Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2016; 7(11):1431-1440. doi:10.7150/jca.15497 This issue Cite
Research Paper
Centrosomal Protein of 55 Regulates Glucose Metabolism, Proliferation and Apoptosis of Glioma Cells via the Akt/mTOR Signaling Pathway
Guangzhi Wang1,2, Mingna Liu3, Hongjun Wang1, Shan Yu4, Zhenfeng Jiang5, Jiahang Sun1, Ke Han6, Jia Shen7, Minwei Zhu5, Zhiguo Lin5, Chuanlu Jiang1 ![]() *, Mian Guo1
*, Mian Guo1 ![]() *
*
1. Department of Neurosurgery, the Second Affiliated Hospital of Harbin Medical University, Harbin, Heilongjiang 150086, China;
2. Department of Medical Service Management, the Second Affiliated Hospital of Harbin Medical University, Harbin, Heilongjiang 150086, China;
3. Department of Gastroenterology, the Second Affiliated Hospital of Harbin Medical University, Harbin, Heilongjiang 150086, China;
4. Department of Pathology, the Second Affiliated Hospital of Harbin Medical University, Harbin, Heilongjiang 150086, China;
5. Department of Neurosurgery, the First Affiliated Hospital of Harbin Medical University, Harbin, Heilongjiang 150086, China;
6. School of Computer and Information Engineering, Harbin University of Commerce, Harbin, Heilongjiang 150086, China.
7. Division of Growth and Development, Section of Orthodontics, School of Dentistry, University of California, Los Angeles, California 90095, USA.
*These authors contributed equally to this work.
Received 2016-3-10; Accepted 2016-5-18; Published 2016-7-4
Citation:
Wang G, Liu M, Wang H, Yu S, Jiang Z, Sun J, Han K, Shen J, Zhu M, Lin Z, Jiang C, Guo M. Centrosomal Protein of 55 Regulates Glucose Metabolism, Proliferation and Apoptosis of Glioma Cells via the Akt/mTOR Signaling Pathway. J Cancer 2016; 7(11):1431-1440. doi:10.7150/jca.15497. https://www.jcancer.org/v07p1431.htm
Other stylesAbstract
Introduction: Glioma is one of the most common and most aggressive brain tumors in humans. The molecular and cellular mechanisms responsible for the onset and the progression of glioma are elusive and controversial. Centrosomal protein of 55 (CEP55) was initially described as a highly coiled-coil protein that plays critical roles in cell division, but was recently identified as being overexpressed in many human cancers. The function of CEP55 has not previously been characterized in glioma. We aim to discover the effect and mechanism of CEP55 in glioma development.
Method: qRT-PCR and immunohistochemistry were used to analyze CEP55 expression. Glucose uptake, western blot, MTS, CCK-8, Caspase-3 activity and TUNEL staining assays were performed to investigate the role and mechanism of CEP55 on glioma cell process.
Results: We found that the levels of CEP55 expression were upregulated in glioma. In addition, CEP55 appeared to regulate glucose metabolism of glioma cells. Furthermore, knockdown of CEP55 inhibited cell proliferation and induced cell apoptosis in glioma. Finally, we provided preliminary evidence that knockdown of CEP55 inhibited glioma development via suppressing the activity of Akt/mTOR signaling.
Conclusions: Our results demonstrated that CEP55 regulates glucose metabolism, proliferation and apoptosis of glioma cells via the Akt/mTOR signaling pathway, and its promotive effect on glioma tumorigenesis can be a potential target for glioma therapy in the future.
Keywords: Glioma, CEP55, Akt, mTOR, proliferation, apoptosis.
Introduction
Glioma is the most is the most common and most aggressive primary brain tumor in humans. The overall incidence rates account for gliomas is 6.03 per 100,000. The pathophysiology of glioma involves deregulated proliferation and migration of glial cells [1, 2]. Despite recent advancement in clinical prevention and surgical treatment, the prognosis of glioma, especially for (GBM) remains poor. The cause of this situation is attributable to the difficulty in removing the tumor tissue in its entirety, and to the development of chemoresistance of tumor cells [3, 4]. Unfortunately, the mechanism responsible for the onset and the progression of glioma on molecular and cellular levels is still not well understood. A series of recent studies suggested that, like malignant tumors in other tissues, malfunction of signaling pathways that govern proliferation and apoptosis of neuron cells plays important roles in glioma [5-8]. Better understanding of the pathogenesis of glioma will lead to improved diagnosis and targeted therapies for effective treatment of the disease.
Centrosomal protein of 55 (CEP55) was initially described as a highly coiled-coil protein that plays critical roles in cell division [9-11]. CEP55 remains associated with the centrosome throughout the cell cycle and is recruited to the mitotic spindle during mitosis [11]. CEP55 is also found to localize to the central spindle during anaphase and the midbody during cytokinesis [9, 12]. Recently, CEP55 has been identified as being overexpressed in many human cancers by mRNA microarray analysis [13-18]. Analysis of patient samples has revealed a number of pathways are dysregulated in CEP55 overexpressing tumors [19-25]. A series of studies demonstrated that the function of CEP55 in cancer development was achieved through its regulation of the PI3K/Akt pathway. For example, in lung cancer cell lines, VEGF induces CEP55 expression in a dose-dependent manner [20]. VEGF is a potent stimulator of angiogenesis, endothelial cell vascular permeability, motility and proliferation, with a well-established role in cancer [26]. Interestingly, in these cell lines, VEGF also stimulates the PI3K/AKT pathway in a CEP55-dependent manner [20]. Similarly, in hepatocellular carcinoma, the overexpression of CEP55 promotes cell migration and invasion via upregulation of the PI3K/Akt pathway, possibly by binding of CEP55 to p110, which increases the stability of this subunit, resulting in increased Akt activation [21].
