Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2016; 7(8):947-956. doi:10.7150/jca.14441 This issue Cite
Research Paper
Effects of CDK4/6 Inhibition in Hormone Receptor-Positive/Human Epidermal Growth Factor Receptor 2-Negative Breast Cancer Cells with Acquired Resistance to Paclitaxel
Adriana Priscila Trapé1, Shuying Liu1, Andrea Carolina Cortes2, Naoto T. Ueno1 ![]() , Ana Maria Gonzalez-Angulo1
, Ana Maria Gonzalez-Angulo1
1. Department of Breast Medical Oncology, The University of Texas MD Anderson Cancer Center, Houston, TX;
2. Institute for Personalized Cancer Therapy, The University of Texas MD Anderson Cancer Center, Houston, TX.
Received 2015-11-16; Accepted 2016-3-21; Published 2016-5-12
Citation:
Trapé AP, Liu S, Cortes AC, Ueno NT, Gonzalez-Angulo AM. Effects of CDK4/6 Inhibition in Hormone Receptor-Positive/Human Epidermal Growth Factor Receptor 2-Negative Breast Cancer Cells with Acquired Resistance to Paclitaxel. J Cancer 2016; 7(8):947-956. doi:10.7150/jca.14441. https://www.jcancer.org/v07p0947.htm
Other stylesAbstract
Among patients with hormone receptor (HR)-positive breast cancer, those with residual disease after neoadjuvant chemotherapy have a higher risk of relapse and poorer survival than those with a complete response. Previous studies have revealed a correlation between activation of cell cycle-regulating pathways in HR-positive breast cancer, particularly cyclin-dependent kinase (CDK) 4 and 6/cyclin D1 signaling, and resistance to standard therapies. Although CDK4/6 inhibition by palbociclib in combination with endocrine therapy has shown potent antiproliferative effects in HR-positive/human epidermal growth factor receptor 2 (HER2)-negative breast cancer, the potential role of palbociclib in re-sensitizing chemotherapy-resistant HR-positive breast cancer is not well defined. We hypothesized that CDK4/6 inhibition by palbociclib re-sensitizes HR-positive/HER2-negative residual breast cancer to taxane-based adjuvant therapy. Using cell counting, flow cytometry, and western blotting, we evaluated the efficacy of palbociclib alone and in concurrent or sequential combination with paclitaxel in parental and paclitaxel-resistant T47D HR-positive/HER2-negative breast cancer cells. The CDK4/6 pathway was constitutively active in both parental and paclitaxel-resistant T47D cells; thus, both cell types were highly sensitive to the inhibitory effects of single-agent palbociclib on cell growth and cell cycle progression. However, palbociclib did not re-sensitize resistant cells to paclitaxel-induced G2/M arrest and cell death in any of the combinations tested. Our results suggest that CDK4/6 inhibition by palbociclib does not re-sensitize HR-positive/HER2-negative residual breast cancer to chemotherapy. Nevertheless, the fact that CDK4/6 activation remained intact in paclitaxel-resistant cells indicates that patients who have HR-positive/HER2-negative residual disease after taxane-based neoadjuvant chemotherapy may still benefit from palbociclib in combination with other regimens, such as endocrine therapies, for adjuvant therapy.
Keywords: paclitaxel, resistance, palbociclib, HR-positive/HER2-negative, breast cancer.
Introduction
In patients with breast cancer, neoadjuvant chemotherapy is used to reduce tumor burden and enable patients to choose breast-conserving surgery for tumor resection. Importantly, the response to neoadjuvant chemotherapy has prognostic implications and might be used to improve decision making during treatment1. Patients with breast cancer that is hormone receptor (HR) positive and human epidermal growth factor receptor 2 (HER2) negative are less likely than patients with HR-negative disease to achieve a pathologic complete response after neoadjuvant chemotherapy, and the lack of a pathologic complete response correlates with a higher risk of relapse and poorer outcome2. In patients with HR-positive/HER2-negative breast cancer and residual disease after neoadjuvant chemotherapy, additional adjuvant chemotherapy may be excessively toxic and negatively affect patients' quality of life3. However, adding targeted therapies to adjuvant chemotherapy may increase the sensitivity of residual disease to adjuvant chemotherapy and, consequently, reduce the dose of chemotherapy necessary to kill the remaining tumor cells, thereby minimizing the toxicity of the prolonged treatment.
Using functional proteomics for the molecular characterization of residual disease, our group identified a two-marker model based on cyclin E1 and CD31 that predicted relapse-free survival in patients with HR-positive tumors that were resistant to taxane- and anthracycline-based neoadjuvant chemotherapy, i.e., patients with residual disease that overexpressed cyclin E1 had a high risk of relapse. Since cyclin E1 is required for cell cycle progression through the G1/S transition, we interpreted our results as indicating that dysregulation of cell cycle progression through G1/S is associated with resistance to neoadjuvant chemotherapy 4.
