Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2016; 7(11):1421-1430. doi:10.7150/jca.15480 This issue Cite
Review
Gut Microbiota Imbalance and Base Excision Repair Dynamics in Colon Cancer
Debolina Ray, Dawit Kidane ![]()
Division of Pharmacology and Toxicology, College of Pharmacy, The University of Texas at Austin, Dell Pediatric Research Institute, 1400 Barbara Jordan Blvd. R1800, Austin, TX 78723, United States.
Received 2016-3-8; Accepted 2016-5-18; Published 2016-7-4
Abstract
Gut microbiota are required for host nutrition, energy balance, and regulating immune homeostasis, however, in some cases, this mutually beneficial relationship becomes twisted (dysbiosis), and the gut flora can incite pathological disorders including colon cancer. Microbial dysbiosis promotes the release of bacterial genotoxins, metabolites, and causes chronic inflammation, which promote oxidative DNA damage. Oxidized DNA base lesions are removed by base excision repair (BER), however, the role of this altered function of BER, as well as microbiota-mediated genomic instability and colon cancer development, is still poorly understood. In this review article, we will discuss how dysbiotic microbiota induce DNA damage, its impact on base excision repair capacity, the potential link of host BER gene polymorphism, and the risk of dysbiotic microbiota mediated genomic instability and colon cancer.
Keywords: Microbiota, Base excision repair, Colon cancer.
Introduction
The human gut is a complex consortium of trillions of microbes, which contain at least 100 times as many genes as the human genome [1]. The composition of intestinal microbiota is determined by various factors including host genetics [2], environment [3, 4], and diet [5]. Dietary habits are considered one of the main factors contributing to the diversity of human gut microbiota [5]. The main functions of the gut microflora include metabolic activities that result in the salvage of energy and the protection of the host against invasion by pathogenic microbes [6]. However, perturbation of the epithelial lining and mucous layer of the human intestinal tract probably causes alteration of the physiological and immunological reactions of the host to promote an inflammatory response and genomic instability.
What determines the colon microbiome's contribution to the pathogenesis of colon cancer? One of the challenges in identifying a microbe as the cause of a disease is the possibility that the inciting microbe is no longer present at the time the disease is diagnosed. Because of this change in the microenvironment, the inciting microbes may have been eliminated or the microbe might have conducted a “hit and run” mechanism to trigger the disease. Previously published work from Sears, et al, proposed three different models to show how microbes provoke colon cancer. The first model represents a specific group of microbes that possess sufficient virulence mechanisms that may induce disease. The second model requires a host genetic factor that permits the virulence of a single microbe to initiate the disease. The third model requires a microbial community or two, acting sequentially and/or in synergy, that causes dysbiosis and influences colon carcinogenesis [7]. All three models could provoke chronic inflammation. Chronic inflammation may promote the release of reactive oxygen and nitrogen species (RONs) that are thought to damage DNA. The cellular consequences of DNA oxidation by RONs can lead to a number of different types of damage such as generation of 7, 8-hydroxy-2'-deoxyguanosine (8-Oxod-G), thymine glycol (Tg), abasic sites (AP), and oxidized deoxyribose sugars which lead to single and double strand DNA breaks (SSBs and DSBs) [8], crosslinking of DNA, and mutation [9-13]. These kinds of DNA damage can be repaired by a variety of DNA repair mechanisms including base excision repair (BER) [14-16]. BER repair systems are thought to play a significant role in survival and adaptation of microbiota, repair of microbiota, or bacterial pathogen induced DNA damage [17]. BER is the major repair pathway of DNA damage induced by RONs and is critical for maintaining genome stability during chronic inflammation and/or bacterial infection [18]. Aberrant function of BER significantly increases chronic inflammation to trigger genomic instability and colon cancer [18, 19]. BER is initiated by DNA glycosylases that recognize and cleave the microbiota mediated base lesions: including the removal of 8-Oxo-G paired with C which involves the bifunctional DNA glycosylase OGG1 [20-22], MUTYH that removes adnine mispaired with 8-oxoG lesions, and TDG that removes thymine glycol paired with G that eventually creates abasic (AP) sites [23] which are cytotoxic and mutagenic [24-26] and further processed by DNA glycosylase with AP-lyase activity or by APE-1 [27]. The single-nucleotide gap is filled by DNA polymerase beta (Pol β) and the nick is sealed by DNA ligase to complete the repair [27].
Microbiota dynamics and oxidative DNA damage
The healthy human gastrointestinal tract is dominated by obligate anaerobic bacteria, the Firmicutes, Bacteroidetes (Bacteroides, Bifidobacterium, Eubacterium, Clostridium, Peptococcus, Peptostreptococcus, and Ruminococcus) and Actinobacteria. However, during microbiota dysbiosis, there is an increase in facultative anaerobic bacteria (Escherichia, Enterobacter, Enterococcus, Klebsiella, Lactobacillus, and Proteus) which are prone to induce inflammatory processes [28]. Moreover, some of the bacteria including enterotoxigenic B. fragilis [29], Fusobacterium nucleatum [30, 31], and colibactin-producing E. coli [32, 33] promote intestinal carcinogenesis. The possible mechanism of DNA damage induced by dysbiotic microbiota may be explained in four different ways. The first potential mechanism is involved when the dysbiotic microbiota release mitogen that possesses the capacity to enter into the cell and induce the expression of enzymes that enhance RONs, which causes DNA damage. The second mechanism will likely occur when the dysbiotic microbiota provoke chronic inflammation to initiate DNA damage and cellular transformation [34-36]. In this case, dysbiotic microbiota induce a constant influx of antigens, such as bacterial cell components, to stimulate host pathogen recognition receptors, provoking chronic inflammation [37, 38]. For example, Fusobacterium nucleatum increases tumor multiplicity by the infiltration of immune cells and NF-κB activation that potentiate intestinal tumorigenesis [30]. With the third mechanism, the dysbiotic microbiota directly releases RONs in the gastrointestinal tracts to induce DNA damage and BER intermediates in the gut [18]. The wide spectrum of DNA damage including single and double strand breaks, abasic sites, nucleotide modification, and an increase in mutations which activate oncogenes or suppress tumor-suppressor genes [39] can accelerate genomic instability and result in colon cancer [40-43] (Figure 1). Finally, the dysbiotic microbiota release specific toxins that have the capacity to directly induce DNA damage and modulate tumorigenesis. For example, bacterial toxins such as cytolethal distending toxin (CDT), cytotoxic necrotizing factor 1, B. fragilis toxin, and colibactin induce DNA double strand breaks that are implicated in genomic instability and tumorigenesis [32, 44-46]. Few studies have suggested that bacterial toxins that are released from many gram-negative bacteria, such as Escherichia coli, Shigella dysenteriae, Actinobacillus actinomycetemcomitans, Campylobacter spp., Helicobacter spp., Salmonella typhi, and H. ducreyi, cause DNA damage, cell cycle arrest, and/or apoptosis [47-50]. However, the mechanism of how microbial toxins induce base excision repair intermediate and its effect on base excision repair capacity is not yet clearly understood.
Possible mechanism of gut microbial impact on genomic instability and Cancer : Many factors including antibiotic use, psychological and physical stress, radiation, and dietary changes can change beneficial members of the gastrointestinal flora (symbiont) and cause disequilibrium of the normal function of (microbiota dysbiosis). The altered bacterial community (pathobiont) in gastrointestinal tract likely release bacterial toxin and /mitogen /bacterial metabolite directly to induce reactive oxygen and nitrogen species (RONs) or indirectly induce chronic inflammation that could result in base damage (represented as red circle). The base damage could be removed by OGG1/MUTYH/TDG/AAG/NEIL1, 2,3, resulting in the formation of AP sites (represented by blue cross mark). AP sites are clustered in close proximity on the opposite strands of the DNA, and processed with AP lyase activity that cleaves the 3' side of AP sites [22] or with APE1 that cleaves the phosphate backbone and generate DSBs that eventually leads to genomic instability and cancer. Furthermore, if DSBs repaired by non-homologous end joining likely trigger genomic instability, cellular transformation and cancer.