Although the involvement of CEP55 in the development of multiple cancer types was reported, the role of CEP55 in glioma development was not previously investigated. In this study, for the first time we reported that CEP55 expression was upregulated in glioma. In addition, CEP55 appeared to regulate glucose metabolism of glioma cells. Furthermore, knockdown of CEP55 inhibited cell proliferation and induced cell apoptosis in glioma. Finally, we provided preliminary evidence that knockdown of CEP55 inhibited glioma development via suppressing the activity of Akt/mTOR signaling.
Materials and methods
Patient samples
This study was approved by the institutional review board (IRB) at Harbin Medical University in accordance with the principles of Declaration of Helsinki. Surgically excised tumor specimens from 50 patients with World Health Organization (WHO) grades II-IV glioma (14 patients with grade II and 31 with grade III-IV gliomas) and 5 normal brain tissues were obtained from the Department of Neurosurgery, Hospital of Harbin Medical University, China. Collected tissues were immediately snap frozen and stored at -80°C. Written informed consent was obtained from all patients. None of the patients in the study received any chemotherapy or radiation therapy prior to surgery.
Immunohistochemistry
Immunohistochemistry was performed as described previously [27]. Anti-CEP55 primary antibody was purchased from Santa Cruz (Santa Cruz, CA, USA).
Cell culture, treatment and transfection
The human glioma cell lines U87 and T98G were obtained from the American Type Culture Collection (ATCC, Rockville, MD, USA). All cells were maintained in Dulbecco's modified Eagle medium (DMEM; Invitrogen, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (FBS; Invitrogen). These cells were grown at 37 °C in an atmosphere of 5 % CO2 and the medium was replaced every 3 days. Akt signaling inhibitor MK-2206 and mTOR signaling inhibitor rapamycin were purchased from Selleckchem (Houston, TX, USA). All cells were cultured in MK-2206 (1 µM) or rapamycin (100 nM) for signaling inhibition. CEP55 siRNA and scrambled siRNA were purchased from Santa Cruz. X-tremeGENE (Roche, Mannheim, Germany) was used for siRNA transfection.
qRT-PCR
Total mRNA from cultured cells and frozen surgical glioma tissues was extracted using the TRIzol reagent (Invitrogen) according to the manufacturer's instructions. SuperScript First-Stand Synthesis System (Invitrogen) was used for reverse transcription. RT-PCR was performed with Power SYBRR Green PCR Master Mix (Applied Biosystems) and a 7500 sequence detection system (Applied Biosystems) using the 2-∆∆Ct method. Sequences of primers were listed as follows: CEP55 forward 5'-TTGGAACAACAGATGCAGGC-3'and reverse 5'-GAGTGCAGCAGTGGGACTTT-3'; GAPDH forward, 5'-TGGACTCCACGACGTACTCAG-3' and reverse, 5'-CGGGAAGCTTGTCATCAATGGAA-3'.
Western blot
U87 and T98G cells were lysed in radioimmunoprecipitation assay buffer [150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS), 50 mM Tris, pH 8.0, 5.0 mM ethylenediaminetetraacetic acid, pH 8.0, 0.5 mM dithiothreitol and 1 mM phenylmethylsulfonyl fluoride]. Protein concentrations were determined using the bicinchoninic acid method (Thermo Scientific, Rockford, IL, USA). Protein lysates were separated by SDS-polyacrylamide gel electrophoresis and transferred onto polyvinylidene difluoride membranes (Millipore, Billerica, MA, USA) by electroblotting. Primary antibodies for immunodetection were anti-CEP55 (Santa Cruz), anti-GLUT1 (Santa Cruz), anti-p-AktS473 (Santa Cruz), anti-p-AktT308(Santa Cruz), anti-Akt (Santa Cruz), anti-p-mTOR (Santa Cruz), anti-mTOR (Santa Cruz), anti-BAD (Santa Cruz), anti-caspase-9 (Santa Cruz), anti-GSK3-β (Santa Cruz), anti-p27 (Abcam) and anti-GAPDH (Santa Cruz). Subsequent to being incubated with Horseradish peroxidase (HRP) conjugated anti-rabbit or anti-mouse secondary antibodies (1: 10000, Santa Cruz) for 1 h, the immune complexes were detected using the enhanced chemiluminescence method.
Glucose uptake assay
The glucose uptake was determined using a 2-Deoxyglucose (2DG) Glucose Uptake Assay Kit (Fluorometric) from Abcam (Cambridge, MA, USA) according to the manufacturer's instructions. Briefly, U87 and T98G cells were gently seeded into 96-well plates (1 × 103 cells/well) overnight. After treatment with reagents for 24 h, the cells were incubated in the darkness with 2DG (10 mM) for 20 min at 37°C in 5% CO2 humidified atmosphere and subjected to the measurement of the 2DG uptake using fluorescence micro-plate reader (Bio-Rad) at Ex/Em=535/587 nm.
MTS assay
The cell proliferation and viability was assessed by 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium inner salt (MTS; Promega, Madison, WI, USA) assay. Cells were plated at a density of 2000 cells per well in 96-well plates overnight. After treatment, Twenty microliters of MTS was added into each well containing 100 µl medium and the cells were then incubated at 37°C for 2 h in a humidified 5% CO2 incubator. Absorbance was detected at 490 nm with amicroplate reader (Bio-Rad, Hercules, CA, USA).
CCK-8 assay
The number of viable cells was quantified using a CCK-8 detection kit (Sigma, Milwaukee, WI, USA) according to the manufacturer's instructions. Briefly, glioma cells were seeded in a 96-well microplate at a density of 5×104/ml. After treatment, 20 µl CCK-8 solution was added to each well and the plate was incubated at 37 °C for 2 h. The viable cells were counted by absorbance measurements at a wavelength of 450 nm with a microplate reader (Bio-Rad).