Cyclin D1 is another important regulator of the G1/S transition and is overexpressed in approximately 50% of breast cancers5. In response to diverse oncogenic stimuli, cyclin D1 activates cyclin-dependent kinases 4 and 6 (CDK4/6), which phosphorylate and inactivate the retinoblastoma tumor suppressor protein (pRb), releasing the transcription factor E2F to initiate the expression of genes required for cell cycle progression through the G1/S transition6.
Palbociclib (PD0332991) is a CDK inhibitor with high selectivity for CDK4/6-cyclin D1 activity and leads to efficient dephosphorylation of pRb and subsequent cell cycle arrest at the G1/S transition7. Palbociclib has been shown to reestablish cell cycle control in breast cancer cells that are resistant to tamoxifen8,9. A phase II clinical trial (PALOMA-1) showed that palbociclib in combination with letrozole as first-line treatment improved the outcome of postmenopausal women with HR-positive/HER2-negative advanced breast cancer compared to letrozole monotherapy10. These results led the United States Food and Drug Administration to approve palbociclib combined with letrozole as first-line treatment for metastatic disease. Recently, results from a phase III clinical trial (PALOMA-3) indicated that palbociclib in combination with fulvestrant significantly improved progression-free survival of women with HR-positive/HER2-negative breast cancer resistant to prior endocrine therapy compared to fulvestrant alone11.
Although palbociclib significantly improves disease response to endocrine therapy, the effects of adding palbociclib to chemotherapy seem to be more complex. Preclinical studies have shown that palbociclib administered concurrently with doxorubicin or paclitaxel antagonizes chemotherapy-induced cytotoxicity12. In contrast, treatment with palbociclib prior to paclitaxel exposure has been shown to inhibit cell growth more efficiently than paclitaxel alone13. However, these studies did not focus on HR-positive/HER2-negative breast cancer or chemotherapy-resistant models, which more closely resemble the residual disease detected in patients after neoadjuvant chemotherapy.
Given that overexpression of cell cycle-regulatory proteins has been associated with resistance of HR-positive/HER2-negative tumors to neoadjuvant chemotherapy, we hypothesized that targeting cell cycle progression with CDK4/6 inhibitors re-sensitizes HR-positive/HER2-negative residual tumors to taxane therapy, thus decreasing the toxicity of additional adjuvant chemotherapy and improving the therapeutic response. We assumed that the G1/S arrest caused by palbociclib would increase the percentage of cells progressing synchronously through G2/M transition, making them more susceptible to the effects of paclitaxel and, therefore, reversing paclitaxel resistance in our model.
Materials and Methods
Cell lines, culture, and reagents
Parental T47D (T47DPAR) breast cancer cells were authenticated and provided by the Characterized Cell Line Core facility at The University of Texas MD Anderson Cancer Center (Houston, TX). T47DPAR and paclitaxel-resistant (T47DPAC) cells were cultured in phenol-red containing RPMI with 10% heat-inactivated fetal bovine serum (FBS), 100 IU/mL penicillin, and 100 μg/mL streptomycin. The cells were maintained at 37°C in a humidified incubator in an atmosphere of 5% CO2 and periodically tested for mycoplasma contamination. The antibodies used were as follows: p-pRb (Ser780) (#9307), pRb (#9309), p-NPM (Thr199) (#3541), p-JNK1/2 (p-Thr183/Tyr185) (#4671), JNK1 (2C6) (#3708), and JNK2 (#4672) purchased from Cell Signaling Technology; cyclin A (H-432) (#751), cyclin D1 (H-295) (#753), cyclin B1 (H-433) (#752), and p55CDC (H-175) (#8358) purchased from Santa Cruz Biotechnology; β-actin (AC-15) (#A1978) purchased from Sigma Aldrich; PCNA (#ab29) purchased from Abcam; and estrogen-receptor α (ERα) (SP1) (#RM-9101-S) purchased from Thermo Scientific Lab Vision.