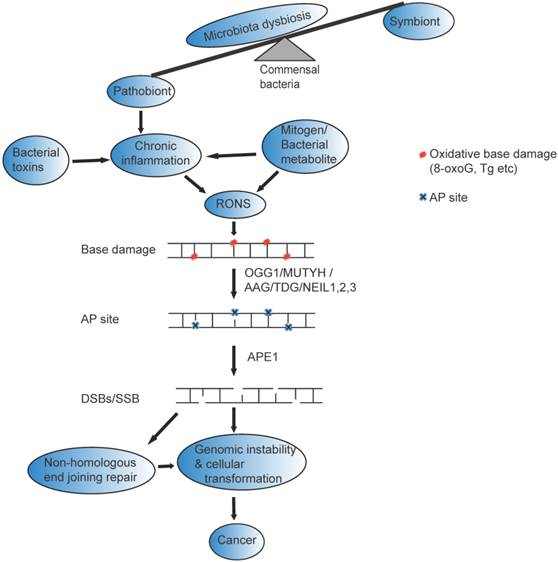
Base excision repair dynamics and microbiota
Base excision repair systems play a significant role in maintaining the genomic integrity of the host cells. BER removes base damage via a number of coordinated sequential reactions that detect and process the damaged base [51]. Modulation of the levels of individual BER enzymes leads to altered phenotypes in mouse model [52]. Evidence from several laboratories indicates that BER imbalances are detrimental [53, 54]. Dysbiotic microbiota may allow the accumulation of unrepaired DNA breaks and BER intermediates that can lead to genomic instability and carcinogenesis. The aberrant function of BER decreases the tumor latency in host carriers of dysbiotic microbiota. The dysbiotic microbiota may induce different types of DNA base lesions that are likely recognized and removed by different types of DNA glycosylases. Mammalian cells contain 11 different glycosylases each with a specialized function [55]. DNA glycosylase initiates the BER process by removing the modified base, which generates an apurinic/apyrimidinic site. If the microbiota induces base damage recognized by bi-functional glycosylases that have an associated apurinic/apyrimidinic lyase activity, this further catalyzes the cleavage of the sugar-phosphate chain and the excision of the abasic residue leaving a single nucleotide gap. This gap is filled by DNA polymerase beta (Pol β) and the nick is sealed by the DNA ligase III/x-ray repair cross-complementing group 1 (XRCC1) complex. In contrast, if microbiota induces base damage and is recognized by monofunctional DNA glycosylases that lack lyase activity which leaves the phosphodiester bond at the 5′ side of the intact apurinic/apyrimidinic site, this will be incised by apurinic/apyrimidinic endonuclease (APE1/APEX1). Finally, Pol β, DNA ligase III, and XRCC1 will complete the repair process.
Does base excision repair polymorphism increase the risk of microbiota induced colon cancer?
Host genetics play an important role in the establishment and shaping of the gut microbiota [3]. Genetic association studies on cancer risk have focused on identifying the functional effects of single nucleotide polymorphisms (SNP) in candidate genes. Sequence variants in DNA repair genes are thought to modulate DNA repair capacity and consequently are suggested to be associated with an altered cancer risk [56]. Several genetic epidemiological studies have linked SNP variant in BER genes to human cancer [57]. However, the link between dysbiotic microbiota and SNP variant of BER genes that cause the loss of biological function has not yet been clearly documented. Several studies show that the impaired function of BER gives rise to genomic instability and cellular transformation [58]. Polymorphisms in BER genes could affect the accumulation of DNA lesions in colorectal mucosa, thus influencing colon cancer risk [59]. BER genes are the most common and well-studied [60], though some of the associations of BER gene polymorphisms and colon cancer risk have been inconsistent [61-63]. However, the characterization of dysbiotic microbiota as a risk factor to induce DNA damage for those who carry BER polymorphism has yet to be studied. The altered function of microbiota likely leads to a different type of DNA base damage that causes a substrate for different types of DNA glycosylase. The biological significance of genetic polymorphism of DNA glycosylases, APE1 and DNA polymerase beta, that are involved in colon cancer (Table 1) and its impact on dysbiotic microbiota are discussed below. However, the biological significance of BER SNPs variant and its impact on gastrointestinal tract microbiota dynamics in promoting the risk of colon cancer have not yet been understood.
Single nucleotide polymorphisms likely a target risk for microbiota dysbiosis to initiate colon cancer. Germline variants of base excision repair enzymes, which are found in specific populations that are susceptible to colon cancer.
| Gene | SNPID | Polymorphism | Amino Acid Change | Population susceptible to CRC | References |
|---|---|---|---|---|---|
| TDG | rs3829300 | A:G | Intron | Asian | [123] |
| TDG | rs3751209 | G:A | Intron | European | [123] |
| TDG | rs2888805 | G:A | V367M | Caucasian | [123] |
| MBD4 | rs140693 | C:T | E346K | East Asian, Korean | [78] |
| MGMT | rs2308321 | A:G | I143V | N. America, European | http://browser.1000genomes.org |
| MGMT | rs2308327 | G:A | K178R | N. America | |
| MGMT | rs12917 | C:T | L84F | N. America | |
| MGMT | rs2308318 | G:A | G160R | Japanese | |
| NEIL1 | rs5745909 | C:T | intron | European | http://www.1000genomes.org. [124] |
| NEIL2 | rs8191683 | delCA | Intron | European, African | http://www.ncbi.nlm.nih.gov/snp http://browser.1000genomes.org |
| NEIL2 | rs8191664 | A:G | R257L | East Asian, European | |
| AAG | rs2259275 | C:G | Intron | European, East Asian | http://www.ncbi.nlm.nih.gov/snp. [93] |
| AAG | rs2308312 | G:A | R141Q | European | |
| AAG | rs2308313 | C:T | R120C | East Asian | |
| OGG1 | rs1052133 | C:G | S326C | Asian, Caucasian | [61] |
| MUTYH | rs3219489 | C:G | Q324H | African American, European | [150] |
| MUTYH | rs36053993 | C:T | G382D | American, European | [107, 108] |
| MUTYH | rs34612342 | G:A | Y165C | Peruvian | [107, 108] |
| MUTYH | rs34612342 | G:A | Y179C | Peruvian | http://www.LOVD.nl/MUTYH. |
| MUTYH | rs36053993 | G:A | G396D | European, Australia, N. America | [105] |
| APEX1 | rs1760944 | T:G | D148E | Turkish, Polish | [151] |
| POLB | rs12678588 | A:G | R137Q | Asians& North Americans | [144] |
| POLB | rs313679 | C:G | P242R | Europeans, Asians | [144] |
TDG: Thymine DNA glycosylase (TDG) is a monofunctional DNA glycosylase and preferentially catalyzes the removal of thymine and uracil paired with guanine, and is also active on 5-fluorouracil (5-FU) paired with adenine or guanine [64, 65]. TDG also interacts with activation-induced deaminase and works with 5-methylcytosine hydroxylases TETs (ten-eleven translocation) to regulate active DNA demethylation [66, 67]. In addition, TDG interacts with transcription factors [68] and plays essential roles in epigenetic regulation [69]. In the human genome a heterozygous mutation in the TDG gene was identified in a rectal cancer patient [70, 71] suggesting that TDG may function as a tumor suppressor. The rs4135113 SNP variant of TDG, where G is mutated to A at position 818, has a minor allele frequency of approximately 10%; it is most commonly found in African and East Asian populations and is usually heterozygous [72]. Even though there is not any scientific evidence, it is possible to predict that the bacterial metabolites and toxins that are produced from gut microbiota in African and East Asian populations may induce base damage that causes more G/T mismatched pairs that increase mutagenesis in this rs4135113 SNP variant carrier population which contributes to the initiation of carcinogenesis.
