Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2016; 7(9):1133-1141. doi:10.7150/jca.14519 This issue Cite
Research Paper
Proteasome Inhibitor YSY01A Enhances Cisplatin Cytotoxicity in Cisplatin-Resistant Human Ovarian Cancer Cells
Wei Huang1,*, Quan Zhou1,*, Xia Yuan1, Ze-mei Ge2, Fu-xiang Ran1, Hua-yu Yang3, Guang-liang Qiang4, Run-tao Li2 ![]() , Jing-rong Cui1
, Jing-rong Cui1 ![]()
1. State Key Laboratory of Natural and Biomimetic Drugs, School of Pharmaceutical Sciences, Peking University, Beijing 100191, China
2. Department of Medicinal Chemistry, School of Pharmaceutical Sciences, Peking University, Beijing 100191, China
3. Department of Liver Surgery, Peking Union Medical College (PUMC) Hospital, Chinese Academy of Medical Sciences and PUMC, Beijing 100730, China
4. Department of Thoracic Surgery, China-Japan Friendship Hospital, Beijing 100029, China
* W. H and Q. Z. contributed equally to this work.
Received 2015-11-24; Accepted 2016-4-26; Published 2016-6-6
Abstract
Cisplatin is one of the most common drugs used for treatment of solid tumors such as ovarian cancer. Unfortunately, the development of resistance against this cytotoxic agent limits its clinical use. Here we report that YSY01A, a novel proteasome inhibitor, is capable of suppressing survival of cisplatin-resistant ovarian cancer cells by inducing apoptosis. And YSY01A treatment enhances the cytotoxicity of cisplatin in drug-resistant ovarian cancer cells. Specifically, YSY01A abrogates regulatory proteins important for cell proliferation and anti-apoptosis including NF-κB p65 and STAT3, resulting in down-regulation of Bcl-2. A dramatic increase in cisplatin uptake was also observed by inductively coupled plasma-mass spectrometry following exposure to YSY01A. Taken together, YSY01A serves as a potential candidate for further development as anticancer therapeutics targeting the proteasome.
Keywords: proteasome inhibitor, YSY01A, ovarian cancer, cisplatin resistance, apoptosis, NF-κB and STAT3
Introduction
Ovarian cancer is the seventh most common cancer in women worldwide, with approximately 239,000 new cases reported in 2012 [1]. The standard therapy for ovarian cancer consists of surgery followed by chemotherapy. Despite early-stage ovarian cancer may be curable, 75% of ovarian cancer patients are diagnosed in advanced stages with short overall survival and eventually develop resistance to platinum-based first-line chemotherapy with each recurrence [2, 3]. A variety of mechanisms are involved in the emergence of a chemoresistant phenotype including decreased drug accumulation, increased drug efflux, elevated repair of DNA damage induced by chemotherapy and impaired ability to undergo apoptosis.
Proteasomes are large proteolytic complexes that are responsible for degradation of more than 80% of intracellular proteins involved in growth and differentiation, cell cycle, signal transduction and apoptosis [4]. In cancer cells its homeostatic function is dysregulated leading to a malignant phenotype. Proteasome inhibition enables stabilization and accumulation of its substances and consequently results in activation of apoptotic pathways via various cellular processes, particularly in rapidly proliferating cells. Thus the inhibition of proteasome activity promises to be an attractive strategy for development of novel antineoplastic drugs. Bortezomib (also called PS-341) is the first proteasome inhibitor approved by United States Food Drug Administration for initial treatment of patients with multiple myeloma or relapsed/refractory mantle cell lymphoma [5]. However, some side effects related to bortezomib have been observed. Approximately 60% of the patients treated with bortezomib developed resistance within an average of one year from the beginning of the treatment [6]. In 2012, carfilzomib, the second-generation proteasome inhibitor, was approved for treatment of multiple myeloma with milder toxicity [7, 8]. Some proteasome inhibitors are also under investigation such as marizomib and ixazomib [9, 10]. Nevertheless, owing to unsatisfactory efficacy in the treatment of solid tumors, severe side effects and poor pharmacodynamic and pharmacokinetic properties, there is considerable interest in extending the benefits of proteasome inhibitors to the treatment of solid tumor malignancies.
YSY01A (N-(2-pyrazinecarbonyl)-L-leucine-L-(2-naphthyl)-alanine-L-leucine boronic acid) is a novel proteasome inhibitor that exhibits antitumor effects in vitro and in vivo as previously described [11]. Further studies have demonstrated that YSY01A modulates various signaling pathways involved in the control of cell proliferation, cell cycle, apoptosis and autophagy [12, 13]. In this study, we investigated the anti-proliferative effects of YSY01A as a single agent and in combination with cisplatin, a commonly-used drug for the treatment of ovarian cancer. The present study revealed the mechanism by which YSY01A overcomes cisplatin resistance including anti-apoptosis and drug uptake. Thus YSY01A may serve as a potential lead candidate for further development of anticancer therapeutics by inhibition of the proteasome.
Materials and Methods
Chemicals and reagents
YSY01A, a pale yellow powder, was synthesized as we previously described [11] (Fig.1A). The compound was dissolved in dimethyl sulfoxide (DMSO) to a concentration of 10 mM and preserved at -20°C. Cis-diammineplatinum (II) dichloride (cisplatin/DDP) was purchased from Tokyo Chemical Industry Co. Ltd. (Tokyo, Japan). For all compounds, the final DMSO level was less than 0.1%. Antibodies against nuclear factor κ-light-chain-enhancer of activated B cells p65 (NF-κB p65; Cat. No. 8242), signal transducer and activator of transcription 3 (STAT3; Cat. No. 9139), pSTAT3 (Cat. No. 9145), B-cell lymphoma 2 (Bcl-2; Cat. No. 2870) and cleaved caspase-3 (Cat. No. 9664) were obtained from Cell Signaling Technology (Danvers, MA, USA). Antibody against glyceraldehyde 3-phosphate dehydrogenase (GAPDH; Cat. No. sc-25778) was purchased from Santa Cruz Biotechnologies (Santa Cruz, CA, USA).