Establishment of paclitaxel-resistant cell line
T47DPAC cells were derived from T47DPAR cells via continuous exposure of these cells to increasing doses of paclitaxel. The medium was replaced with fresh drug-containing medium 2-3 times a week. Resistance to 5 nM paclitaxel was observed after 6 months, and the cells were allowed to recover from treatment in drug-free medium for at least 1 month before experiments were performed. To confirm the acquisition of a resistant phenotype in T47DPAC cells, we seeded both T47DPAR and T47DPAC cells in 6-well plates at a density of 50,000 cells/well. After 24 hours, both T47DPAR and T47DPAC cells were treated with DMSO (control) or 2 nM paclitaxel for 6 days. Cells were counted using a Cellometer cell counter (Nexcelom Bioscience) for 6 consecutive days. For clonogenic assays, cells were seeded in 60-mm dishes at a density of 1000 cells/dish and treated with 2 nM paclitaxel for 96 hours. Drug-containing medium was then removed, and the cells were allowed to recover in drug-free medium. Dishes were scanned and the number of colonies was observed after 12 days.
Figure 1
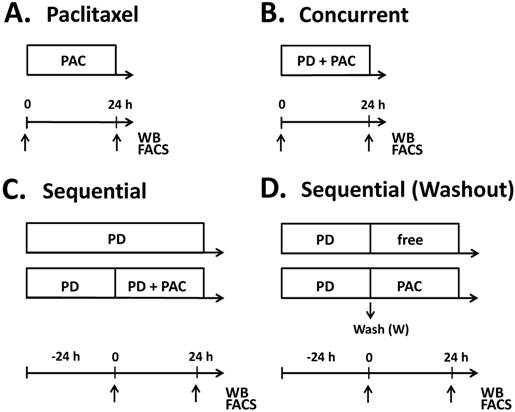
Treatment schedules for parental (T47DPAR) and paclitaxel-resistant T47D cells (T47DPAC). Cells were treated with paclitaxel alone (PAC) up to 24 hours (A), concurrent combination therapy up to 24 hours (B), palbociclib (PD) alone for up to 48 hours (C) and sequential therapy (C, D). In the sequential therapy, cells were treated with palbociclib (PD) for 24 hours followed by addition of paclitaxel for an additional 24 hours (C), or followed by exposure to drug-free (free) medium or paclitaxel for 24 hours after PD washout (D). Samples were collected (arrows) for fluorescence-activated cell sorting (FACS) and western blotting (WB) at 24 hours after addition of paclitaxel in the monotherapy experiments. In the concurrent combination, samples were collected at 24 hours after simultaneous addition of palbociclib and paclitaxel into the cell culture medium. For sequential experiments, samples were collected at 24 hours and 48 hours after palbociclib treatment and 24 hours after addition of drug-free medium or paclitaxel.
Drugs and general drug treatment scheme
Paclitaxel was purchased from Sigma-Aldrich (#T7402) and palbociclib (PD0332991) was purchased from Selleck Chemicals (#S1116). Stock solutions were prepared in dimethylsulfoxide (DMSO) and stored at -20°C.
To evaluate the effects of adding palbociclib to paclitaxel in T47DPAR and T47DPAC cells, treatment was administered as shown in Figure 1. Control cells were treated with vehicle (DMSO) for the same period of time as the other cells were treated with drugs. For concurrent treatment, palbociclib and paclitaxel were added simultaneously to the cell culture medium, whereas for sequential therapy, the cells were exposed to palbociclib for 24 hours prior to the addition of paclitaxel in the presence of or after washout of palbociclib. The effects of each combination treatment were compared to those of control and each drug alone.
Western blotting procedure
For western blotting, cells were lysed in ice-cold X-100 lysis buffer (1% Triton X-100, 50 mM HEPES [pH 7.4], 150 mM NaCl, 1.5 mM MgCl2, 1 mM ethylene glycol tetraacetic acid, 10% glycerol, 100 mM NaF, 10 mM sodium pyrophosphate, phosphatase, and a proteinase inhibitor mixture [Roche Applied Science]). After the samples were diluted in sodium dodecyl sulfate (SDS) buffer (2% SDS, 62.5 mM Tris [pH 6.8], 10% glycerol, and 2.5% β-mercaptoethanol), cell lysates were resolved via SDS/polyacrylamide gel electrophoresis, transferred to polyvinylidene fluoride membranes, and stained with antibodies following the protocols provided by the manufacturers. The bands were visualized via an ECL detection kit (Amersham Biosciences).
Effects of drug treatment on JNK and ERK activation
To assess the effects of drug treatment on JNK and ERK activation, T47DPAR and T47DPAC cells were serum-starved overnight and then treated with 500 nM palbociclib, 4 nM paclitaxel, or both concurrently for 6 hours. Using western blotting, the activation status of the JNK and ERK signaling pathways was evaluated in the cells before and after stimulation with 10% FBS for 10 minutes.