MBD4: A methyl-CpG-binding DNA glycosylase (MBD4) is involved in the repair of mismatches arising from deamination of methyl-C in mammalian cells. In vitro experiments have shown that MBD4 excises mismatched thymine (T) bases from oligo templates [73, 74]. In addition, the mutation frequency at methyl-CpG sites is significantly increased in MDB4 knockout mouse [75] and shows reduced apoptosis in response to DNA damage [76]. Absence of MBD4 in mice also increases tumorigenicity in the tumor-susceptible APCMin/+ background [75]. The tumorigenic effect may be due to an increase in mutation frequency, decreased apoptosis, or a combination of both. Previous studies have suggested that gut microbiota derived antigens promote inflammation that may cause intestinal epithelial disruption and activate signaling pathways to enhance intestinal tumor load in the APCMin/+ mouse model of colon cancer [77]. However, the mechanism of how dysbiotic microbiota induce genomic instability and carcinogenesis in MBD4/APCMin/+ compound mutant as well was how dysbiotic microbiota impact those who carry the MBD4 SNP variant is still unclear and would be an interesting scientific question to explore in the future. For example, a single nucleotide polymorphism in the MBD4 gene (rs140693) has been associated with increased risk of colorectal cancer in Korean populations [78]. This polymorphism results in an amino acid change from Glutamine to Lysine at position 346 driving microsatellite instability [78]. However, whether the altered function of gut microbiota contributes for early onset genomic instability and cancer in this population that carries MBD4 SNP variants remains unknown. Thus, we have hypothesized that microbiota dysbiosis and MBD4 genetic polymorphism may decrease tumor latency and promote an early onset of colon cancers.
MGMT: The DNA repair gene O6-methylguanine-DNA methyltransferase (MGMT) is responsible of the elimination of alkyl groups from the O6-position of guanine and the O4-position of thymine. Altered function of MGMT may be involved in early steps of colorectal tumorigenesis through an increase of the mutational rate particularly with G-to-A point mutations of KRAS gene [79, 80]. Moreover, epigenetic silencing of MGMT during colon tumorigenesis is associated with hypermethylation of the CpG island in its promoter [81]. This transcriptional gene silencing is responsible for diminished DNA-repair of O6-alkylguanine adducts with the consequence of enhancing chemosensitivity to alkylating agents in particular temozolomide [82]. For colon cancer, the MGMT Ile143Val polymorphism may confer an increased or decreased risk depending on dietary exposure [83]. Individuals who carry this variant genotype have a higher risk for colon cancer with a higher intake of red or processed meat. However, whether this dietary exposure alters the function of microbiota to increase the risk of inflammation or metabolite product in a host that carries MGMT genetic polymorphism remains unknown. For example, L84F variant is found in up to 20% of the worldwide population and rescue MGMT-deficient cells in survival assays with alkylating drugs [84, 85]. However, L84F is not inactivated by O6BG treatment [86, 87] suggesting that the active site cysteine is not affected by O6-BG. In contrast, G160R is a rare variant found in Japanese populations and cannot fully rescue MGMT-deficient cells treated with an alkylating agent and resistant to O6BG [88].
AAG: The Alkyladenine DNA glycosylase (AAG) has a wide substrate specificity excising numerous structurally diverse lesions, some of which exert a mild effect (7-methylguanine), while others can be replication blocking and cytotoxic (3-methyladenine) [89]. The absence of AAG results in unrepaired alkylated DNA bases that block DNA replication, thus increasing cytotoxicity, and etheno adducts levels in the colon's DNA [18]. In addition, inflamed colon tissue from ulcerative colitis patients have increased levels of AAG compared with un-inflamed tissue and is presumably induced to repair the DNA base damage that is inflicted by RONs during the inflammatory response [18]. It would be interesting to compare the microbiota dynamics altered in the gastrointestinal tract of AAG deficient mice to see if this promotes genomic instability and/or chronic inflammation. Western diet has been implicated to induce inflammatory immune response through multiple mechanisms and subsequently lead to colon cancer. Western diet has been proven to perturb the gut microbial population and increase colonic permeability to microbial products such as lipopolysaccharides (LPS), which promotes chronic inflammation in the colon [90, 91]. Repeated cycles of Dextran Sulfate Sodium feeding to animal models is an accepted mode of inciting chronic inflammation mimicking consumption of western diet. Previous studies have shown that AAG deficient mouse are more prone to DSS induced colitis upon co-treatment with azoxymethane (AOM) than wild type counterpart [18]. In addition, the AAG deficient mice displayed enhanced tumor multiplicity and development. In contrast, overexpression of AAG promotes microsatellite instability and colon cancer risk [92]. In addition, SNPs variant R141Q and R120C enhance non-specific binding of DNA that may promote genomic instability and cancer [93]. Therefore, the delicate balance of AAG and healthy diets are critical to maintain genomic integrity and prevent colon cancer.
OGG1: Human Oxoguanine Glycosylase 1 (OGG1) is a member of BER repair protein that is involved in the removal of a specific oxidative DNA damaged base 8-oxo-G. Previous studies reported that the inflammatory response in Ogg1-/- mice is lower than that of wild type mice and suggested that OGG1 is an inflammatory/immune system modulator [94]. Therefore, deficiency of OGG1 promotes a protective role against inflammatory lesions [95] and reduces the level of BER intermediates (AP sites) that are generated during bacterial infection [17]. Some of the mechanisms concerning how the lack of OGG1 protects the cells from inflammation or reduces BER intermediate mediated responses may be correlated with an inhibition of the T helper 1 type (Th1) response. This is known to promote inflammation, bacterial load, and epithelial cell damage [95]. Microbiota dysbiosis may induce inflammation that potentially causes 8-oxo-G lesions. Intriguing recent studies have shown that OGG1 binds the 8-oxoG base with a high affinity resulting in immune response [96, 97]. If the host is devoid of functional OGG1, then cells probably accumulate 8-oxoG or other BER intermediates to induce genomic instability and cancer. In contrast, OGG1 deficient mice that are protected from this inflammatory response [94] may suggest that the loss of OGG1 function does not cause immunological disequilibrium incited by microbiota dysbiosis. However, OGG deficient mice treated with DSS significantly increased adenocarcinoma development in the colon with a high incidence of tumor [19]. Furthermore, several reports have been published regarding the association of OGG1 SNP variant rs1052133 polymorphism and cancer [98, 99]. Some epidemiological studies have indicated that this particular polymorphism poses a greater risk for colon cancer in the Caucasian and Asian population [99]. The amino acid change accompanying this single nucleotide polymorphism occurs at position 326 from Serine to Cytosine and has impaired glycosylase activity [100, 101]. The oxidative stress caused by inflammatory cytokines such as TNF-α induces 8-oxoG and inactivate S326C variant of OGG1 increases the risk of cancer among homozygous individuals [102].
MUTYH: The MUTYH protein is a BER glycosylase involved in repair by excising adenine opposite 8-oxoguanine and 2-hydroxyadenine opposite guanine, thereby preventing G: C to T: A transversion caused by oxidative stress. The MUTYH protein directly interacts with various proteins involved in other DNA repair pathways [103, 104]. Several different mutations, mainly missense mutations, have been found. The two most common mutations in Western populations are Y179C and G396D, with probable different effects on MUTYH glycosylase function [105]. Interestingly, MUTYH was expressed at higher levels in the normal colon, and are directly involved in oxidative DNA damage repair. An increased susceptibility to spontaneous carcinogenesis in the intestine was observed in MUTYH-null mice [106]. Loss of MUTYH function may result in an increase of mutations in oncogenes or tumor suppressor genes due to the accumulation of 8-oxoG and this could promote tumorigenesis. Moreover, germline variant of MUTYH increases the susceptibility of European populations to multiple adenomas or polyposis [107]. For example, Y165C and G382D are the most common variants of the MUTYH variant [107, 108]. Both of these MUTYH residues have important roles in the recognition of 8-oxoG in A:8-oxoG mispairs [109-111]. In addition, other variants such as Y179C and G396D lost the ability of substrate recognition [112] and suggested that those germline variants may result in an increase of mutation on oncogene or tumor suppressor genes to initiate genomic instability and carcinogenesis. Thus, microbiota dysbiosis may increase the number of oxidative DNA damage (8-oxoG) that likely increase mutation to induce genomic instability colon cancer.