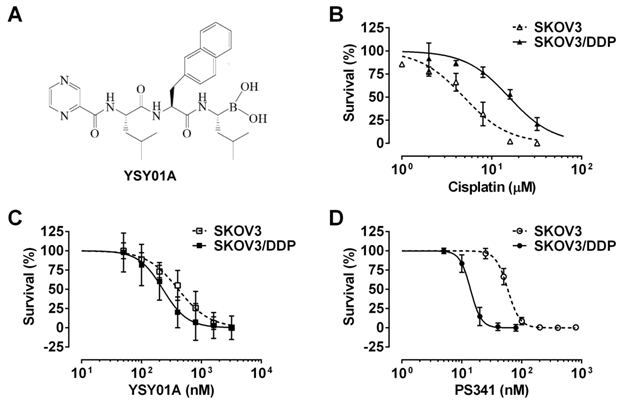
Effect of YSY01A on survival of cisplatin-sensitive and -resistant ovarian cancer cells. (A) Chemical structure of YSY01A. N-(2-pyrazinecarbonyl)-L-leucine-L-(2-naphthyl)-alanine-L-leucine boronic acid, formula: C29H38BN5O5, molecular mass: 547.45 mg/mol. (B) Cisplatin-sensitive SKOV3 and cisplatin-resistant SKOV3/DDP cells were subjected to SRB assay after treatment with increasing concentrations of cisplatin for 48 hours. SKOV3/DDP cells showed ~4-fold resistance in cisplatin than the parental cell line. (C&D) The effects of YSY01A (C) and PS-341 (D) on survival of SKOV3 and SKOV3/DDP cells were detected by SRB assay. IC50 values were computed and dose-response curves were created using GraphPad software.
Cell viability assay
Human ovarian cancer cell line SKOV3 and its cisplatin-resistant strain SKOV3/DDP were maintained in RPMI 1640 medium (Macgene, Beijing, China) containing 10% fetal bovine serum (PAN-Biotech GmbH, Aidenbach, Germany) and 1% penicillin/streptomycin (Macgene) in a 5% CO2 incubator at 37°C. To determine the cytotoxicity of YSY01A on both cell lines, cells were seeded at 5,000 cells/well in a 96-well microplate. 24 hours later, cultures were treated with increasing concentrations of YSY01A for 72 hours followed by sulforhodamine B (SRB) assay as we previously described [14]. To determine cellular sensitivity to cisplatin after treatment with YSY01A, 5,000 cells/well of SKOV3/DDP cells were plated in a 96-well plate overnight and then pretreated with 0 (vehicle control), 50 or 100 nM YSY01A for 24 hours followed by 48-hour combination treatment with cisplatin. The cell viability was also determined using SRB assay as mentioned above. IC50 values and dose-response curves were derived using GraphPad software (GraphPad Software, Inc., La Jolla, CA, USA). The coefficient of drug interaction (CDI) was used to analyze the synergistically inhibitory effect of drug combination as described previously [15, 16]. CDI was calculated as follows: CDI=AB/(A×B). According to the absorbance of each group, AB is the ratio of the combination groups to the control group; A or B is the ratio of the single agent group to the control group. The drug interaction is considered synergistic, additive or antagonistic when CDI is less than, equal to or greater than 1. CDI less than 0.7 indicates a significant synergistic effect.
Flow cytometric analysis of apoptosis
Annexin V/propidium iodide (PI) staining for flow cytometry was used to quantify YSY01A-induced apoptosis as previously described [17]. Cells were labeled with Annexin V-PI Apoptosis Detection Kit (KeyGene, Nanjing, China) according to the manufacturer's instructions followed by flow cytometric analysis using FACSCalibur flow cytometer (BD Biosciences, San Jose, CA, USA). The apoptosis rate was determined as the percentage of Annexin V-positive cells.
Enzyme-linked immunosorbent assay (ELISA) for quantification of apoptosis
Apoptosis was also analyzed using Cell Death Detection ELISAPLUS Photometric Enzyme Immunoassay Kit (Roche Diagnostics GmbH, Mannheim, Germany). In brief, cells were harvested and lyzed in lysis buffer provided by the kit after treatment. Cytoplasmic histone-associated DNA fragments after induced apoptosis were finally quantified according to the manufacturer's instructions.
Western blotting
Western blotting analysis was performed to determine protein levels as previously described [17]. Briefly, cells were harvested and suspended in lysis buffer containing 50 mM Tris·HCl, pH 7.4, 150 mM NaCl, 0.5% NP-40, 20 mM EDTA, 50 mM NaF, 1 mM Na3VO4, 2 mM PMSF and 1 mM DTT. After 10-minitue incubation on ice, the cell lysates were sonicated briefly and centrifuged at 15,000 ×g using Heraeus Multifuge X1R centrifuge (Thermo Scientific, Osterode, Germany) at 4°C for 20 minutes. The supernatants were collected and their protein concentrations were measured using Bicinchoninic Acid Protein Assay Kit (Dingguo Changsheng Biotechnology, Beijing, China). Cell lysates were then separated on sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes (EMD Millipore, Billerica, MA, USA) that were probed using antibodies against different proteins and visualized by using ChemiDoc XRS+ imaging system (Bio-Rad, Hercules, CA, USA) with enhanced chemiluminescence (Dingguo Changsheng Biotechnology).