Effects of drug treatment on cell growth
To assess the effects of drug treatment on the growth of T47DPAR and T47DPAC cells, we seeded cells in 6-well plates at a density of 50,000 cells/well and treated them with 500 nM palbociclib, 2 nM paclitaxel, or both concurrently for 6 days or with 500 nM palbociclib for 1 day followed by 2 nM paclitaxel for 6 days in the presence of or after washout of palbociclib. Remaining cells were counted using a Cellometer cell counter.
Effects of drug treatment on cell cycle progression
The effects of drug treatments on cell cycle progression in T47DPAR and T47DPAC cells were evaluated by seeding cells at a density of 300,000 cells per 25-cm2 flask or 60-mm dish followed by treatment with 4 nM paclitaxel alone for 24hs (Fig. 1A); 4nM paclitaxel in concurrent combination with 500 nM palbociclib for 24hs (Fig. 1B) or with 500 nM palbociclib for 24 hours followed by 4 nM paclitaxel for 24 hours in the presence of or after washout of palbociclib (Fig. 1C, Fig. 1D). Cells were harvested at 24 hours after the addition of paclitaxel as a single drug or in sequential combination with palbociclib. For cell cycle analyses, cells were fixed in 70% ethanol, stained with propidium iodide, and analyzed using a Gallios Flow Cytometer (Beckman Coulter) and FCS Express Version 4 (De Novo Software). Cell cycle-promoting proteins were analyzed by western blotting.
Statistical analyses
Statistical analyses were carried out using GraphPad Prism, version 6.0, for Windows (GraphPad Software). Two-way analysis of variance (ANOVA) was used in growth curve experiments to compare the number of cells in the control versus paclitaxel-treated cells. For cell counting experiments, the effects of treatments were compared among all groups using one-way ANOVA. The results were considered statistically significant when the p value was less than 0.05 and only differences between each treatment and the control are shown in the figures, unless otherwise indicated.
Results
T47DPAC cells are more resistant to the growth inhibitory effects of paclitaxel than are T47DPAR cells
To validate our model of paclitaxel resistance, we performed cell counting assays for T47DPAR and T47DPAC cells treated with paclitaxel (Fig. 2A, left panel). The growth of T47DPAR cells was strongly inhibited by 2 nM paclitaxel, since the number of cells in the control and treated groups was significantly different, particularly after 72 hours. On the other hand, the growth of T47DPAC cells treated with paclitaxel was indistinguishable from that of control cells. Similar results were obtained via clonogenic assays (Fig. 2A, right panel), confirming the acquisition of paclitaxel-resistant phenotype by T47DPAC cells.
ERα expression levels are similar in T47DPAC and T47DPAR cells
Some reports have demonstrated that in a significant percentage of HR-positive tumors, exposure to taxane-based neoadjuvant chemotherapy causes a loss of ERα expression14. Therefore, we used western blotting assays to determine whether long-term exposure to paclitaxel caused changes in the expression levels of ERα in T47DPAC cells (Fig. 2B). Constitutive levels of this protein were similar in T47DPAR and T47DPAC cells, indicating that paclitaxel did not affect ERα expression in T47DPAC cells.
JNK signaling activation is higher in T47DPAC cells than in T47DPAR cells
Given that both JNK signaling and ERK signaling play important roles in cell cycle progression, we evaluated the degree of activation of these pathways by assessing the phosphorylation status of JNK1/2 and ERK1/2 proteins in T47DPAR and T47DPAC cells before and after treatment with palbociclib and/or paclitaxel (Fig. 2C, Fig. 2D). The T47DPAC cells showed higher constitutive activation of the JNK pathway than the T47DPAR cells did, whereas activation of the ERK pathway did not seem to differ between the two cell lines (control cells; C). Treatment with palbociclib, paclitaxel, or both seemed to exert no effect on the activation of these pathways: both T47DPAR and T47DPAC cells showed similar levels of phosphorylated JNK1/2 and ERK1/2 between treated and control groups.
Palbociclib inhibits cell growth and G1/S transition in T47DPAR cells and T47DPAC cells
Next, we performed cell counting experiments and flow cytometry to evaluate the antiproliferative effects of palbociclib on T47DPAR and T47DPAC cells (Figure 3). Interestingly, palbociclib alone remarkably inhibited cell growth (Fig. 3A) and induced G1 arrest (Fig. 3B) in both T47DPAR and T47DPAC cells, thus indicating that both cell lines were sensitive to this drug. This response might be explained by the fact that the two cell lines had high constitutive levels of phosphorylated pRb protein (Fig. 3C, control cells; C), suggesting that constitutive activation of this pathway occurs in T47DPAR cells and persists after acquisition of resistance to paclitaxel in T47DPAC. Palbociclib-induced G1 arrest was associated with a decrease in phosphorylation and subsequent activation of the pRb protein, which resulted in downregulation of four proteins involved in cell cycle progression through S, G2 and M phases: cyclin A, cyclin B1, p-NPM and p55CDC. Additionally, palbociclib treatment resulted in increased expression of cyclin D1 (Fig. 3C).