NEIL 1,2,3: The DNA endonuclease eight-like (NEIL) glycosylases have broader substrate specificity and are associated with the repair of different types of base lesions. NEIL1 repair pyrimidine lesions such as Tg and 5-hydroxyuracil (5-OHU) in duplex DNA [113] and single stranded and bubble DNA structures [114]. Both NEIL2 and NEIL3 prefer oxidized pyrimidine and some purine damages in single stranded DNA [114, 115]. NEIL1 is involved in removing lesions that block replication forks before they are encountered by the replicative DNA polymerases [116] while NEIL2 appears to function during transcription-coupled repair [114, 117]. NEIL3, which is expressed at higher levels in the normal colon, is directly involved in oxidative stress repair and removes lesions from quadruplex DNA [118] and further oxidation products, specifically guanidinohydantoin (Gh) and spiroiminodihydantoin (Sp) substrates [115, 119, 120]. Furthermore, Neil3 is highly expressed in embryonic stem cells as well as pluripotent cells in the brain [121] and human cancer cells [122]. Genome wide association study has shown that NEIL1, 2, 3 are often found to carry mutations and are prevalent in different types of cancers. A specific germline variant of NEIL1, rs5745908, has been noted to contribute to the development of colon cancer in European populations [123] (Table 1). In addition, biochemical characterization of the G83D germline variant of NEIL1 revealed less glycosylase activity [124] that may contribute to the accumulation of an oxidative damaged base. Similarly, SNP variants of Neil 2 (rs8191683 and rs8191664) result in nucleotide changes: A: G and G: T respectively [123] and also contribute to familial colon cancers (Table 1). However, the functional impact of the SNP variant of NEIL3 has not yet been studied. In addition, microbiota dysbiosis mediated DNA damage and DNA repair capacities of NEIL1, 2, 3 SNP variants remain unknown.
APE1: Apurinic endonuclease I (APE1) is the key enzyme responsible for the removal of the apurinic/apyrmidinic sites created at the regions of single base DNA damage and subsequent generation of the 3'-OH termini [125]. In addition to its role in DNA repair, APE1 also functions as a reduction/oxidation (redox) factor in mammalian cells maintaining transcription factors (TFs) in an active reduced state, thus stimulating the DNA binding capacity of several TFs such as AP-1 (activator protein-1-fos/Jun), NFκB (nuclear factor-κB), HIF-1α (hypoxia-inducible factor 1-α), TP53, and others [126, 127]. Furthermore, high expression levels of APE1 enhance cellular resistance to chemotherapeutic agents in several tumor cells [128-130] as well as regulating inflammatory response [131]. It is possible that the dysbiotic microbiota enhance the level of APE1 to provoke redox role or DNA repair activity similar to pathogenic bacteria induced APE1 activity [132]. Even though genetic polymorphism of APE1 is not detected at the active site of endonuclease or the site required for acetylation that impairs redox function or DNA repair activity respectively, some reports have suggested that APE1 Asp148Glu polymorphism is associated with an increased risk of colorectal cancer in Turkish and Polish populations [133, 134]. The mutation of an aspartate residue to a glutamate residue potentially affects the stability of the protein structure and thus could affect the functionality of the enzyme in BER [135] (Table 1).
DNA polymerase beta: Pol β is a key enzyme during BER of oxidative DNA lesions. A number of studies suggest that approximately 30% of human tumors characterized to date express Pol β variant proteins [136]. Excision of damaged bases by DNA glycosylases generates a single nucleotide gap and a 5'-dRP group. The 8 kDa dRP lyase domain of Pol β functions in removing the 5'-dRP group and Pol β then fills in the gap. The regulation of cellular Pol β protein levels is vital as haploinsufficiency resulting in reduced BER capacity has increased susceptibility to cancer [137]. Furthermore, increased expression of Pol β also leads to increased spontaneous mutagenesis in mammalian cells [138], and points to evidence that tumor cell lines manifest increased Pol β expression [139, 140]. DNA polymerase variant that is found in colon cancer likely has slow polymerase activity or high infidelity in selection of the correct nucleotide [141, 142]. Two germline SNPs of the POLB gene (rs12678588 and rs3136797) have been previously identified and the variant alleles have been shown to be present in specific populations [143, 144]. The rs12678588 SNP results in a nonsynonymous amino acid substitution of glutamine for arginine at residue 137 (R137Q). Arg137 is methylated by the protein arginine N-methyltransferase 1 (PRMT1), leading to a reduction in proliferating cell nuclear antigen (PCNA) binding [145]. R137Q is a slow polymerase with decreased BER activity in cell extracts, and cells expressing this variant have increased formation of AP sites following methyl methanesulfonate (MMS) exposure [146]. However, the P242R variant of Pol β induces genomic instability and cellular transformation [144].
Does chemoprevention strategy of colon cancer reduce DNA base damage?
The pros and cons of the colonic microbiota may be a promising target for the development of a colon cancer therapeutic [147]. Using probiotics to modulate the altered function of microbiota or scavenge bacterial metabolites may prevent inflammation induced genomic instability and cancer. In addition, designing the strategy to increase beneficial gastrointestinal microbiota, stimulates innate and adaptive immunity likely critical for future chemoprevention approaches in colon cancer [148]. These strategies may be successful if probiotics include the host genetic and environmental factors. Alternatively, few studies have demonstrated that suppression of the inflammatory response by BER inhibitors likely promotes genomic integrity and/or delays the onset of cancer initiation [149]. Therefore, using BER inhibitor (such as PARP1 inhibitor) may possibly be an attractive target to suppress DNA damage response and excessive inflammation, acting potentially as a preventative agent for genomic instability and carcinogenesis. Therefore, the future focus will likely target inflammatory response and BER intermediates generated from the crosstalk between host base excision repair and the microbiota dynamics which will help to enable development and a more rigorous testing of probiotics as more natural and less disruptive treatments to prevent colon cancer.
Concluding remarks
The significance of many recent observations still needs to be established. For instance, many factors can contribute to microbiota dysbiosis, including host genetics, lifestyle, and exposure to microorganisms. Microbiota dysibiosis has the potential to cause immunological disequilibrium response that exert chronic inflammation or release RONs directly to initiate the accumulation of BER intermediates [17]. The normal function of BER is critical for the maintenance of genomic integrity and likely influences the response of the host to microbiota dysbiosis. In contrast, loss of BER function leads to accumulation of BER intermediates and inflammation in the intestine [18]. During the last few years, remarkable progress has been made in our understanding of the molecular mechanism of BER role in colon cancer. Nevertheless, many questions about the missing link of microbiota dysbiosis and genetic polymorphism of BER gene remain unanswered. Does microbiota dysbiosis first induce immunological disequilibrium or genomic instability in host gastrointestinal track to drive cancer? Does microbiota dysbiosis decrease tumor latency for those who are the carriers of the genotype of BER germline variant? The future will uncover how microbiota dysbiosis manipulates the hosts' genetics, and the quest for answers to these questions will occupy the field for years to come.
Acknowledgements
We would like to thank Michael McKinley for editing the manuscript. DK is supported by United States National Institutes of Health (NIH/National Cancer Institute (NCI)) K01 CA15485401 and start-up funds from The University of Texas at Austin, College of Pharmacy.
Authors' contribution
All authors contributed to the writing of the review manuscript. DK initiated, conceptualized and wrote the manuscript. DR conducted and organized the BER SNP data and contributes on writing up the manuscript.
Competing Interests
The authors have no conflict of interest.