Bromodeoxyuridine (BrdU) labeling of cultured cells
U87 and T98G cells (5×104 per well) were cultured in 4-well Millicell EZ SLIDE (Millipore, Billerica, MA, USA) overnight in growing medium. The cells were then incubated with 10 µM bromodeoxyuridine (BrdU; Invitrogen) for 2 h after treatment. The glioma cells were then fixed and labeled with anti-BrdU antibody (Invitrogen) for 12 h, as per the manufacturer's instruction. Secondary antibody was added. DAPI was used for nuclear staining. The number of BrdU positive cells was counted under six random microscopic fields by NIH Image J software.
Caspase-3 activity assay
Caspase-3 activity was measured using a Caspase-3 activity fluorescence detection kit (Beyotime, Beijing, China) following the manufacturer's protocol. Briefly, 1×104 cells were seeded in 96-well plates overnight. The cells were lysed and mixed with mixture reaction solution (containing Ac-DEVD-pNA) was added in each well. The plate was incubated in dark and the fluorescence was read at 405 nm. Absorbance of samples was measured by subtraction of mean absorbance of the blank and corrected by the protein concentration of the cell lysate. The results were relative to activity of control group.
TUNEL staining
Glioma cell apoptosis were measured through TUNEL staining with the staining kit (Boster, Wuhan, Hubei, China) according to the manufacturer's instructions. Briefly, 5×104 per well were cultured in 4-well Millicell EZ SLIDE (Millipore) for each group. After treatment, the cells were fixed by 4% paraformaldehyde for 30 minutes, TUNEL staining was followed. The positive cells were counted under a microscopy (Olympus).
Statistical Analysis
Statistical analysis was done with the GraphPad Prism 6.0 statistical software package (La Jolla, CA, USA). All results were presented as mean ± standard deviation and analyzed using Student's t-test and one-way analysis of variance (ANOVA) analysis to determine the levels of significance. A P-value less than 0.05 were considered statistically significant.
Results
CEP55 expression is upregulated in glioma
CEP55 has been shown by previous study to be involved in the tumorigenesis of a variety of cancers [28], but its function in glioma development has not been characterized. In this study, we first analyzed the levels of CEP55 expression in database GDS1962 / 218542_at / CEP55. We found that compared to 23 non-tumor samples, the levels of CEP55 expression were substantially increased in all glioma tissues analyzed (including 7 astrocytomas grade II, 19 astrocytomas grade III, 81 glioblastoma, 38 oligodendrogliomas grade II and 12 oligodendrogliomas grade III samples) (Fig.1A). We next examined CEP55 expression in 14 glioma grade II and 31 grade III-IV tissue samples. Compared to 5 non-tumor brain tissues, CEP55 expression was significantly upregulated in glioma tissues (Fig.1B). The increased levels of CEP55 expression in glioma was also confirmed by immunohistochemical staining, which demonstrated that the protein levels of CEP55 were much higher in glioma tissues than those in non-tumor brain tissues (Fig.1C). Taken together, these results indicated that the levels of CEP55 expression were upregulated in glima.
CEP55 regulates glioma cell glucose metabolism
Since enhanced glucose metabolism is a hallmark of tumor cells [29], we first sought to determine the regulation of glucose metabolism of glioma cells by CEP55. To this end, we treated two independent glioma cell lines U87 and T98G with increasing concentrations of glucose (0, 1, 5, 10 or 20mM) for 12 h, and examined the levels of CEP55 in these cells. We found that in both cell lines, the protein levels of CEP55 were markedly upregulated in response to glucose treatment in a dose dependent manner (Fig.2A). We next exposed U87 and T98G cells to 5mM glucose for increasing amount of time (0, 4, 8, 12 or 24h). We determined that the protein levels of CEP55 were increased with treatment time in both cell lines (Fig.2B). We then reduced the expression of CEP55 in U87 and T98G cells using the RNA interference technique. The efficacy of CEP55 siRNA was confirmed by Western blot analysis (Fig.2C). Significantly, knockdown of CEP55 expression significantly reduced GLUT1 levels in both cell lines (Fig.2C). Consistently, knockdown of CEP55 expression also significantly inhibited glucose uptake by glioma cell lines (Fig.2D). Interestingly, overexpression of CEP55 failed to regulate glucose metabolism (data not shown), which might be due to high expression level of CEP55 in glioma cells. Ectopic CEP55 did not further affect glucose metabolism. These results collectively suggested that CEP55 might be involved in the regulation of glucose metabolism in glioma.
Figure 1
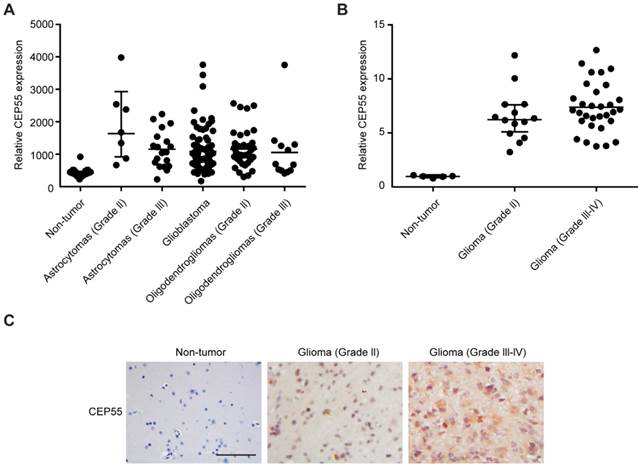
CEP55 expression is upregulated in glioma. (A) The levels of CEP55 mRNA expression in glioma tissue samples (7 astrocytomas grade II, 19 astrocytomas grade III, 81 glioblastoma, 38 oligodendrogliomas grade II and 12 oligodendrogliomas grade III samples) compared to those in 23 non-tumor samples in microarray database (GDS1962 / 218542_at / CEP55, p < 0.01). (B) mRNA levels of CEP55 as determined by qRT-PCR analysis in 14 glioma grade II and 31 grade III-IV tissue samples compared to those in 5 non-tumor brain tissue samples (p < 0.01). (C) Representative immunohistochemical staining of CEP55 in non-tumor, glioma grade II and grade III-IV samples.