Figure 2
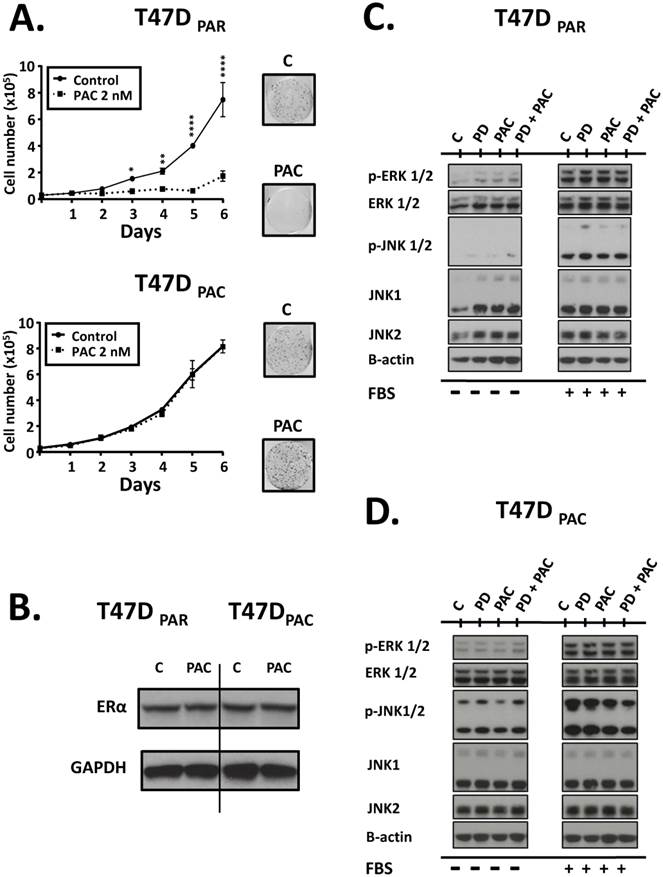
Effects of paclitaxel on parental (T47DPAR) and paclitaxel-resistant T47D cells (T47DPAC). A (left panel): Growth curves of cells treated for 6 days with DMSO (control; C) or 2 nM paclitaxel (PAC). For each cell line, the results were compared between control and treatment using two-way ANOVA. *p < 0.5; **p < 0.01; ****p < 0.0001. A (right panel): Clonogenic assays after treatment with 2 nM paclitaxel for 96 hours followed by 12 days of recovery in drug-free medium. B: Western blotting to evaluate the effects of long-term exposure to paclitaxel on the expression levels of ERα. Cells were treated with DMSO (control; C) or were treated with 2 nM paclitaxel for 24 hours. C, D: Western blotting of activation status of JNK and ERK pathways in T47DPAR and T47DPAC cells. After overnight serum starvation, cells were treated with DMSO (C) or 500 nM palbociclib (PD), 4 nM paclitaxel, or concurrent combination therapy for 6 hours and then stimulated with 10% FBS for 10 minutes (+) or not (-).
Figure 3
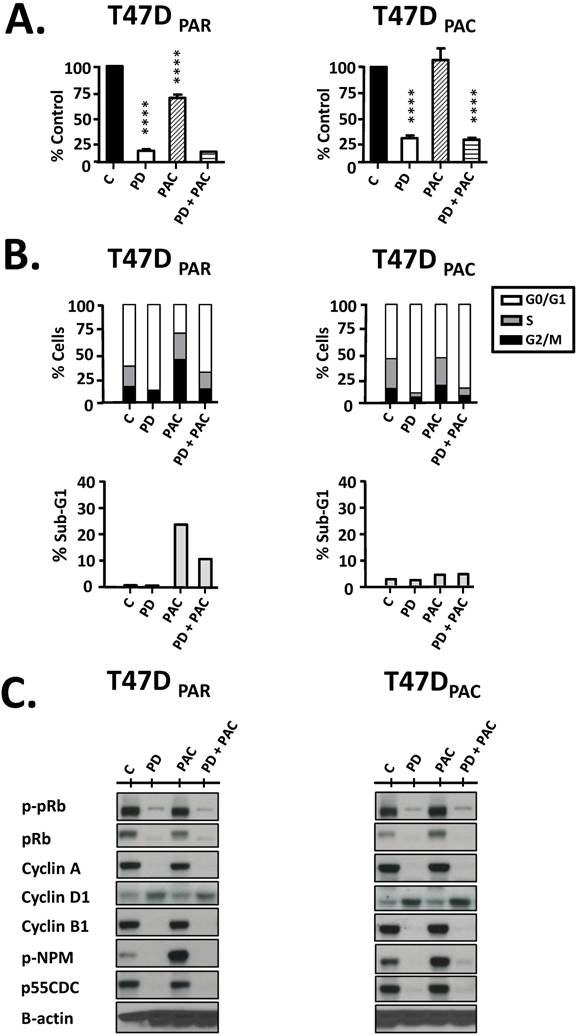
Effects of palbociclib, paclitaxel, and concurrent combination therapy on cell growth and cell cycle progression of parental (T47DPAR) and paclitaxel-resistant T47D cells (T47DPAC). A: Cells were treated with DMSO (control), 500 nM palbociclib (PD), 2 nM paclitaxel (PAC), or concurrent combination therapy. Cell counting was performed after 6 days of treatment and presented as percentages of control. ****p < 0.0001. B: Cells were treated with DMSO (control), 500 nM palbociclib, 4 nM paclitaxel, or concurrent combination therapy. The effects of each treatment on cell cycle distribution and cell death (sub-G1) were evaluated by fluorescence-activated cell sorting (FACS) after 24 hours. C: Western blotting analyses to evaluate the status of activation of cell cycle-regulatory proteins.