References
1. Gill SR, Pop M, Deboy RT, Eckburg PB, Turnbaugh PJ, Samuel BS. et al. Metagenomic analysis of the human distal gut microbiome. Science. 2006;312:1355-9
2. Turnbaugh PJ, Gordon JI. The core gut microbiome, energy balance and obesity. J Physiol. 2009;587:4153-8
3. Benson AK, Kelly SA, Legge R, Ma F, Low SJ, Kim J. et al. Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors. Proc Natl Acad Sci U S A. 2010;107:18933-8
4. Campbell JH, Foster CM, Vishnivetskaya T, Campbell AG, Yang ZK, Wymore A. et al. Host genetic and environmental effects on mouse intestinal microbiota. ISME J. 2012;6:2033-44
5. Backhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI. Host-bacterial mutualism in the human intestine. Science. 2005;307:1915-20
6. Sekirov I, Russell SL, Antunes LC, Finlay BB. Gut microbiota in health and disease. Physiol Rev. 2010;90:859-904
7. Sears CL, Garrett WS. Microbes, microbiota, and colon cancer. Cell Host Microbe. 2014;15:317-28
8. Cooke MS, Evans MD, Dizdaroglu M, Lunec J. Oxidative DNA damage: mechanisms, mutation, and disease. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2003;17:1195-214
9. Halliwell B, Aruoma OI. DNA damage by oxygen-derived species. Its mechanism and measurement in mammalian systems. FEBS Lett. 1991;281:9-19
10. Kim JJ, Tao H, Carloni E, Leung WK, Graham DY, Sepulveda AR. Helicobacter pylori impairs DNA mismatch repair in gastric epithelial cells. Gastroenterology. 2002;123:542-53
11. Park DI, Park SH, Kim SH, Kim JW, Cho YK, Kim HJ. et al. Effect of Helicobacter pylori infection on the expression of DNA mismatch repair protein. Helicobacter. 2005;10:179-84
12. Touati E, Michel V, Thiberge JM, Wuscher N, Huerre M, Labigne A. Chronic Helicobacter pylori infections induce gastric mutations in mice. Gastroenterology. 2003;124:1408-19
13. Koshiji M, To KK, Hammer S, Kumamoto K, Harris AL, Modrich P. et al. HIF-1alpha induces genetic instability by transcriptionally downregulating MutSalpha expression. Molecular cell. 2005;17:793-803
14. Radicella JP, Dherin C, Desmaze C, Fox MS, Boiteux S. Cloning and characterization of hOGG1, a human homolog of the OGG1 gene of Saccharomyces cerevisiae. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:8010-5
15. Aburatani H, Hippo Y, Ishida T, Takashima R, Matsuba C, Kodama T. et al. Cloning and characterization of mammalian 8-hydroxyguanine-specific DNA glycosylase/apurinic, apyrimidinic lyase, a functional mutM homologue. Cancer Res. 1997;57:2151-6
16. Demple B, Harrison L. Repair of oxidative damage to DNA: enzymology and biology. Annu Rev Biochem. 1994;63:915-48
17. Kidane D, Murphy DL, Sweasy JB. Accumulation of abasic sites induces genomic instability in normal human gastric epithelial cells during Helicobacter pylori infection. Oncogenesis. 2014;3:e128
18. Meira LB, Bugni JM, Green SL, Lee CW, Pang B, Borenshtein D. et al. DNA damage induced by chronic inflammation contributes to colon carcinogenesis in mice. J Clin Invest. 2008;118:2516-25
19. Liao J, Seril DN, Lu GG, Zhang M, Toyokuni S, Yang AL. et al. Increased susceptibility of chronic ulcerative colitis-induced carcinoma development in DNA repair enzyme Ogg1 deficient mice. Mol Carcinog. 2008;47:638-46
20. Fortini P, Parlanti E, Sidorkina OM, Laval J, Dogliotti E. The type of DNA glycosylase determines the base excision repair pathway in mammalian cells. The Journal of biological chemistry. 1999;274:15230-6
21. Nishimura S. Involvement of mammalian OGG1(MMH) in excision of the 8-hydroxyguanine residue in DNA. Free radical biology & medicine. 2002;32:813-21
22. Bjoras M, Luna L, Johnsen B, Hoff E, Haug T, Rognes T. et al. Opposite base-dependent reactions of a human base excision repair enzyme on DNA containing 7,8-dihydro-8-oxoguanine and abasic sites. EMBO J. 1997;16:6314-22
23. Barnes DE, Lindahl T, Sedgwick B. DNA repair. Curr Opin Cell Biol. 1993;5:424-33
24. Gentil A, Cabral-Neto JB, Mariage-Samson R, Margot A, Imbach JL, Rayner B. et al. Mutagenicity of a unique apurinic/apyrimidinic site in mammalian cells. J Mol Biol. 1992;227:981-4
25. Guillet M, Boiteux S. Origin of endogenous DNA abasic sites in Saccharomyces cerevisiae. Mol Cell Biol. 2003;23:8386-94
26. Loeb LA, Preston BD. Mutagenesis by apurinic/apyrimidinic sites. Annu Rev Genet. 1986;20:201-30
27. Fortini P, Pascucci B, Parlanti E, D'Errico M, Simonelli V, Dogliotti E. The base excision repair: mechanisms and its relevance for cancer susceptibility. Biochimie. 2003;85:1053-71
28. Winter SE, Lopez CA, Baumler AJ. The dynamics of gut-associated microbial communities during inflammation. EMBO Rep. 2013;14:319-27
29. Wu N, Yang X, Zhang R, Li J, Xiao X, Hu Y. et al. Dysbiosis signature of fecal microbiota in colorectal cancer patients. Microb Ecol. 2013;66:462-70
30. Kostic AD, Chun E, Robertson L, Glickman JN, Gallini CA, Michaud M. et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe. 2013;14:207-15
31. Rubinstein MR, Wang X, Liu W, Hao Y, Cai G, Han YW. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/beta-catenin signaling via its FadA adhesin. Cell Host Microbe. 2013;14:195-206
32. Arthur JC, Perez-Chanona E, Muhlbauer M, Tomkovich S, Uronis JM, Fan TJ. et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science. 2012;338:120-3
33. Bonnet M, Buc E, Sauvanet P, Darcha C, Dubois D, Pereira B. et al. Colonization of the human gut by E. coli and colorectal cancer risk. Clin Cancer Res. 2014;20:859-67
34. Elinav E, Nowarski R, Thaiss CA, Hu B, Jin C, Flavell RA. Inflammation-induced cancer: crosstalk between tumours, immune cells and microorganisms. Nat Rev Cancer. 2013;13:759-71
35. Grivennikov SI. Inflammation and colorectal cancer: colitis-associated neoplasia. Semin Immunopathol. 2013;35:229-44
36. Schwabe RF, Jobin C. The microbiome and cancer. Nat Rev Cancer. 2013;13:800-12
37. Levi L, Toyooka T, Patarroyo M, Frisan T. Bacterial genotoxins promote inside-out integrin beta1 activation, formation of focal adhesion complexes and cell spreading. PLoS One. 2015;10:e0124119
38. Landskron G, De la Fuente M, Thuwajit P, Thuwajit C, Hermoso MA. Chronic inflammation and cytokines in the tumor microenvironment. J Immunol Res. 2014;2014:149185
39. Jackson AL, Loeb LA. The contribution of endogenous sources of DNA damage to the multiple mutations in cancer. Mutat Res. 2001;477:7-21
40. Esteban-Jurado C, Garre P, Vila M, Lozano JJ, Pristoupilova A, Beltran S. et al. New genes emerging for colorectal cancer predisposition. World J Gastroenterol. 2014;20:1961-71
41. Watson IR, Takahashi K, Futreal PA, Chin L. Emerging patterns of somatic mutations in cancer. Nat Rev Genet. 2013;14:703-18
42. Kawanishi S, Hiraku Y, Pinlaor S, Ma N. Oxidative and nitrative DNA damage in animals and patients with inflammatory diseases in relation to inflammation-related carcinogenesis. Biol Chem. 2006;387:365-72
43. Mantovani A, Pierotti MA. Cancer and inflammation: a complex relationship. Cancer Lett. 2008;267:180-1
44. Wu S, Rhee KJ, Albesiano E, Rabizadeh S, Wu X, Yen HR. et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat Med. 2009;15:1016-22
45. Nesic D, Hsu Y, Stebbins CE. Assembly and function of a bacterial genotoxin. Nature. 2004;429:429-33