Figure 2
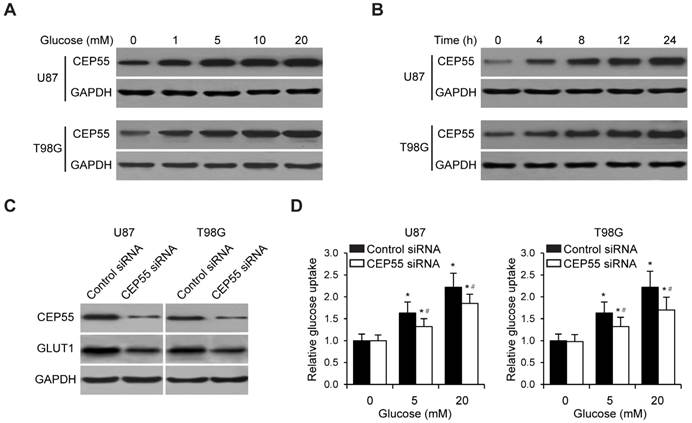
CEP55 regulates glioma cell glucose metabolism. (A) Protein levels of CEP55 as determined by Western blot analysis in U87 and T98G cells treated with the indicated concentrations of glucose (0, 1, 5, 10 or 20 mM). GAPDH was used as the loading control. (B) Protein levels of CEP55 as determined by Western blot analysis in U87 and T98G cells treated with 5 mM glucose for the indicated time periods (0, 4, 8, 12 or 24 h). (C) Protein levels of CEP55 and GLUT1 in U87 and T98G cells transfected with control siRNA or siRNA targeting CEP55. (D) Relative glucose uptake in U87 and T98G cells transfected with control siRNA or siRNA targeting CEP55 and treated with the indicated concentrations of glucose (0, 5, or 20 mM). *p < 0.5 compared to cells treated with 0 mM glucose, #p < 0.05 compared to control siRNA transfected cells treated with the same glucose concentration.
Knockdown of CEP55 inhibits cell proliferation and induces cell apoptosis in glioma
To examine the effects of CEP55 on glioma cell proliferation, we performed MTS assay in U87 and T98G cells (Fig.3A). We found that in both cell lines, knockdown of CEP55 led to decreased levels of cell proliferation compared to the control cells. This result was confirmed by CCK-8 assay (Fig.3B). Additionally, BrdU assay further confirmed the inhibition of cell proliferation by CEP55 knockdown (Fig.3C). To assess whether CEP55 regulates glioma cell apoptosis, we cultured glioma cells in no glucose medium and measured cell viability by MTS assay every 24 h. We found that knockdown of CEP55 significantly decreased the number of viable cells (Fig.4A). In addition, knockdown of CEP55 increased the activity of cleaved caspase-3 in U87 and T98G cells (Fig.4B). We also assessed cell apoptosis by Tunel staining after glucose deprivation for 24 h. We found that compared to control cells, knockdown of CEP55 resulted in significantly more apoptotic cells (Fig.4C). In addition, CEP55 was found related to poor condition in clinical glioma patients (Table 1). These results indicated that CEP55 played roles in the regulation of proliferation and apoptosis of glioma cells.
Table 1
Clinicopathological characteristics of patients and expression of CEP55 in glioma.
| Variables | Total patients | CEP55 low expression (%) | CEP55 high expression (%) | p-value |
|---|---|---|---|---|
| Gender | 0.130 | |||
| Male | 35 | 15 (42.9) | 20 (57.1) | |
| Female | 10 | 7 (70.0) | 3 (30.0) | |
| Age (years) | 0.465 | |||
| ≤ 40 | 27 | 12 (44.4) | 15 (55.6) | |
| > 40 | 18 | 10 (55.6) | 8 (44.4) | |
| Preoperative KPS | 0.001 | |||
| ≤ 70 | 26 | 7 (26.9) | 19 (73.1) | |
| > 70 | 19 | 15 (78.9) | 4 (21.1) | |
| Histopathology | 0.165 | |||
| WHO II | 14 | 9 (64.3) | 5 (35.7) | |
| WHO III-IV | 31 | 13 (41.9) | 18 (58.1) |
Knockdown of CEP55 inhibits glioma development via suppressing Akt/mTOR signaling
Since CEP55 has been reported to regulate embryonic growth and cancer progression by promoting Akt signaling [25, 30], we hypothesized that CEP55 might employ the same mechanism to regulate glioma development. To test this hypothesis, we first examined the status of a series of proteins reflecting the activity of Akt signaling by Western blot analysis. We found that when CEP55 was knocked down, the levels of both phospho-Akt and phosphor-mTOR were decreased in glioma cells (Fig.5A), indicating decreased Akt signaling activity. The levels of the downstream targets of Akt signaling, including BAD, caspase-9, p27 and GSK-3β were also modulated by CEP55 knockdown. Knockdown of CEP55 increased BAD, caspase-9 and p27 expression, and decreased GSK-3β levels (Fig.5B). These Akt signaling downstream genes involve in programmed cell death, cell cycle and energy metabolism, and determine the process of cell apoptosis, proliferation and glucose metabolism [31-33]. If CEP55 regulates glioma development via modulating the activity of the Akt signaling pathway, CEP55 would not be able to regulate glioma development when the Akt signaling pathway is pharmacologically blocked. We treated U87 and T98G cells with Akt inhibitor MK-2206 or mTOR inhibitor rapamycin, and examined the effects of CEP55 knockdown in regulating glucose uptake (Fig.6A), cell proliferation (Fig.6B) and cell viability (Fig.6C). While CEP55 knockdown induced significantly decreased glucose uptake, cell proliferation and cell viability in U87 and T98G cells not treated with Akt signaling inhibitors, CEP55 knockdown was no longer able to cause reduced glucose uptake, cell proliferation or cell viability in cells treated with either Akt inhibitor or mTOR inhibitor. These results suggested that Akt/mTOR signaling activity was required for CEP55 to regulate glucose metabolism, proliferation and apoptosis of glioma cells.