Palbociclib does not re-sensitize T47DPAC cells to paclitaxel and antagonizes the effects of paclitaxel on T47DPAR
Cell counting experiments showed that the growth inhibitory effects of concurrent combination therapy with palbociclib and paclitaxel were similar to those of single-agent palbociclib (Fig. 3A) in both T47DPAR and T47DPAC cells. These results may be explained by the fact that paclitaxel treatment usually leads to mitotic arrest in sensitive cells. Accordingly, our data showed that paclitaxel caused significant G2/M arrest in the parental cells, whereas the resistant cells were able to bypass the mitotic blockade and re-enter the cell cycle (Fig. 3B). Palbociclib-induced G1/S arrest prevented cell cycle progression through S and G2/M phases in both T47DPAC and T47DPAR cells treated with concurrent combination therapy. Consequently, palbociclib did not improve the sensitivity of T47DPAC cells to paclitaxel-induced G2/M arrest and actually attenuated the occurrence of cell death caused by paclitaxel in T47DPAR cells (Fig. 3B). Likewise, concurrent combination therapy and single-agent palbociclib caused similar decreases in pRb phosphorylation and in the expression of cell cycle-promoting proteins cyclin A, p-NPM, cyclin B1, and p55CDC (Fig. 3C).
Since concurrent combination therapy did not improve the sensitivity of T47DPAR and T47DPAC cells to paclitaxel, we sought to determine whether brief exposure to palbociclib prior to paclitaxel treatment would increase the sensitivity to paclitaxel (Figure 4). However, in both T47DPAR cells and T47DPAC cells, the inhibitory effects of the sequential therapy on proliferation and cell cycle progression were similar to the effects of single-agent palbociclib as demonstrated by cell growth assays (Fig. 4A), flow cytometry (Fig. 4B) and western blotting for cell cycle-promoting proteins (Figure 5).
These results suggested that palbociclib-induced G1 arrest blocks cell cycle progression toward the mitotic phase, which impairs paclitaxel activity. Therefore, we next sought to determine whether palbociclib washout improves the efficacy of paclitaxel in the sequential therapy. Addition of drug-free medium after palbociclib treatment allowed both T47DPAR cells and T47DPAC cells to re-enter the cell cycle, as indicated by the fact that the number of surviving cells (Fig. 4A) and the percentage of cells in S phase (Fig. 4B) were higher after palbociclib washout than after continuous exposure to single-agent palbociclib. However, T47DPAR cells seemed to be more sensitive to palbociclib-induced G1/S arrest, as they showed a delay in cell cycle progression after addition of drug-free medium: the expression levels of cyclin A, PCNA, cyclin B1, and p-NPM were lower in the T47DPAR cells exposed to drug-free medium than in the control cells (Figure 5). Consequently, pre-exposure to palbociclib actually seemed to attenuate the inhibitory effects of paclitaxel on growth (Fig. 4A) and cell cycle (Fig. 4B) in T47DPAR cells: treatment with paclitaxel after palbociclib washout decreased the percentage of T47DPAR cells in the sub-G1 and G2/M phases, which correlated with reduced levels of G2 and M proteins (cyclin B, p55CDC and p-NPM) compared to single-agent paclitaxel (Fig. 5).