46. Cuevas-Ramos G, Petit CR, Marcq I, Boury M, Oswald E, Nougayrede JP. Escherichia coli induces DNA damage in vivo and triggers genomic instability in mammalian cells. Proc Natl Acad Sci U S A. 2010;107:11537-42
47. Pickett CL, Whitehouse CA. The cytolethal distending toxin family. Trends Microbiol. 1999;7:292-7
48. De Rycke J, Oswald E. Cytolethal distending toxin (CDT): a bacterial weapon to control host cell proliferation? FEMS Microbiol Lett. 2001;203:141-8
49. Lara-Tejero M, Galan JE. Cytolethal distending toxin: limited damage as a strategy to modulate cellular functions. Trends Microbiol. 2002;10:147-52
50. Frisan T, Cortes-Bratti X, Thelestam M. Cytolethal distending toxins and activation of DNA damage-dependent checkpoint responses. Int J Med Microbiol. 2002;291:495-9
51. Krokan HE, Nilsen H, Skorpen F, Otterlei M, Slupphaug G. Base excision repair of DNA in mammalian cells. FEBS Lett. 2000;476:73-7
52. Larsen E, Meza TJ, Kleppa L, Klungland A. Organ and cell specificity of base excision repair mutants in mice. Mutation research. 2007;614:56-68
53. Hofseth LJ, Khan MA, Ambrose M, Nikolayeva O, Xu-Welliver M, Kartalou M. et al. The adaptive imbalance in base excision-repair enzymes generates microsatellite instability in chronic inflammation. The Journal of clinical investigation. 2003;112:1887-94
54. Bergoglio V, Pillaire MJ, Lacroix-Triki M, Raynaud-Messina B, Canitrot Y, Bieth A. et al. Deregulated DNA polymerase beta induces chromosome instability and tumorigenesis. Cancer Res. 2002;62:3511-4
55. Barnes DE, Lindahl T. Repair and genetic consequences of endogenous DNA base damage in mammalian cells. Annu Rev Genet. 2004;38:445-76
56. Spitz MR, Wei Q, Dong Q, Amos CI, Wu X. Genetic susceptibility to lung cancer: the role of DNA damage and repair. Cancer epidemiology, biomarkers & prevention: a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2003;12:689-98
57. Goode EL, Ulrich CM, Potter JD. Polymorphisms in DNA repair genes and associations with cancer risk. Cancer epidemiology, biomarkers & prevention: a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2002;11:1513-30
58. Yamtich J, Nemec AA, Keh A, Sweasy JB. A germline polymorphism of DNA polymerase beta induces genomic instability and cellular transformation. PLoS Genet. 2012;8:e1003052
59. Brevik A, Joshi AD, Corral R, Onland-Moret NC, Siegmund KD, Le Marchand L. et al. Polymorphisms in base excision repair genes as colorectal cancer risk factors and modifiers of the effect of diets high in red meat. Cancer epidemiology, biomarkers & prevention: a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2010;19:3167-73
60. Wilson DM 3rd, Kim D, Berquist BR, Sigurdson AJ. Variation in base excision repair capacity. Mutat Res. 2011;711:100-12
61. Zhang Y, He BS, Pan YQ, Xu YQ, Wang SK. Association of OGG1 Ser326Cys polymorphism with colorectal cancer risk: a meta-analysis. Int J Colorectal Dis. 2011;26:1525-30
62. Gsur A, Bernhart K, Baierl A, Feik E, Fuhrlinger G, Hofer P. et al. No association of XRCC1 polymorphisms Arg194Trp and Arg399Gln with colorectal cancer risk. Cancer Epidemiol. 2011;35:e38-41
63. Tomlinson IP, Houlston RS, Montgomery GW, Sieber OM, Dunlop MG. Investigation of the effects of DNA repair gene polymorphisms on the risk of colorectal cancer. Mutagenesis. 2012;27:219-23
64. Aravind L, Koonin EV. The alpha/beta fold uracil DNA glycosylases: a common origin with diverse fates. Genome Biol. 2000;1:RESEARCH0007
65. Cortazar D, Kunz C, Saito Y, Steinacher R, Schar P. The enigmatic thymine DNA glycosylase. DNA Repair (Amst). 2007;6:489-504
66. Cortellino S, Xu J, Sannai M, Moore R, Caretti E, Cigliano A. et al. Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell. 2011;146:67-79
67. Gong Z, Zhu JK. Active DNA demethylation by oxidation and repair. Cell research. 2011;21:1649-51
68. Cobellis G, Missero C, Simionati B, Valle G, Di Lauro R. Immediate early genes induced by H-Ras in thyroid cells. Oncogene. 2001;20:2281-90
69. Tini M, Benecke A, Um SJ, Torchia J, Evans RM, Chambon P. Association of CBP/p300 acetylase and thymine DNA glycosylase links DNA repair and transcription. Molecular cell. 2002;9:265-77
70. Vasovcak P, Krepelova A, Menigatti M, Puchmajerova A, Skapa P, Augustinakova A. et al. Unique mutational profile associated with a loss of TDG expression in the rectal cancer of a patient with a constitutional PMS2 deficiency. DNA repair. 2012;11:616-23
71. Sjolund AB, Senejani AG, Sweasy JB. MBD4 and TDG: multifaceted DNA glycosylases with ever expanding biological roles. Mutation research. 2013;743-744:12-25
72. Sjolund A, Nemec AA, Paquet N, Prakash A, Sung P, Doublie S. et al. A germline polymorphism of thymine DNA glycosylase induces genomic instability and cellular transformation. PLoS Genet. 2014;10:e1004753
73. Hendrich B, Hardeland U, Ng HH, Jiricny J, Bird A. The thymine glycosylase MBD4 can bind to the product of deamination at methylated CpG sites. Nature. 1999;401:301-4
74. Petronzelli F, Riccio A, Markham GD, Seeholzer SH, Stoerker J, Genuardi M. et al. Biphasic kinetics of the human DNA repair protein MED1 (MBD4), a mismatch-specific DNA N-glycosylase. J Biol Chem. 2000;275:32422-9
75. Millar CB, Guy J, Sansom OJ, Selfridge J, MacDougall E, Hendrich B. et al. Enhanced CpG mutability and tumorigenesis in MBD4-deficient mice. Science. 2002;297:403-5
76. Cortellino S, Turner D, Masciullo V, Schepis F, Albino D, Daniel R. et al. The base excision repair enzyme MED1 mediates DNA damage response to antitumor drugs and is associated with mismatch repair system integrity. Proc Natl Acad Sci U S A. 2003;100:15071-6
77. Li Y, Kundu P, Seow SW, de Matos CT, Aronsson L, Chin KC. et al. Gut microbiota accelerate tumor growth via c-jun and STAT3 phosphorylation in APCMin/+ mice. Carcinogenesis. 2012;33:1231-8
78. Song JH, Maeng EJ, Cao Z, Kim SY, Nam SW, Lee JY. et al. The Glu346Lys polymorphism and frameshift mutations of the Methyl-CpG Binding Domain 4 gene in gastrointestinal cancer. Neoplasma. 2009;56:343-7
79. Shen L, Kondo Y, Rosner GL, Xiao L, Hernandez NS, Vilaythong J. et al. MGMT promoter methylation and field defect in sporadic colorectal cancer. J Natl Cancer Inst. 2005;97:1330-8
80. Esteller M, Toyota M, Sanchez-Cespedes M, Capella G, Peinado MA, Watkins DN. et al. Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation is associated with G to A mutations in K-ras in colorectal tumorigenesis. Cancer Res. 2000;60:2368-71
81. Qian X, von Wronski MA, Brent TP. Localization of methylation sites in the human O6-methylguanine-DNA methyltransferase promoter: correlation with gene suppression. Carcinogenesis. 1995;16:1385-90
82. Esteller M, Herman JG. Generating mutations but providing chemosensitivity: the role of O6-methylguanine DNA methyltransferase in human cancer. Oncogene. 2004;23:1-8
83. Loh YH, Mitrou PN, Bowman R, Wood A, Jeffery H, Luben RN. et al. MGMT Ile143Val polymorphism, dietary factors and the risk of breast, colorectal and prostate cancer in the European Prospective Investigation into Cancer and Nutrition (EPIC)-Norfolk study. DNA Repair (Amst). 2010;9:421-8