Discussion
CEP55 was initially identified as a pivotal component of cytokinesis, serving to regulate the physical separation of two daughter cells. However, over the past decade, several studies have illuminated additional roles of CEP55 in the context of cancers including those of the lung [20], bladder [24] and liver [21]. CEP55 overexpression has been found to significantly correlate with tumor stage, aggressiveness, metastasis and poor prognosis across multiple tumor types and therefore has been included as part of several prognostic 'gene signatures' for cancer [13-18]. The role of CEP55 in the development of glioma was nonetheless not carefully studied.
Figure 3
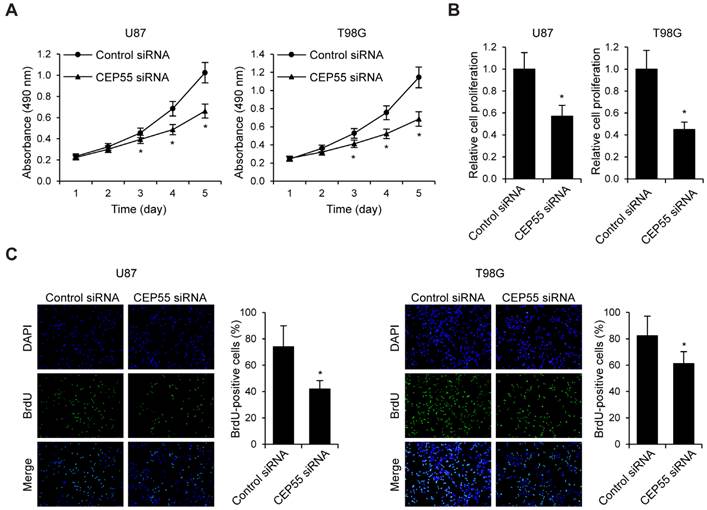
Knockdown of CEP55 inhibits glioma cell proliferation. (A) Cell proliferation rates as determined by MTS assay in U87 and T98G cells transfected with control siRNA or siRNA targeting CEP55. (B) Cell proliferation rates as determined by CCK-8 assay in U87 and T98G cells transfected with control siRNA or siRNA targeting CEP55. (C) BrdU staining and quantification in U87 and T98G cells transfected with control siRNA or siRNA targeting CEP55. *p < 0.5 compared to control siRNA transfected cells.
Figure 4
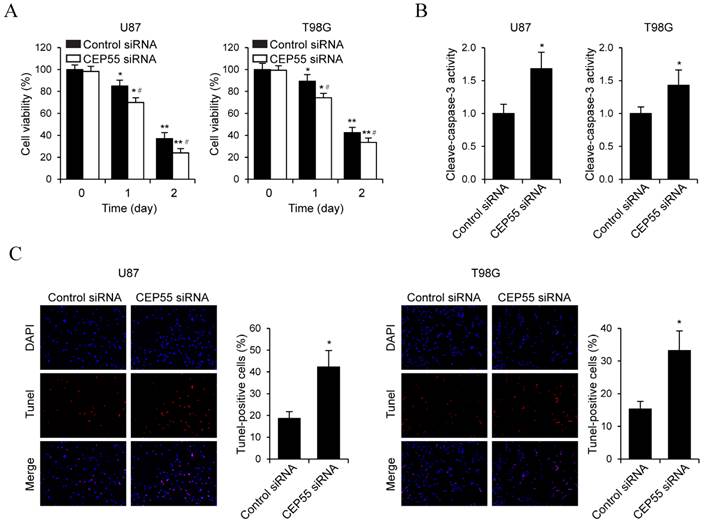
Knockdown of CEP55 induces glioma cell apoptosis. (A) Cell viability assay in U87 and T98G cells transfected with control siRNA or siRNA targeting CEP55. (B) Caspase-3 activity assay in U87 and T98G cells transfected with control siRNA or siRNA targeting CEP55. (C) TUNEL staining and quantification in U87 and T98G cells transfected with control siRNA or siRNA targeting CEP55. *p < 0.5 compared to control siRNA transfected cells.
Figure 5
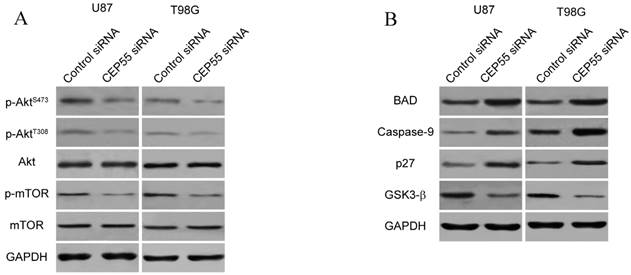
CEP55 regulates the Akt/mTOR signaling pathway. (A) Protein levels of p-Akt, total Akt, p-mTOR and total mTOR as determined by Western blot analysis in U87 and T98G cells transfected with control siRNA or siRNA targeting CEP55. GAPDH was used as the loading control. (B) Protein levels of Akt downstream targets (BAD, Caspase-9, p27 and GSK-3β) as determined by Western blot analysis in U87 and T98G cells transfected with control siRNA or siRNA targeting CEP55.