On the other hand, T47DPAC cells showed full recovery from palbociclib-induced cell cycle arrest after palbociclib washout, since the expression levels of G2/M-regulatory proteins were similar between T47DPAC cells exposed to drug-free medium and control cells. Nevertheless, palbociclib pretreatment did not improve the growth inhibitory effects of paclitaxel (Fig. 4A) and did not re-sensitize T47DPAC cells to paclitaxel-induced cell cycle arrest and death, since no significant differences were observed either in the proportion of T47DPAC cells in sub-G1 and G2/M phases (Fig. 4B) or in the expression levels of G2/M-regulatory proteins between single and sequential therapy with paclitaxel (Fig. 5).
Taken together, these data suggested that the effects of palbociclib on G1 arrest prevailed over those of paclitaxel in the combination therapy, irrespective of scheduling, which explains why the combination therapy did not re-sensitize T47DPAC cells to paclitaxel and actually antagonized paclitaxel-induced G2/M arrest and cell death in T47DPAR cells.
Discussion
In this study, we sought to investigate whether CDK4/6 inhibition by palbociclib re-sensitizes HR-positive/HER2-negative breast cancer cells to paclitaxel. We demonstrated that palbociclib has a greater effect on growth and cell cycle progression than paclitaxel: palbociclib delayed cell cycle progression and antagonized the effects of paclitaxel in both parental and paclitaxel-resistant cells treated with concurrent combination and sequential therapy in the presence of palbociclib. Paclitaxel is a microtubule-stabilizing agent and, therefore, relies on active cell cycle progression to induce mitotic arrest and cell death. Accordingly, previous studies have shown that pRb deficiency increases cell cycle progression and sensitivity of breast cancer cells to DNA-damaging agents15 and neoadjuvant chemotherapy16. Therefore, our next step was to evaluate whether the release of cells from palbociclib treatment improves the efficacy of paclitaxel in the sequential therapy. Our washout experiments showed that pretreatment of T47DPAR with palbociclib had antagonistic effects on paclitaxel-induced cell cycle arrest and cell death. Moreover, palbociclib pretreatment did not re-sensitize T47DPAC cells to paclitaxel; refuting the hypothesis that palbociclib in combination with paclitaxel re-sensitizes HR-positive/HER2-negative residual tumors to additional taxane adjuvant therapy.
Figure 4
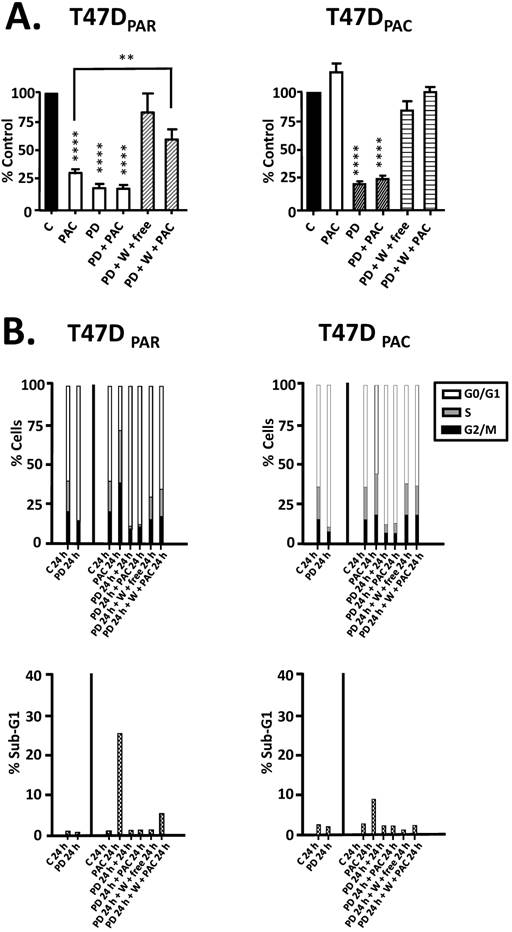
Effects of palbociclib, paclitaxel, and sequential combination therapy on cell growth and cell cycle progression of parental (T47DPAR) and paclitaxel-resistant T47D cells (T47DPAC). A: Cells were treated with DMSO (control; C) for 6 days; 2 nM paclitaxel (PAC) for 6 days; 500 nM palbociclib (PD) for 7 days; 500 nM palbociclib for 1 day followed by addition of 2 nM paclitaxel for 6 days; 500 nM palbociclib for 1 day followed by addition of drug-free medium (free) or 2 nM paclitaxel for 6 days after PD washout (W). Cell counting was performed and the results for each treatment were presented as percentages of control. ****p < 0.0001; **p < 0.01. B: Cells were treated with DMSO (control; C) for up to 24 hours; 4 nM paclitaxel for up to 24 hours; 500 nM palbociclib for up to 48 hours; 500 nM palbociclib for 24 hours followed by 4 nM paclitaxel for 24 hours; 500 nM palbociclib for 24 hours followed by drug-free medium or 4nM paclitaxel for 24 hours after PD washout. The effects of each treatment on cell cycle distribution and cell death (sub-G1) were evaluated by flow cytometry.
