2007;25:1960-1966
223. Carter LG, D'Orazio JA, Pearson KJ. Resveratrol and cancer: focus on in vivo evidence. Endocr Relat Cancer. 2014;21:R209-225
224. Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: Mission Possible? Nat Rev Drug Discov. 2014;13:828-851
225. Nandy D, Mukhopadhyay D. Growth factor mediated signaling in pancreatic pathogenesis. Cancers (Basel). 2011;3:841-871
226. Wolfgang CL, Herman JM, Laheru DA, Klein AP, Erdek MA, Fishman EK, Hruban RH. Recent progress in pancreatic cancer. CA Cancer J Clin. 2013;63:318-348
227. Li C, Lee CJ, Simeone DM. Identification of human pancreatic cancer stem cells. Methods Mol Biol. 2009;568:161-173
228. Gupta PB, Chaffer CL, Weinberg RA. Cancer stem cells: mirage or reality? Nat Med. 2009;15:1010-1012
229. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D. et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med. 2015;373:23-34
230. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A. et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371:1507-1517
231. Kotteas E, Saif MW, Syrigos K. Immunotherapy for pancreatic cancer. J Cancer Res Clin Oncol. 2016
Author contact
![]() Corresponding author: Qi Chen, PhD. Department of Pharmacology, Toxicology and Therapeutics, University of Kansas Medical Center, 3901 Rainbow Blvd., Kansas City, KS, USA 66160. Tel: 913-588-3690 Fax: 913-945-6874 Email: qchenedu.
Corresponding author: Qi Chen, PhD. Department of Pharmacology, Toxicology and Therapeutics, University of Kansas Medical Center, 3901 Rainbow Blvd., Kansas City, KS, USA 66160. Tel: 913-588-3690 Fax: 913-945-6874 Email: qchenedu.