Real-time reverse transcription polymerase chain reaction (PCR)
The mRNA expression levels were analyzed by quantitative PCR as we previously described [17]. Total RNAs were extracted using E.Z.N.A.TM Total RNA Kit I (Omega Bio-tek, Norcross, GA, USA) followed by reverse transcription to cDNA using All-In-One RT MasterMix Kit (Applied Biological Materials, Richmond, BC, Canada). Real-time PCR was performed with EvaGreen qPCR MaxterMix (Applied Biological Materials) on Stratagene Mx3000 system (Agilent, Santa Clara, CA, USA). The gene-specific primers are shown in Supplemental Table S1. The cycling conditions for NF-κB p65, STAT3, Bcl-2 and GAPDH were as follows: initial denaturation at 95°C for 10 minutes, followed by 40 cycles of 95°C for 15 seconds and 60°C for 1 minute. The threshold cycles (Ct) of indicated genes were determined and normalized against that of the internal control GAPDH. The relative mRNA levels were shown as the value of 2-ΔΔCt.
Measurement of intracellular platinum content
Cells were plated in Petri dish at a density of 1.5×106/dish followed by pretreatment with 400 nM YSY01A or vehicle control for 24 hours and further incubation with 50 μM cisplatin for 4 hours. To determine the intracellular platinum content, 2×106 cells were collected and digested in 100 μl of concentrated nitric acid for 4 hours, and then diluted with absolutely pure water until the platinum contents were in the range of 1~100 ng/ml. Platinum contents were then measured by inductively coupled plasma-mass spectrometry (ICP-MS) (PerkinElmer ICP-MS Elan DRC2, PerkinElmer, Waltham, MA, USA).
Statistical analysis
All experiments were repeated in triplicate. Data were presented as mean ± standard deviation. Statistical differences between two groups were compared using the Student's t-test. The differences among multiple groups were determined by one-way analysis of variance (ANOVA) followed by post hoc pairwise comparisons. Statistical significance was considered at p<0.05.
Results
YSY01A inhibits cancer cell survival by inducing apoptosis
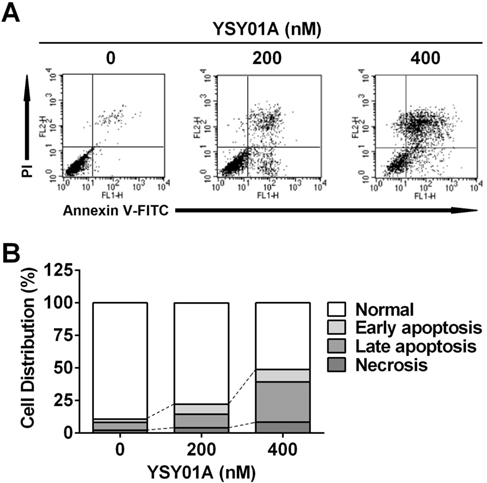
We firstly verified drug resistance between cisplatin-sensitive (SKOV3) and -resistant (SKOV3/DDP) ovarian cancer cell lines. Following exposure to increasing concentrations of cisplatin for 48 hours, SKOV3 cells showed more sensitive to cisplatin than their resistant counterparts, while the IC50 values for cisplatin were 5.7±1.5 μM and 20.3±1.3 μM in SKOV3 and SKOV3/DDP cells, respectively, suggesting ~4-fold resistance between two cell lines (p<0.001) (Fig.1B). To determine the effect of YSY01A on survival of SKOV3 and SKOV3/DDP cells, we subsequently performed a dose-response study using SRB assay following YSY01A treatment for 72 hours. Results showed that the IC50 values of YSY01A were 573.0±286.0 nM and 206.9±84.5 nM in SKOV3 and SKOV3/DDP cells, respectively (Fig.1C). SKOV3/DDP cells appeared to be more sensitive to YSY01A whereas no significant difference was found between both cell lines. IC50 values of PS-341 were 130.8±96.5 nM and 11.0±2.8 nM in SKOV3 and SKOV3/DDP cells, respectively (Fig.1D). YSY01A, although not quite as potent as the FDA-approved drug in both cells, showed comparable inhibition on survival of tumor cells including A549 (lung cancer), MCF-7 (breast cancer), MGC-803 (gastric cancer) and PC-3M (prostate cancer) cells [12, 13 and data to be published]. To determine whether apoptosis contributes to YSY01A-induced loss of cancer cell viability, we performed flow cytometry by labeling with Annexin V/PI. As evident from Fig.2, early and late apoptotic cells accounted for 7.7% and 9.5% upon treatment of SKOV3/DDP cells with 200 nM YSY01A for 48 hours, respectively. The percentages of early and late apoptotic cells increased to 10.4% and 30.7% at 400 nM, respectively. Apparently YSY01A treatment led to significant induction of apoptosis in SKOV3/DDP cells as compared to untreated cells (p<0.05). Overall, our data support that apoptosis contributes to loss of cancer cell viability induced by YSY01A.
Induction of apoptosis by YSY01A in cisplatin-resistant SKOV3/DDP cells. Following exposure to YSY01A for 72 hours, apoptotic cells were detected by using dual staining with Annexin V and PI by flow cytometry analysis. (A) Shown is a representative of flow cytometry data. Left bottom quadrant (Annexin V-/PI-, viable cells), right bottom quadrant (Annexin V+/PI-, early apoptotic cells), right upper quadrant (Annexin V+/PI+, late apoptotic cells) and left upper quadrant (Annexin V-/PI+, necrotic cells). (B) Percentages of cells in each quadrant are shown and represent a set of five experiments.