84. Coulter R, Blandino M, Tomlinson JM, Pauly GT, Krajewska M, Moschel RC. et al. Differences in the rate of repair of O6-alkylguanines in different sequence contexts by O6-alkylguanine-DNA alkyltransferase. Chem Res Toxicol. 2007;20:1966-71
85. Inoue R, Abe M, Nakabeppu Y, Sekiguchi M, Mori T, Suzuki T. Characterization of human polymorphic DNA repair methyltransferase. Pharmacogenetics. 2000;10:59-66
86. Fang Q, Noronha AM, Murphy SP, Wilds CJ, Tubbs JL, Tainer JA. et al. Repair of O6-G-alkyl-O6-G interstrand cross-links by human O6-alkylguanine-DNA alkyltransferase. Biochemistry. 2008;47:10892-903
87. Margison GP, Heighway J, Pearson S, McGown G, Thorncroft MR, Watson AJ. et al. Quantitative trait locus analysis reveals two intragenic sites that influence O6-alkylguanine-DNA alkyltransferase activity in peripheral blood mononuclear cells. Carcinogenesis. 2005;26:1473-80
88. Edara S, Kanugula S, Goodtzova K, Pegg AE. Resistance of the human O6-alkylguanine-DNA alkyltransferase containing arginine at codon 160 to inactivation by O6-benzylguanine. Cancer Res. 1996;56:5571-5
89. Engelward BP, Weeda G, Wyatt MD, Broekhof JL, de Wit J, Donker I. et al. Base excision repair deficient mice lacking the Aag alkyladenine DNA glycosylase. Proc Natl Acad Sci U S A. 1997;94:13087-92
90. Gabele E, Dostert K, Hofmann C, Wiest R, Scholmerich J, Hellerbrand C. et al. DSS induced colitis increases portal LPS levels and enhances hepatic inflammation and fibrogenesis in experimental NASH. Journal of hepatology. 2011;55:1391-9
91. Agus A, Denizot J, Thevenot J, Martinez-Medina M, Massier S, Sauvanet P. et al. Western diet induces a shift in microbiota composition enhancing susceptibility to Adherent-Invasive E. coli infection and intestinal inflammation. Sci Rep. 2016;6:19032
92. Hofseth LJ. The adaptive imbalance to genotoxic stress: genome guardians rear their ugly heads. Carcinogenesis. 2004;25:1787-93
93. Adhikari S, Chetram MA, Woodrick J, Mitra PS, Manthena PV, Khatkar P. et al. Germ line variants of human N-methylpurine DNA glycosylase show impaired DNA repair activity and facilitate 1,N6-ethenoadenine-induced mutations. J Biol Chem. 2015;290:4966-80
94. Mabley JG, Pacher P, Deb A, Wallace R, Elder RH, Szabo C. Potential role for 8-oxoguanine DNA glycosylase in regulating inflammation. FASEB J. 2005;19:290-2
95. Fan X, Crowe SE, Behar S, Gunasena H, Ye G, Haeberle H. et al. The effect of class II major histocompatibility complex expression on adherence of Helicobacter pylori and induction of apoptosis in gastric epithelial cells: a mechanism for T helper cell type 1-mediated damage. J Exp Med. 1998;187:1659-69
96. Aguilera-Aguirre L, Bacsi A, Radak Z, Hazra TK, Mitra S, Sur S. et al. Innate inflammation induced by the 8-oxoguanine DNA glycosylase-1-KRAS-NF-kappaB pathway. J Immunol. 2014;193:4643-53
97. Boldogh I, Hajas G, Aguilera-Aguirre L, Hegde ML, Radak Z, Bacsi A. et al. Activation of ras signaling pathway by 8-oxoguanine DNA glycosylase bound to its excision product, 8-oxoguanine. The Journal of biological chemistry. 2012;287:20769-73
98. Park J, Chen L, Tockman MS, Elahi A, Lazarus P. The human 8-oxoguanine DNA N-glycosylase 1 (hOGG1) DNA repair enzyme and its association with lung cancer risk. Pharmacogenetics. 2004;14:103-9
99. Su Y, Xu A, Zhu J. The effect of oxoguanine glycosylase 1 rs1052133 polymorphism on colorectal cancer risk in Caucasian population. Tumour Biol. 2014;35:513-7
100. Dherin C, Radicella JP, Dizdaroglu M, Boiteux S. Excision of oxidatively damaged DNA bases by the human alpha-hOgg1 protein and the polymorphic alpha-hOgg1(Ser326Cys) protein which is frequently found in human populations. Nucleic Acids Res. 1999;27:4001-7
101. Hill JW, Evans MK. Dimerization and opposite base-dependent catalytic impairment of polymorphic S326C OGG1 glycosylase. Nucleic Acids Res. 2006;34:1620-32
102. Morreall J, Limpose K, Sheppard C, Kow YW, Werner E, Doetsch PW. Inactivation of a common OGG1 variant by TNF-alpha in mammalian cells. DNA Repair (Amst). 2015;26:15-22
103. Guan Y, Manuel RC, Arvai AS, Parikh SS, Mol CD, Miller JH. et al. MutY catalytic core, mutant and bound adenine structures define specificity for DNA repair enzyme superfamily. Nat Struct Biol. 1998;5:1058-64
104. Parker AR, Eshleman JR. Human MutY: gene structure, protein functions and interactions, and role in carcinogenesis. Cell Mol Life Sci. 2003;60:2064-83
105. Poulsen ML, Bisgaard ML. MUTYH Associated Polyposis (MAP). Curr Genomics. 2008;9:420-35
106. Sakamoto K, Tominaga Y, Yamauchi K, Nakatsu Y, Sakumi K, Yoshiyama K. et al. MUTYH-null mice are susceptible to spontaneous and oxidative stress induced intestinal tumorigenesis. Cancer Res. 2007;67:6599-604
107. Sieber OM, Lipton L, Crabtree M, Heinimann K, Fidalgo P, Phillips RK. et al. Multiple colorectal adenomas, classic adenomatous polyposis, and germ-line mutations in MYH. N Engl J Med. 2003;348:791-9
108. Al-Tassan N, Chmiel NH, Maynard J, Fleming N, Livingston AL, Williams GT. et al. Inherited variants of MYH associated with somatic G:C->T:A mutations in colorectal tumors. Nat Genet. 2002;30:227-32
109. Volk DE, House PG, Thiviyanathan V, Luxon BA, Zhang S, Lloyd RS. et al. Structural similarities between MutT and the C-terminal domain of MutY. Biochemistry. 2000;39:7331-6
110. Fromme JC, Banerjee A, Huang SJ, Verdine GL. Structural basis for removal of adenine mispaired with 8-oxoguanine by MutY adenine DNA glycosylase. Nature. 2004;427:652-6
111. Chmiel NH, Golinelli MP, Francis AW, David SS. Efficient recognition of substrates and substrate analogs by the adenine glycosylase MutY requires the C-terminal domain. Nucleic Acids Res. 2001;29:553-64
112. Pope MA, Chmiel NH, David SS. Insight into the functional consequences of hMYH variants associated with colorectal cancer: distinct differences in the adenine glycosylase activity and the response to AP endonucleases of Y150C and G365D murine MYH. DNA Repair (Amst). 2005;4:315-25
113. Bandaru V, Sunkara S, Wallace SS, Bond JP. A novel human DNA glycosylase that removes oxidative DNA damage and is homologous to Escherichia coli endonuclease VIII. DNA Repair (Amst). 2002;1:517-29
114. Dou H, Mitra S, Hazra TK. Repair of oxidized bases in DNA bubble structures by human DNA glycosylases NEIL1 and NEIL2. J Biol Chem. 2003;278:49679-84
115. Liu M, Bandaru V, Bond JP, Jaruga P, Zhao X, Christov PP. et al. The mouse ortholog of NEIL3 is a functional DNA glycosylase in vitro and in vivo. Proc Natl Acad Sci U S A. 2010;107:4925-30
116. Hegde ML, Hegde PM, Bellot LJ, Mandal SM, Hazra TK, Li GM. et al. Prereplicative repair of oxidized bases in the human genome is mediated by NEIL1 DNA glycosylase together with replication proteins. Proc Natl Acad Sci U S A. 2013;110:E3090-9
117. Banerjee D, Mandal SM, Das A, Hegde ML, Das S, Bhakat KK. et al. Preferential repair of oxidized base damage in the transcribed genes of mammalian cells. J Biol Chem. 2011;286:6006-16
118. Zhou J, Liu M, Fleming AM, Burrows CJ, Wallace SS. Neil3 and NEIL1 DNA glycosylases remove oxidative damages from quadruplex DNA and exhibit preferences for lesions in the telomeric sequence context. J Biol Chem. 2013;288:27263-72