We found in this study that the expression levels of CEP55 were upregulated in glioma tissues. The correlation between upregulation of CEP55 and glioma occurrence suggested that CEP55 plays important roles in supporting tumor development under physiological conditions. In support of this hypothesis, we found that knockdown of CEP55 inhibited glucose uptake and cell proliferation of glioma cells, while in the same time induced glioma cell apoptosis. Further investigation in this study revealed that the function of CEP55 in regulating glucose metabolism, cell proliferation apoptosis of glioma cells was dependent on the activity of the Akt signaling pathway, as pharmacological blockade of Akt signaling abolished CEP55 effects. Our results are consistent with previous studies with regard to the function of the Akt signaling pathway in cancer development. The Akt pathway plays critical roles in the regulation of apoptosis [34]. Deregulation of apoptosis leads to the expansion of neoplastic cells and accumulation of genetic mutations [35], and therefore constitutes one of the key mechanisms in the progression of cancers. The Akt signaling pathway was also demonstrated to play diverse roles in cancer development [36, 37], including agiogenesis [38], cell growth [39] and mesenchymal transition [40]. In this context, the regulation of proliferation and apoptosis of glioma cells by CEP55 could be explained, at least in part, by the modulation of the Akt signaling pathway.
Interestingly, the regulation of Akt signaling by CEP55 is not restricted in cancer development. A CEP55 mutant zebrafish line exhibited abnormal development and larval lethality largely because of massively increased apoptosis through Akt destabilization [30]. CEP55 was also implicated in vertebrate neural function. CEP55 mediated an increase in the activation of the Akt pathway, resulting in upregulation of the nitric oxide synthase II pathway, progressive hypotension and a reduction of smooth muscle tension in the blood vessels [41].
Figure 6
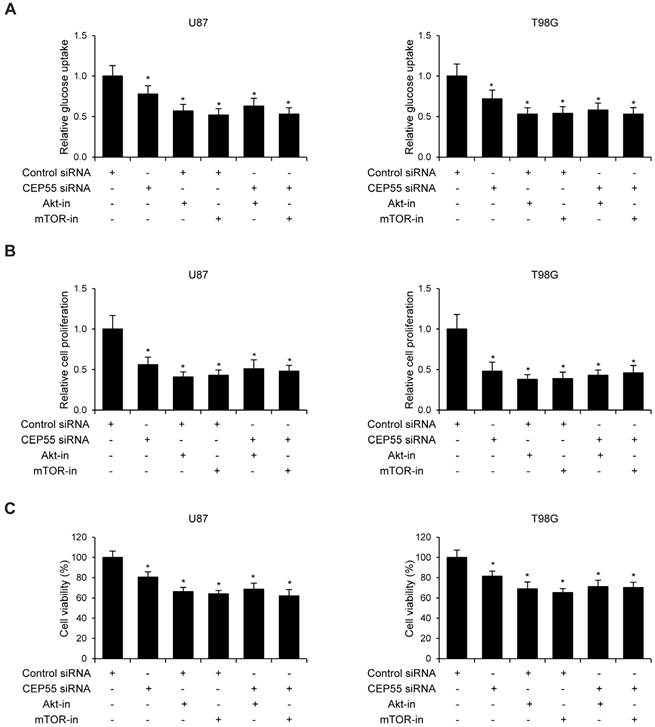
Knockdown of CEP55 inhibits glioma development via suppressing Akt/mTOR signaling. (A) Relative glucose uptake, (B) cell proliferation and (C) cell viability in U87 and T98G cells transfected with control siRNA or siRNA targeting CEP55 treated with or without Akt inhibitor MK-2206 or mTOR inhibitor rapamycin.
Previous studies have shed light on the mechanism by which CEP55 regulates the Akt signaling pathway. It has been found that the Akt signaling pathway is deregulated in multiple cancer types mainly through mutation of the catalytic subunit of PI3K, p110. Mutation of p110 leads to constitutive PI3K activity or loss of phosphatase, resulting in increased pool of PIP3, and consequently to Akt overactivity [42]. CEP55 has been reported to bind p110. This interaction increases the stability of this subunit, resulting in increased Akt activation as evidenced by an increase in S473 phosphorylation [21]. Whether or not CEP55 regulates Akt signaling through p110 in glioma is not currently clear, and it remains an ongoing topic that we are actively pursuing to address.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Nos. 81572472, 81571646 and 61402138), the National High Technology Research and Development Program of China (863) (No. 2012AA02A508) and the International S&T Cooperation Program of China (Nos. 2014DFA31630 and 2012DFA30470).
Conflict of interest
The authors have declared no competing interest exists.