YSY01A abrogates regulatory proteins important for cell proliferation and anti-apoptosis in cisplatin-resistance cancer cells
DNA damage and subsequent induction of apoptosis are the primary cytotoxic mechanism of cisplatin whereas aberrant signaling transduction usually contributes to apoptosis resistance, for example, the transcription factors NF-κB p65 and STAT3 associate with overexpression of anti-apoptotic gene, Bcl-2, in tumor cells [18, 19]. To explore the mechanism of YSY01A to suppress cell survival, we examined the cellular effect of YSY01A treatment on the proteins favoring cell proliferation and anti-apoptosis. SKOV3/DDP cells were treated with 0 (vehicle control), 200 and 400 nM YSY01A for 48 hours or 400 nM YSY01A for 0, 12, 24 and 48 hours, respectively. As evident from Fig.3A, NF-κB p65 was reduced by YSY01A treatment in dose- and time-dependent manners and a reduction in phosphorylated STAT3 at Tyr705 was also detected with decreased total protein level of STAT3. However, the mRNA levels of NF-κB p65 and STAT3 were not changed substantially as determined by quantitative real-time PCR analysis, suggesting a protein-level regulation on these proteins (Fig.3B).
Effect of YSY01A on regulatory proteins important for cell proliferation and anti-apoptosis in cisplatin-resistance cancer cells. (A) Expression levels of specific proteins that are involved in cell proliferation and anti-apoptosis were analyzed by Western blotting with antibodies indicated after SKOV3/DDP cells were exposed to 0 (vehicle control), 200 or 400 nM YSY01A. GAPDH was used as a loading control. (B) SKOV3/DDP cells were treated as indicated followed by extracting total RNA and real-time PCR analysis of selected genes. GAPDH was used as an internal control. (N=3; ** p<0.01, by Student's t-test as compared with vehicle control)
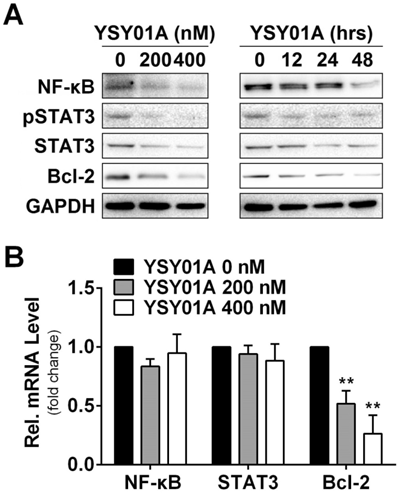
Anti-apoptotic genes are prominent targets for NF-κB p65 and STAT3, e.g. Bcl-2 are regulated by both factors [20, 21]. We also found that the protein level of Bcl-2 was reduced following YSY01A treatment (Fig.3A). This observation was further confirmed on mRNA level as well (Fig.3B). Treatment with 200 nM YSY01A diminished the Bcl-2 mRNA level by 48% and it further decreased by 73% after treatment with 400 nM YSY01A, as compared with vehicle control. Collectively, our data suggest that YSY01A modulates the NF-κB and STAT3 at the protein level and their anti-apoptotic downstream, Bcl-2, was down-regulated accordingly.
YSY01A enhances the sensitivity of drug-resistant ovarian cancer cells to cisplatin
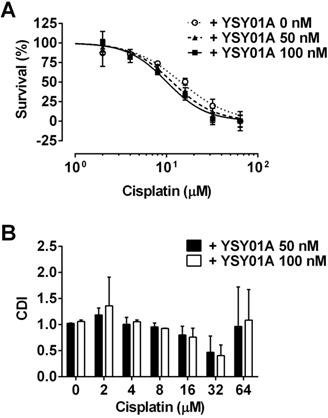
Considering YSY01 alone can cause loss of cell viability and induction of apoptosis, we hypothesized that the addition of YSY01A may increase cisplatin sensitivity in cisplatin-resistant cancer cells, SKOV3/DDP cells were pretreated with 0 (vehicle control), 50 or 100 nM YSY01A for 24 hours followed by 48-hour combination treatment with cisplatin. The IC50 of cisplatin was reduced to 13.4±1.3 μM in combination with 50 nM YSY01A. YSY01A at 100 nM further reduced the IC50 value to 11.7±1.3 μM. The change in sensitivity was reflected as a left shift of the dose-response curve in combination with YSY01A (Fig.4A). We also analyzed the nature of the interaction between YSY01A and cisplatin using CDI, which quantitatively measures the interaction of two drugs. As shown in Fig.4B, the synergistic effect was most prominent when 32 µM cisplatin was combined with 100 nM YSY01A. Thus our findings indicate that YSY01A enhances the cytotoxicity of cisplatin in cisplatin-resistant ovarian cells.
Enhancement of cisplatin cytotoxicity in SKOV3/DDP cells after YSY01A treatment. (A) SKOV3/DDP cells were treated with cisplatin alone or in combination with YSY01A followed by SRB assay. GraphPad software was used to generate cell survival curves and compute IC50 values of cisplatin in the presence or absence of YSY01A. (B) Drug interaction was analyzed using the coefficient of drug interaction (CDI). CDI<1 indicates a synergistic effect, CDI=1 indicates an additive effect, CDI>1 indicates an antagonistic effect.
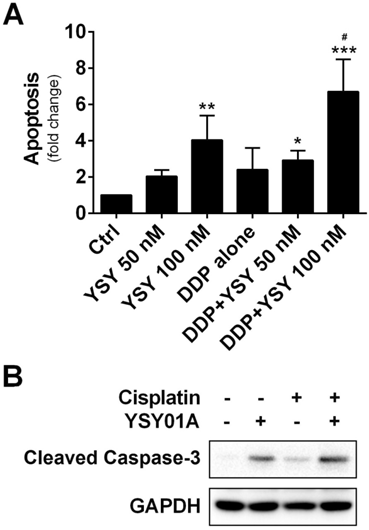
We further investigated whether combination of YSY01A with cisplatin can induce apoptosis in cisplatin-resistant cells by using ELISA which determines DNA fragmentation and histone release from the nucleus during the apoptosis process. As shown in Fig.5A, YSY01A treatment alone induced apoptosis in a dose-dependent manner. Moreover, when cells were pretreated for 24 hours with 100 nM YSY01A prior to exposure to the combination, the apoptosis effect was improved to 6.7-fold whereas cisplatin alone only induced 4-fold increase in apoptosis. And the combined treatment induced significantly higher apoptosis than either agent alone (p<0.05 compared with YSY01A or cisplatin alone). This finding was also confirmed by cleavage of caspase 3, a key executioner of apoptosis resulting from proteolytic processing of its inactive zymogen, as determined by Western blotting analysis (Fig.5B). Thus YSY01A and cisplatin combined treatment also suppresses cell survival by inducing apoptosis in SKOV3/DDP cells.