119. Krishnamurthy N, Zhao X, Burrows CJ, David SS. Superior removal of hydantoin lesions relative to other oxidized bases by the human DNA glycosylase hNEIL1. Biochemistry. 2008;47:7137-46
120. David SS, O'Shea VL, Kundu S. Base-excision repair of oxidative DNA damage. Nature. 2007;447:941-50
121. Hildrestrand GA, Neurauter CG, Diep DB, Castellanos CG, Krauss S, Bjoras M. et al. Expression patterns of Neil3 during embryonic brain development and neoplasia. BMC Neurosci. 2009;10:45
122. Kauffmann A, Rosselli F, Lazar V, Winnepenninckx V, Mansuet-Lupo A, Dessen P. et al. High expression of DNA repair pathways is associated with metastasis in melanoma patients. Oncogene. 2008;27:565-73
123. Broderick P, Bagratuni T, Vijayakrishnan J, Lubbe S, Chandler I, Houlston RS. Evaluation of NTHL1, NEIL1, NEIL2, MPG, TDG, UNG and SMUG1 genes in familial colorectal cancer predisposition. BMC Cancer. 2006;6:243
124. Roy LM, Jaruga P, Wood TG, McCullough AK, Dizdaroglu M, Lloyd RS. Human polymorphic variants of the NEIL1 DNA glycosylase. J Biol Chem. 2007;282:15790-8
125. Zhang SH, Wang LA, Li Z, Peng Y, Cun YP, Dai N. et al. APE1 polymorphisms are associated with colorectal cancer susceptibility in Chinese Hans. World J Gastroenterol. 2014;20:8700-8
126. Abbotts R, Madhusudan S. Human AP endonuclease 1 (APE1): from mechanistic insights to druggable target in cancer. Cancer Treat Rev. 2010;36:425-35
127. Luo M, He H, Kelley MR, Georgiadis MM. Redox regulation of DNA repair: implications for human health and cancer therapeutic development. Antioxid Redox Signal. 2010;12:1247-69
128. Bobola MS, Blank A, Berger MS, Stevens BA, Silber JR. Apurinic/apyrimidinic endonuclease activity is elevated in human adult gliomas. Clin Cancer Res. 2001;7:3510-8
129. Kelley MR, Cheng L, Foster R, Tritt R, Jiang J, Broshears J. et al. Elevated and altered expression of the multifunctional DNA base excision repair and redox enzyme Ape1/ref-1 in prostate cancer. Clin Cancer Res. 2001;7:824-30
130. Robertson KA, Bullock HA, Xu Y, Tritt R, Zimmerman E, Ulbright TM. et al. Altered expression of Ape1/ref-1 in germ cell tumors and overexpression in NT2 cells confers resistance to bleomycin and radiation. Cancer Res. 2001;61:2220-5
131. Jedinak A, Dudhgaonkar S, Kelley MR, Sliva D. Apurinic/Apyrimidinic endonuclease 1 regulates inflammatory response in macrophages. Anticancer Res. 2011;31:379-85
132. Futagami S, Hiratsuka T, Shindo T, Horie A, Hamamoto T, Suzuki K. et al. Expression of apurinic/apyrimidinic endonuclease-1 (APE-1) in H. pylori-associated gastritis, gastric adenoma, and gastric cancer. Helicobacter. 2008;13:209-18
133. Canbay E, Cakmakoglu B, Zeybek U, Sozen S, Cacina C, Gulluoglu M. et al. Association of APE1 and hOGG1 polymorphisms with colorectal cancer risk in a Turkish population. Curr Med Res Opin. 2011;27:1295-302
134. Jelonek K, Gdowicz-Klosok A, Pietrowska M, Borkowska M, Korfanty J, Rzeszowska-Wolny J. et al. Association between single-nucleotide polymorphisms of selected genes involved in the response to DNA damage and risk of colon, head and neck, and breast cancers in a Polish population. J Appl Genet. 2010;51:343-52
135. AlMutairi F, Pathan AA, Alanazi M, Shalaby M, Alabdulkarim HA, Alamri A. et al. Association of DNA Repair Gene APE1 Asp148Glu Polymorphism with Breast Cancer Risk. Dis Markers. 2015;2015:869512
136. Sweasy JB, Lang T, DiMaio D. Is base excision repair a tumor suppressor mechanism? Cell cycle. 2006;5:250-9
137. Cabelof DC, Ikeno Y, Nyska A, Busuttil RA, Anyangwe N, Vijg J. et al. Haploinsufficiency in DNA polymerase beta increases cancer risk with age and alters mortality rate. Cancer Res. 2006;66:7460-5
138. Canitrot Y, Cazaux C, Frechet M, Bouayadi K, Lesca C, Salles B. et al. Overexpression of DNA polymerase beta in cell results in a mutator phenotype and a decreased sensitivity to anticancer drugs. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:12586-90
139. Bergoglio V, Canitrot Y, Hogarth L, Minto L, Howell SB, Cazaux C. et al. Enhanced expression and activity of DNA polymerase beta in human ovarian tumor cells: impact on sensitivity towards antitumor agents. Oncogene. 2001;20:6181-7
140. Canitrot Y, Laurent G, Astarie-Dequeker C, Bordier C, Cazaux C, Hoffmann JS. Enhanced expression and activity of DNA polymerase beta in chronic myelogenous leukemia. Anticancer Res. 2006;26:523-5
141. Donigan KA, Sun KW, Nemec AA, Murphy DL, Cong X, Northrup V. et al. Human POLB gene is mutated in high percentage of colorectal tumors. The Journal of biological chemistry. 2012;287:23830-9
142. Nemec AA, Donigan KA, Murphy DL, Jaeger J, Sweasy JB. Colon cancer-associated DNA polymerase beta variant induces genomic instability and cellular transformation. The Journal of biological chemistry. 2012;287:23840-9
143. Mohrenweiser HW, Xi T, Vazquez-Matias J, Jones IM. Identification of 127 amino acid substitution variants in screening 37 DNA repair genes in humans. Cancer epidemiology, biomarkers & prevention: a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2002;11:1054-64
144. Yamtich J, Speed WC, Straka E, Kidd JR, Sweasy JB, Kidd KK. Population-specific variation in haplotype composition and heterozygosity at the POLB locus. DNA Repair (Amst). 2009;8:579-84
145. El-Andaloussi N, Valovka T, Toueille M, Hassa PO, Gehrig P, Covic M. et al. Methylation of DNA polymerase beta by protein arginine methyltransferase 1 regulates its binding to proliferating cell nuclear antigen. FASEB J. 2007;21:26-34
146. Guo Z, Zheng L, Dai H, Zhou M, Xu H, Shen B. Human DNA polymerase beta polymorphism, Arg137Gln, impairs its polymerase activity and interaction with PCNA and the cellular base excision repair capacity. Nucleic Acids Res. 2009;37:3431-41
147. Roukos DH, Katsios C, Liakakos T. Genotype-phenotype map and molecular networks: a promising solution in overcoming colorectal cancer resistance to targeted treatment. Expert Rev Mol Diagn. 2010;10:541-5
148. Hammami R, Fernandez B, Lacroix C, Fliss I. Anti-infective properties of bacteriocins: an update. Cell Mol Life Sci. 2013;70:2947-67
149. Nossa CW, Jain P, Tamilselvam B, Gupta VR, Chen LF, Schreiber V. et al. Activation of the abundant nuclear factor poly(ADP-ribose) polymerase-1 by Helicobacter pylori. Proc Natl Acad Sci U S A. 2009;106:19998-20003
150. Picelli S, Zajac P, Zhou XL, Edler D, Lenander C, Dalen J. et al. Common variants in human CRC genes as low-risk alleles. Eur J Cancer. 2010;46:1041-8
151. Gu D, Wang M, Wang M, Zhang Z, Chen J. The DNA repair gene APE1 T1349G polymorphism and cancer risk: a meta-analysis of 27 case-control studies. Mutagenesis. 2009;24:507-12
Author contact
![]() Corresponding author: Dawit Kidane, Division of Pharmacology and Toxicology, College of Pharmacy, The University of Texas at Austin, Dell Pediatric Research Institute, 1400 Barbara Jordan Blvd. Mail code R1800, Austin, TX 78723, United States. Phone: (512) 495-4720 FAX : 512-495-4945 Email: dawit.kidaneutexas.edu.
Corresponding author: Dawit Kidane, Division of Pharmacology and Toxicology, College of Pharmacy, The University of Texas at Austin, Dell Pediatric Research Institute, 1400 Barbara Jordan Blvd. Mail code R1800, Austin, TX 78723, United States. Phone: (512) 495-4720 FAX : 512-495-4945 Email: dawit.kidaneutexas.edu.