References
1. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ. et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. The New England journal of medicine. 2005;352:987-96
2. Dolecek TA, Propp JM, Stroup NE, Kruchko C. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2005-2009. Neuro-oncology. 2012;14(Suppl 5):v1-49
3. Shervington L, Patil H, Shervington A. Could the Anti-Chaperone VER155008 Replace Temozolomide for Glioma Treatment. Journal of Cancer. 2015;6:786-94
4. Alifieris C, Trafalis DT. Glioblastoma multiforme: Pathogenesis and treatment. Pharmacology & therapeutics. 2015;152:63-82
5. Guo M, Zhang X, Wang G, Sun J, Jiang Z, Khadarian K. et al. miR-603 promotes glioma cell growth via Wnt/beta-catenin pathway by inhibiting WIF1 and CTNNBIP1. Cancer letters. 2015;360:76-86
6. Jiang Y, Zhang Q, Bao J, Du C, Wang J, Tong Q. et al. Schisandrin B suppresses glioma cell metastasis mediated by inhibition of mTOR/MMP-9 signal pathway. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2015;74:77-82
7. Jiang L, Wang C, Lei F, Zhang L, Zhang X, Liu A. et al. miR-93 promotes cell proliferation in gliomas through activation of PI3K/Akt signaling pathway. Oncotarget. 2015;6:8286-99
8. Dixit D, Ghildiyal R, Anto NP, Ghosh S, Sharma V, Sen E. Guggulsterone sensitizes glioblastoma cells to Sonic hedgehog inhibitor SANT-1 induced apoptosis in a Ras/NFkappaB dependent manner. Cancer letters. 2013;336:347-58
9. Fabbro M, Zhou BB, Takahashi M, Sarcevic B, Lal P, Graham ME. et al. Cdk1/Erk2- and Plk1-dependent phosphorylation of a centrosome protein, Cep55, is required for its recruitment to midbody and cytokinesis. Developmental cell. 2005;9:477-88
10. Lee HH, Elia N, Ghirlando R, Lippincott-Schwartz J, Hurley JH. Midbody targeting of the ESCRT machinery by a noncanonical coiled coil in CEP55. Science. 2008;322:576-80
11. Martinez-Garay I, Rustom A, Gerdes HH, Kutsche K. The novel centrosomal associated protein CEP55 is present in the spindle midzone and the midbody. Genomics. 2006;87:243-53
12. Zhao WM, Seki A, Fang G. Cep55, a microtubule-bundling protein, associates with centralspindlin to control the midbody integrity and cell abscission during cytokinesis. Molecular biology of the cell. 2006;17:3881-96
13. Fournier MV, Martin KJ, Kenny PA, Xhaja K, Bosch I, Yaswen P. et al. Gene expression signature in organized and growth-arrested mammary acini predicts good outcome in breast cancer. Cancer research. 2006;66:7095-102
14. Jones J, Otu H, Spentzos D, Kolia S, Inan M, Beecken WD. et al. Gene signatures of progression and metastasis in renal cell cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2005;11:5730-9
15. Kikuchi T, Daigo Y, Katagiri T, Tsunoda T, Okada K, Kakiuchi S. et al. Expression profiles of non-small cell lung cancers on cDNA microarrays: identification of genes for prediction of lymph-node metastasis and sensitivity to anti-cancer drugs. Oncogene. 2003;22:2192-205
16. Ma XJ, Salunga R, Tuggle JT, Gaudet J, Enright E, McQuary P. et al. Gene expression profiles of human breast cancer progression. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:5974-9
17. Martin KJ, Patrick DR, Bissell MJ, Fournier MV. Prognostic breast cancer signature identified from 3D culture model accurately predicts clinical outcome across independent datasets. PloS one. 2008;3:e2994
18. Naderi A, Teschendorff AE, Barbosa-Morais NL, Pinder SE, Green AR, Powe DG. et al. A gene-expression signature to predict survival in breast cancer across independent data sets. Oncogene. 2007;26:1507-16
19. Chen CH, Chien CY, Huang CC, Hwang CF, Chuang HC, Fang FM. et al. Expression of FLJ10540 is correlated with aggressiveness of oral cavity squamous cell carcinoma by stimulating cell migration and invasion through increased FOXM1 and MMP-2 activity. Oncogene. 2009;28:2723-37
20. Chen CH, Lai JM, Chou TY, Chen CY, Su LJ, Lee YC. et al. VEGFA upregulates FLJ10540 and modulates migration and invasion of lung cancer via PI3K/AKT pathway. PloS one. 2009;4:e5052
21. Gaur A, Jewell DA, Liang Y, Ridzon D, Moore JH, Chen C. et al. Characterization of microRNA expression levels and their biological correlates in human cancer cell lines. Cancer research. 2007;67:2456-68
22. Chen CH, Shiu LY, Su LJ, Huang CY, Huang SC, Huang CC. et al. FLJ10540 is associated with tumor progression in nasopharyngeal carcinomas and contributes to nasopharyngeal cell proliferation, and metastasis via osteopontin/CD44 pathway. Journal of translational medicine. 2012;10:93
23. Hwang CF, Shiu LY, Su LJ, Yu-Fang Y, Wang WS, Huang SC. et al. Oncogenic fibulin-5 promotes nasopharyngeal carcinoma cell metastasis through the FLJ10540/AKT pathway and correlates with poor prognosis. PloS one. 2013;8:e84218
24. Singh PK, Srivastava AK, Rath SK, Dalela D, Goel MM, Bhatt ML. Expression and clinical significance of Centrosomal protein 55 (CEP55) in human urinary bladder transitional cell carcinoma. Immunobiology. 2015;220:103-8
25. Tao J, Zhi X, Tian Y, Li Z, Zhu Y, Wang W. et al. CEP55 contributes to human gastric carcinoma by regulating cell proliferation. Tumour biology: the journal of the International Society for Oncodevelopmental Biology and Medicine. 2014;35:4389-99
26. Goel HL, Mercurio AM. VEGF targets the tumour cell. Nature reviews Cancer. 2013;13:871-82