Effect of YSY01A on apoptosis in combination with cisplatin. (A) SKOV3/DDP cells were treated with cisplatin alone or in combination with YSY01A followed by determination of apoptosis using ELISA. (N=3; * p<0.05, ** p<0.01, *** p<0.001, compared with vehicle control; # p<0.05, compared with YSY01A or cisplatin alone) (B) Cleavage of caspase-3, one of the key executioners of apoptosis, was analyzed by Western blotting after SKOV3/DDP cells were treated with cisplatin alone or in combination with YSY01A.
YSY01A enhances cisplatin accumulation in cisplatin-resistant ovarian cancer cells
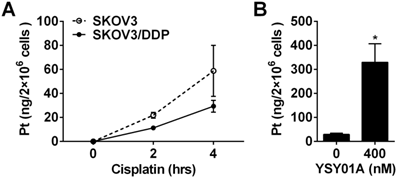
Cisplatin resistance is a complex process in which many factors are involved [22]. As shown above, down-regulation of anti-apoptotic gene, Bcl-2, via NF-κB and STAT3, appears to be one of the mechanisms which explain that YSY01A overcomes cisplatin resistance. On the other hand, the efficacy of cisplatin is associated with its cellular accumulation. Following exposure to 50 μM cisplatin alone for 2 or 4 hours, 21.9±2.4 ng and 58.8±21.3 ng of platinum were detected in 2×106 SKOV3 cells, respectively; however, the platinum contents were reduced to 11.3±1.7 ng and 29.3±4.9 ng in the same amount of SKOV3/DDP cells as determined by ICP-MS analysis (Fig.6A). The results suggest that the chemoresistant phenotype of SKOV3/DDP cells is likely due to decreased cellular accumulation of platinum. To investigate the effect of YSY01A on the uptake of cisplatin in cisplatin-resistant cells, SKOV3/DDP cells were treated for 4 hours with 50 μM cisplatin with or without a prior 24-hour exposure to 400 nM YSY01A. As shown in Fig.6B, the combination of cisplatin with YSY01A induced a dramatic increase in intracellular platinum content up to 329.7±77.5 ng. Thus the ability of YSY01A to increase cisplatin sensitivity may also result from a substantial increase of cisplatin uptake in tumor cells. Nevertheless, how this event is involved awaits further investigation.
Effect of YSY01A on cisplatin delivery to cisplatin-resistant SKOV3/DDP cells. (A) Differences of cisplatin delivery between SKOV3 and SKOV3/DDP cells were analyzed by measurements of platinum content using ICP-MS after cells were exposed to 50 μM cisplatin for 2 or 4 hours. (B) SKOV3/DDP cells were exposed to 50 μM cisplatin for 4 hours with a pretreatment with 400 nM YSY01A or vehicle control for 24 hours. Platinum contents were then measured by ICP-MS. (N=3; * p<0.05, by Student's t-test as compared with vehicle control)
Discussion
Numerous studies have demonstrated that combination therapy with conventional agents may result in greater tumor growth inhibition than treatment with either agent alone [23-25]. The proteasome inhibitor YSY01A is a promising novel agent that exhibits anti-tumor effect via multiple signaling pathways [12, 13]. In an attempt to identifying the potential of YSY01A to enhance the cytotoxicity of cisplatin, particularly in cisplatin-resistant cells, the present study demonstrated for the first time that YSY01A and cisplatin combined treatment inhibited cell survival by inducing apoptosis and enhancing cisplatin uptake in the cisplatin-resistant ovarian cancer cell line. Our findings provide further evidence for a contribution of proteasomal inhibition to overcome conventional drug resistance in tumors.
Platinum-based chemotherapy including cisplatin or carboplatin has been a general standard for treatment of ovarian cancer for decades [26]. Despite most patients with ovarian cancer initially respond to cisplatin, the majority will ultimately die with platinum-resistant disease. Obviously, the development of chemoresistance is one of the major limitations to achieve successful platinum-based therapy regimens. Genetic heterogeneity in cancer may limit effectiveness of single target drugs even if they successfully inhibit or activate a specific target as complex diseases such as cancers are always involved in compensatory regulation. The development of cisplatin resistance in human cancer cells also results from complex genetic and epigenetic changes in gene expression and alternations in protein localization involving in cell proliferation, apoptosis, DNA damage repair and endocytosis [27]. Thus identification of effective and safe anticancer regimens to improve the efficacy of cisplatin is thus a major therapeutic challenge whilst may provide a promising strategy for treatment of platinum-resistant advanced ovarian cancer.
It is noteworthy that the ubiquitin-proteasome pathway regulates multiple events and many protein targets, including the regulatory proteins important for cell proliferation and anti-apoptosis as well as cisplatin transportation in cells [4]. On the one hand, inhibition of proteasome activity results in the accumulation of numerous regulatory proteins within the cell and constitutive endoplasmic reticulum stress blocks cellular growth and division, eventually leading to cell death. Chemoresistance has previously been found to associate with the failure of cisplatin to induce apoptosis [22]. Numerous studies have demonstrated great contribution of NF-κB and STAT3 to cisplatin resistance via suppression of cisplatin-induced apoptosis [28-31]. The activation level of NF-κB is higher in cisplatin-resistant human epidermoid carcinoma cells and curcumin, an inhibitor of NF-κB activation, sensitizes the cisplatin-resistant cells by down-regulation of apoptosis-related proteins such as Bcl-2 [28]. The ectopic expression of constitutively-active STAT3c in the cisplatin-sensitive ovarian cancer cells also induces cisplatin resistance and the inhibition of STAT3 activity represses the colony-forming potential of the cisplatin-resistant ovarian cancer cells and sensitizes them to cisplatin [30]. In the present study, the presence of elevated NF-κB p65 and phospho-STAT3 levels was also observed in cisplatin-resistant SKOV3/DDP cells (Supplemental Fig.S1). Therefore, the two transcriptional factors may make a vital contribution to enhanced expression of anti-apoptotic genes in cancer cells, making SKOV3/DDP cells resistant to chemotherapy. We found that YSY01A treatment leads to a reduction in the two proteins. One possibility is likely due to proteasome inhibitor-induced endoplasmic reticulum stress and resulting shut-down of de novo protein synthesis. Our previous study has shown the phosphorylation of Akt and mTOR decreased after MCF-7 cells were exposed to YSY01A [13]. The direct link between bortezomib-induced endoplasmic reticulum stress and the shutdown of de novo synthesis has been reported [32]. Thus the role of YSY01A in this process awaits further investigation. Additionally, proteasome-independent mechanisms may be also involved in the process as YSY01A has been shown to induce autophagy in prostate cancer cells [33]. Bortezomib and related proteasome inhibitors have been shown to activate autophagy in various cancers [34, 35]. Under conditions of persistent bortezomib stress, some proteins may be sequestered to lysosomes, resulting in degradation. Nevertheless, regardless of the reasons, NF-κB and STAT3 pathways control the expression of various genes, either synergistically or individually [36]. Bcl-2 is one of anti-apoptotic genes activated by both factors [20, 21]. As one of important anti-apoptosis proteins, overexpressed Bcl-2 can protect cells from apoptotic stimuli [18, 19]. Not surprisingly YSY01A alone caused apoptosis by transcriptional down-regulation of the anti-apoptotic gene via NF-κB and STAT3 pathways. Moreover, YSY01A not only suppresses survival of cisplatin-resistant SKOV3/DDP cells but also enhances cellular sensitivity to cisplatin. The effect of YSY01A on apoptosis induction was also observed in combination with cisplatin as evidenced by apoptosis ELISA and up-regulation of cleaved caspase-3. Collectively, our data support that the combination of YSY01A and cisplatin may promise a reasonable strategy for improving therapeutic efficacy in cisplatin-resistant advanced ovarian cancer where little effective regimen exists.
On the other hand, bortezomib has been reported to block the cisplatin-induced down-regulation of copper transporter 1, a major influx transporter of platinum drugs, and increase cisplatin uptake [37]. Although whether YSY01A affects multi-drug resistance medicated drug transportation remains to be determined, e.g. diverse ATP-binding cassette transporters involving in cisplatin resistance, we found a dramatic rise in platinum content by 11.3-fold with a 24-hour pretreatment with 400 nM YSY01A, suggesting that YSY01A may augment the antitumor effect of cisplatin by increasing cisplatin accumulation in drug-resistant cells. If such findings are found to be true in vivo, the results would be significant clinically whereas YSY01A will likely need to be given on a schedule what allows for maximal proteasomal inhibition prior to administration of platinum-based drugs.
As a multi-target agent, proteasome inhibitors induce multiple cellular changes via different signaling pathways. Although questions remain regarding the cellular effects of proteasome inhibitors, clinical studies have demonstrated the efficacy of this class of therapeutics [38]. In fact, combination therapy is also a form of multi-target medication that is widely used in clinical practice. We believe that the multi-target drugs have much potential to affect the complex equilibrium of whole cellular networks than drugs that act on a single target. Proteasome inhibitors undoubtedly represent a promising strategy for treatment of complex diseases such as cancers. Despite the clinical success of bortezomib in multiple myeloma, resistance to this drug remains a clinically significant problem involving gene mutation and overexpression and pharmacokinetic properties [4, 39]. Design of novel proteasome inhibitors might be helpful in partially overcomming bortezomib resistance due to expression of proteasome β5 subunit [39]. Previous study has shown that YSY01A is 5-fold more potent than bortezomib against the trypsin-like site (β2/β2i) of the proteasome with comparable activitiy in inhibiting the chymotrpsin-like site (β5/β5i) and the post-glutamyl peptide hydrolase site (β1/β1i) [12]. YSY01A undoubtedly represents a promising candidate for development of next-generation proteasome inhibitors. Our ongoing animal study shows that YSY01A has a broad spectrum of anti-tumor activity in vivo that is more potent than or comparable to bortezomib whilst YSY01A shows a 4-fold lower toxicity (data not shown). The present study reinforces the potential benefit of YSY01A in cancer therapy and provides extensive description of the mechanism of YSY01A on cisplatin resistance. Further studies directed at this strategy will bring more insights into the approach to interrupt the proteasome for potential antineoplastic therapy.
Supplementary Material
Table S1 and Figure S1.
Abbreviations
ANOVA: analysis of variance; Bcl-2: B-cell lymphoma 2; CDI: coefficient of drug interaction; Ct: threshold cycle; DDP: cisplatin; DMSO: dimethyl sulfoxide; ELISA: enzyme-linked immunosorbent assay; GAPDH: glyceraldehyde 3-phosphate dehydrogenase; ICP-MS: inductively coupled plasma-mass spectrometry; NF-κB: nuclear factor κ-light-chain-enhancer of activated B cells; PCR: polymerase chain reaction; PI: propidium iodide; SRB: sulforhodamine B; STAT3: signal transducer and activator of transcription 3.