27. Guo M, Shen J, Kwak JH, Choi B, Lee M, Hu S. et al. Novel role for cyclophilin A in regulation of chondrogenic commitment and endochondral ossification. Molecular and cellular biology. 2015;35:2119-30
28. Jeffery J, Sinha D, Srihari S, Kalimutho M, Khanna KK. Beyond cytokinesis: the emerging roles of CEP55 in tumorigenesis. Oncogene. 2015
29. Adekola K, Rosen ST, Shanmugam M. Glucose transporters in cancer metabolism. Current opinion in oncology. 2012;24:650-4
30. Jeffery J, Neyt C, Moore W, Paterson S, Bower NI, Chenevix-Trench G. et al. Cep55 regulates embryonic growth and development by promoting Akt stability in zebrafish. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2015;29:1999-2009
31. Patel P, Asbach B, Shteyn E, Gomez C, Coltoff A, Bhuyan S. et al. Brk/Protein tyrosine kinase 6 phosphorylates p27KIP1, regulating the activity of cyclin D-cyclin-dependent kinase 4. Molecular and cellular biology. 2015;35:1506-22
32. Ibata-Ombetta S, Idziorek T, Trinel PA, Poulain D, Jouault T. Role of phospholipomannan in Candida albicans escape from macrophages and induction of cell apoptosis through regulation of bad phosphorylation. Annals of the New York Academy of Sciences. 2003;1010:573-6
33. Zhang P, Li S, Gao Y, Lu W, Huang K, Ye D. et al. Novel benzothiazinones (BTOs) as allosteric modulator or substrate competitive inhibitor of glycogen synthase kinase 3beta (GSK-3beta) with cellular activity of promoting glucose uptake. Bioorganic & medicinal chemistry letters. 2014;24:5639-43
34. Hemmings BA. Akt signaling: linking membrane events to life and death decisions. Science. 1997;275:628-30
35. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-74
36. Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nature reviews Drug discovery. 2009;8:627-44
37. Yu X, Zhen Y, Yang H, Wang H, Zhou Y, Wang E. et al. Loss of connective tissue growth factor as an unfavorable prognosis factor activates miR-18b by PI3K/AKT/C-Jun and C-Myc and promotes cell growth in nasopharyngeal carcinoma. Cell death & disease. 2013;4:e634
38. Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261-74
39. Lau MT, Leung PC. The PI3K/Akt/mTOR signaling pathway mediates insulin-like growth factor 1-induced E-cadherin down-regulation and cell proliferation in ovarian cancer cells. Cancer letters. 2012;326:191-8
40. Wen W, Ding J, Sun W, Fu J, Chen Y, Wu K. et al. Cyclin G1-mediated epithelial-mesenchymal transition via phosphoinositide 3-kinase/Akt signaling facilitates liver cancer progression. Hepatology. 2012;55:1787-98
41. Tsai CY, Chen CH, Chang AY, Chan JY, Chan SH. Upregulation of FLJ10540, a PI3K-association protein, in rostral ventrolateral medulla impairs brain stem cardiovascular regulation during mevinphos intoxication. Biochemical pharmacology. 2015;93:34-41
42. Cheung M, Testa JR. Diverse mechanisms of AKT pathway activation in human malignancy. Current cancer drug targets. 2013;13:234-44
Author contact
![]() Corresponding authors: Chuanlu Jiang, Department of Neurosurgery, the Second Affiliated Hospital of Harbin Medical University, 246 Xuefu Road, Nangang, Harbin, Harbin, Heilongjiang 150086, China. Email: jcl6688com; Tel.: 86-451-86605040; Fax: 86-451-86605041 or Mian Guo, Department of Neurosurgery, the Second Affiliated Hospital of Harbin Medical University, 246 Xuefu Road, Nangang, Harbin, Harbin, Heilongjiang 150086, China. Email: guomianedu.cn; Tel.: 86-451-86605040; Fax: 86-451-86605041.
Corresponding authors: Chuanlu Jiang, Department of Neurosurgery, the Second Affiliated Hospital of Harbin Medical University, 246 Xuefu Road, Nangang, Harbin, Harbin, Heilongjiang 150086, China. Email: jcl6688com; Tel.: 86-451-86605040; Fax: 86-451-86605041 or Mian Guo, Department of Neurosurgery, the Second Affiliated Hospital of Harbin Medical University, 246 Xuefu Road, Nangang, Harbin, Harbin, Heilongjiang 150086, China. Email: guomianedu.cn; Tel.: 86-451-86605040; Fax: 86-451-86605041.
Citation styles
APA
Wang, G., Liu, M., Wang, H., Yu, S., Jiang, Z., Sun, J., Han, K., Shen, J., Zhu, M., Lin, Z., Jiang, C., Guo, M. (2016). Centrosomal Protein of 55 Regulates Glucose Metabolism, Proliferation and Apoptosis of Glioma Cells via the Akt/mTOR Signaling Pathway. Journal of Cancer, 7(11), 1431-1440. https://doi.org/10.7150/jca.15497.
ACS
Wang, G.; Liu, M.; Wang, H.; Yu, S.; Jiang, Z.; Sun, J.; Han, K.; Shen, J.; Zhu, M.; Lin, Z.; Jiang, C.; Guo, M. Centrosomal Protein of 55 Regulates Glucose Metabolism, Proliferation and Apoptosis of Glioma Cells via the Akt/mTOR Signaling Pathway. J. Cancer 2016, 7 (11), 1431-1440. DOI: 10.7150/jca.15497.
NLM
Wang G, Liu M, Wang H, Yu S, Jiang Z, Sun J, Han K, Shen J, Zhu M, Lin Z, Jiang C, Guo M. Centrosomal Protein of 55 Regulates Glucose Metabolism, Proliferation and Apoptosis of Glioma Cells via the Akt/mTOR Signaling Pathway. J Cancer 2016; 7(11):1431-1440. doi:10.7150/jca.15497. https://www.jcancer.org/v07p1431.htm
CSE
Wang G, Liu M, Wang H, Yu S, Jiang Z, Sun J, Han K, Shen J, Zhu M, Lin Z, Jiang C, Guo M. 2016. Centrosomal Protein of 55 Regulates Glucose Metabolism, Proliferation and Apoptosis of Glioma Cells via the Akt/mTOR Signaling Pathway. J Cancer. 7(11):1431-1440.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.