Acknowledgements
This work was primarily supported by the National Major Scientific and Technological Special Project for “Significant New Drugs Development” Program (Ministry of National Science and Technology of China, Grant No. 2009ZX0930010) and the State Key Program of National Natural Science Foundation of China (Grant No. 81172915). The author, Wei Huang, was sponsored by National Natural Science Foundation of China (Grant No. 81503091), General Financial Grant from the China Postdoctoral Science Foundation (Grant No. 2015M570906) and Postdoctoral Grant of Peking University-Tsinghua University Center for Life Science. The author, Hua-yu Yang, was supported by National Natural Science Foundation of China (Grant No. 81201566).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Continuous Update Project Report. Food, Nutrition, Physical Activity, and the Prevention of Ovarian Cancer 2014 World Cancer Research Fund/American Institute for Cancer Research. http://www.dietandcancerreport.org/cup/cup_resources.php
2. Eckstein N. Platinum resistance in breast and ovarian cancer cell lines. J Exp Clin Cancer Res. 2011;30:91
3. Vasey PA. Resistance to chemotherapy in advanced ovarian cancer: mechanisms and current strategies. Br J Cancer. 2003;89(Suppl 3):S23-8
4. McConkey DJ, Zhu K. Mechanisms of proteasome inhibitor action and resistance in cancer. Drug Resist Updat. 2008;11(4-5):164-79
5. Chen D, Frezza M, Schmitt S, Kanwar J, Dou QP. Bortezomib as the first proteasome inhibitor anticancer drug: current status and future perspectives. Curr Cancer Drug Targets. 2011;11(3):239-53
6. Ruschak AM, Slassi M, Kay LE, Schimmer AD. Novel proteasome inhibitors to overcome bortezomib resistance. J Natl Cancer Inst. 2011;103(13):1007-17
7. McBride A, Klaus JO, Stockerl-Goldstein K. Carfilzomib: a second-generation proteasome inhibitor for the treatment of multiple myeloma. Am J Health Syst Pharm. 2015;72(5):353-60
8. Thompson JL. Carfilzomib: a second-generation proteasome inhibitor for the treatment of relapsed and refractory multiple myeloma. Ann Pharmacother. 2013;47(1):56-62
9. Potts BC, Albitar MX, Anderson KC, Baritaki S, Berkers C, Bonavida B. et al. Marizomib, a proteasome inhibitor for all seasons: preclinical profile and a framework for clinical trials. Curr Cancer Drug Targets. 2011;11(3):254-84
10. Offidani M, Corvatta L, Caraffa P, Gentili S, Maracci L, Leoni P. An evidence-based review of ixazomib citrate and its potential in the treatment of newly diagnosed multiple myeloma. Onco Targets Ther. 2014;7:1793-800
11. Zhu Y, Yao S, Xu B, Ge Z, Cui J, Cheng T. et al. Design, synthesis and biological evaluation of tripeptide boronic acid proteasome inhibitors. Bioorg Med Chem. 2009;17(19):6851-61
12. Zhang M, Yuan X, Xu B, Guo W, Ran FX, Li RT. et al. Anticancer Effect of a Novel Proteasome Inhibitor, YSY01A, via G2/M Arrest in PC-3M Cells in vitro and in vivo. J Cancer. 2015;6(8):701-8
13. Xue B, Huang W, Yuan X, Xu B, Lou Y, Zhou Q. et al. YSY01A, a Novel Proteasome Inhibitor, Induces Cell Cycle Arrest on G2 Phase in MCF-7 Cells via ERalpha and PI3K/Akt Pathways. J Cancer. 2015;6(4):319-26
14. Huang W, Dong Z, Chen Y, Wang F, Wang CJ, Peng H. et al. Small-molecule inhibitors targeting the DNA-binding domain of STAT3 suppress tumor growth, metastasis and STAT3 target gene expression in vivo. Oncogene. 2015 [Epub ahead of print]
15. Xu SP, Sun GP, Shen YX, Wei W, Peng WR, Wang H. Antiproliferation and apoptosis induction of paeonol in HepG2 cells. World J Gastroenterol. 2007;13(2):250-6
16. Jin JL, Gong J, Yin TJ, Lu TJ, Xia JJ, Xie YY. et al. PTD4-apoptin protein and dacarbazine show a synergistic antitumor effect on B16-F1 melanoma in vitro and in vivo. Eur J Pharmacol. 2011;654(1):17-25
17. Huang W, Dong Z, Wang F, Peng H, Liu JY, Zhang JT. A small molecule compound targeting STAT3 DNA-binding domain inhibits cancer cell proliferation, migration, and invasion. ACS Chem Biol. 2014;9(5):1188-96
18. Real PJ, Sierra A, De Juan A, Segovia JC, Lopez-Vega JM, Fernandez-Luna JL. Resistance to chemotherapy via Stat3-dependent overexpression of Bcl-2 in metastatic breast cancer cells. Oncogene. 2002;21(50):7611-8
19. Buchholz TA, Garg AK, Chakravarti N, Aggarwal BB, Esteva FJ, Kuerer HM. et al. The nuclear transcription factor kappaB/bcl-2 pathway correlates with pathologic complete response to doxorubicin-based neoadjuvant chemotherapy in human breast cancer. Clin Cancer Res. 2005;11(23):8398-402
20. Viatour P, Bentires-Alj M, Chariot A, Deregowski V, de Leval L, Merville MP. et al. NF-kappa B2/p100 induces Bcl-2 expression. Leukemia. 2003;17(7):1349-56
21. Choi HJ, Han JS. Overexpression of phospholipase D enhances Bcl-2 expression by activating STAT3 through independent activation of ERK and p38MAPK in HeLa cells. Biochim Biophys Acta. 2012;1823(6):1082-91
22. Galluzzi L, Senovilla L, Vitale I, Michels J, Martins I, Kepp O. et al. Molecular mechanisms of cisplatin resistance. Oncogene. 2012;31(15):1869-83
23. Xu Y, Li D, Zeng L, Wang C, Zhang L, Wang Y. et al. Proteasome inhibitor lactacystin enhances cisplatin cytotoxicity by increasing endoplasmic reticulum stress-associated apoptosis in HeLa cells. Mol Med Rep. 2015;11(1):189-95
24. Fribley AM, Evenchik B, Zeng Q, Park BK, Guan JY, Zhang H. et al. Proteasome inhibitor PS-341 induces apoptosis in cisplatin-resistant squamous cell carcinoma cells by induction of Noxa. J Biol Chem. 2006;281(42):31440-7
25. Konac E, Varol N, Kiliccioglu I, Bilen CY. Synergistic effects of cisplatin and proteasome inhibitor bortezomib on human bladder cancer cells. Oncol Lett. 2015;10(1):560-4
26. Muggia F. Platinum compounds 30 years after the introduction of cisplatin: implications for the treatment of ovarian cancer. Gynecol Oncol. 2009;112(1):275-81
27. Shen DW, Pouliot LM, Hall MD, Gottesman MM. Cisplatin resistance: a cellular self-defense mechanism resulting from multiple epigenetic and genetic changes. Pharmacol Rev. 2012;64(3):706-21
28. Oiso S, Ikeda R, Nakamura K, Takeda Y, Akiyama SI, Kariyazono H. Invovlment of NF-κB activation in the cisplatin resistance of human epidermoid carcinoma KCP-4 cells. Oncol Rep. 2012;28(1):27-32
29. Shen W, Liang B, Yin J, Li X, Cheng J. Noscapine Increases the Sensitivity of drug-resistant ovarian cancer cell line SKOV3/DDP to cisplatin by regulating cell cycle and activating apoptotic pathways. Cell Biochem Biophys. 2014 [Epub ahead of print]
30. Yue PB, Zhang XL, Paladino D, Sengupta B, Ahmad S, Holloway H. et al. Heperactive EGF receptor, Jaks and Stat3 signaling promote enhanced colony-forming ability, motility and migration of cisplatin-resistant ovarian cancer cells. Oncogene. 2012;31(18):2309-22
31. Tang YJ, Sun ZL, Wu WG, Xing J, He YF, Xin DM. et al. Inhibition of signal transducer and activator of transcription 3 (STAT3) suppresses ovarian cancer growth, migraiton and invasion and enhances the effect of cisplatin in vitro. Genet Mol Res. 2015;14(1):2350-60
32. Obeng EA, Carlson LM, Gutman DM, Harrington WJ Jr, Lee KP, Boise LH. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood. 2006;107(12):4907-16
33. Wang Z, Yuan X, Ge ZM, Ran FX, Wu J, Li RT. et al. A new proteasome inhibitor YSY01A induced autophagy in PC-3M cells. J Chin Pharm Sci. 2014;23(8):565-71
34. Selimovic D, Porzig BB, El-Khattouti A, Badura HE, Ahmad M, Ghanjati F. et al. Bortezomib/proteasome inhibitor triggers both apoptosis and autophagy-dependent pathways in melanoma cells. Cell Signal. 2013;25(1):308-18
35. Lou Z, Ren T, Peng X, Sun Y, Jiao G, Lu Q. et al. Bortezomib induces apoptosis and autophagy in osteosarcoma cells through mitogen-activated protein kinase pathway in vitro. J Int Med Res. 2013;41(5):1505-19
36. Grivennikov SI, Karin M. Dangerous liaisons: STAT3 and NF-kappaB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev. 2010;21(1):11-9
37. Jandial DD, Farshchi-Heydari S, Larson CA, Elliott GI, Wrasidlo WJ, Howell SB. Enhanced delivery of cisplatin to intraperitoneal ovarian carcinomas mediated by the effects of bortezomib on the human copper transporter 1. Clin Cancer Res. 2009;15(2):553-60
38. Dou QP, Zonder JA. Overview of proteasome inhibitor-based anti-cancer therapies: perspective on bortezomib and second generation proteasome inhibitors versus future generation inhibitors of ubiquitin-proteasome system. Curr Cancer Drug Targets. 2014;14(6):517-36
39. Franke NE, Niewerth D, Assaraf YG, van Meerloo J, Vojtekova K, van Zantwijk CH. et al. Impaired bortezomib binding to mutant beta5 subunit of the proteasome is the underlying basis for bortezomib resistance in leukemia cells. Leukemia. 2012;26(4):757-68
Author contact
![]() Corresponding authors: Jingrong Cui, State Key Laboratory of Natural and Biomimetic Drugs, School of Pharmaceutical Sciences, Peking University, 38 Xueyuan Road, Haidian District, Beijing 100191, China, Tel: 86(10)82802467, Email: jrcuiedu and Runtao Li, Department of Medicinal Chemistry, School of Pharmaceutical Sciences, Peking University, 38 Xueyuan Road, Haidian District, Beijing 100191, China, Tel: 86(10)82801504, Email: lirtedu
Corresponding authors: Jingrong Cui, State Key Laboratory of Natural and Biomimetic Drugs, School of Pharmaceutical Sciences, Peking University, 38 Xueyuan Road, Haidian District, Beijing 100191, China, Tel: 86(10)82802467, Email: jrcuiedu and Runtao Li, Department of Medicinal Chemistry, School of Pharmaceutical Sciences, Peking University, 38 Xueyuan Road, Haidian District, Beijing 100191, China, Tel: 86(10)82801504, Email: lirtedu