Figure 5
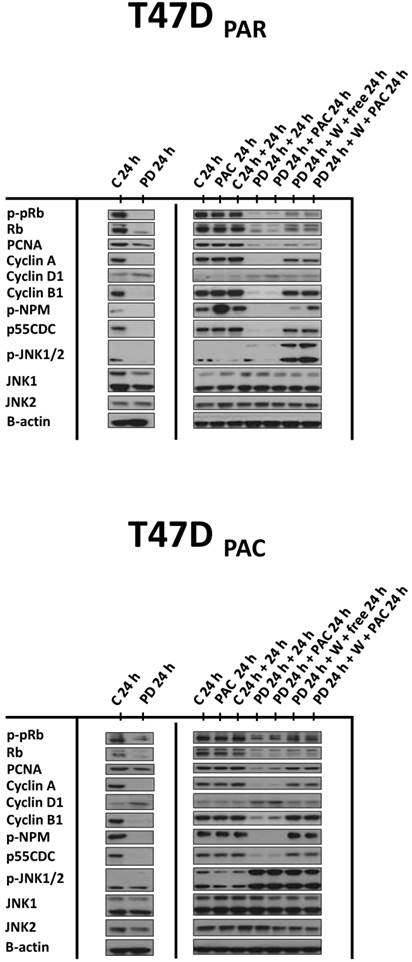
Effects of palbociclib, paclitaxel, and sequential combination therapy on the expression of cell cycle-regulatory proteins in parental (T47DPAR) and paclitaxel-resistant T47D cells (T47DPAC). Cells were treated with DMSO (control; C) for up to 48 hours; 4 nM paclitaxel (PAC) for 24 hours; 500 nM palbociclib (PD) for up to 48 hours; 500 nM palbociclib for 24 hours followed by treatment with 4 nM paclitaxel for 24 hours; 500 nM palbociclib for 24 hours followed by addition of drug-free medium (free) or by treatment with 4 nM paclitaxel for 24 hours after PD washout (W).
On the other hand, we showed that both parental and paclitaxel-resistant cells were characterized by high levels of CDK4/6 activation and, therefore, responded similarly to single-agent palbociclib. Therefore, the CDK4/6 signaling pathway does not seem to represent a molecular mechanism that drives paclitaxel resistance. However, the fact that the T47DPAC cells remained sensitive to palbociclib was critically important for our study, since it indicated that pre-exposure to paclitaxel did not interfere with the functionality of CDK4/6 signaling, making this a targetable pathway for combination therapies intended to re-sensitize residual tumors to taxane-based therapies in the adjuvant setting. Although palbociclib did not reverse paclitaxel resistance, breast cancers that did not respond to neoadjuvant chemotherapy might still be sensitive to the antiproliferative effects of palbociclib in combination with adjuvant endocrine therapies. Indeed, the PENELOPE-B trial is currently investigating whether palbociclib increases the efficacy of hormonal therapy in patients who did not achieve pathologic complete response after neoadjuvant chemotherapy17.
The T47DPAC cells remained resistant to paclitaxel even though pretreatment with palbociclib in the sequential therapy led these cells to progress synchronously through G2/M transition after palbociclib washout. The JNK pathway may play a role in the ability of paclitaxel-resistant cells to escape from paclitaxel-induced mitotic arrest, since paclitaxel-resistant cells showed higher constitutive levels of JNK1/2 activation than the parental cells. Overexpression of JNK proteins has been described to enhance epithelial-mesenchymal transition, invasion, cell cycle progression through G2/M phase, and resistance to paclitaxel in breast cancer cells18,19. Other evidence indicates that the JNK signaling pathway may have a role in the progression of HR-positive tumors: decreased JNK activity has been shown to contribute to apoptotic responses in ER-positive breast cancer cells20, and data from The Cancer Genome Atlas Network revealed that mutations in upstream regulators of JNK proteins occur specifically in this molecular subtype21.
Although further investigation is required to confirm whether the JNK signaling pathway is a key mechanism that contributes to the failure of palbociclib to re-sensitize HR-positive/HER2-negative breast cancer to paclitaxel, our study has relevant translational implications. First, whereas most of the preclinical studies focusing on paclitaxel in combination with palbociclib did not focus on paclitaxel-resistant cells, our work demonstrates a clinically relevant model to test whether targeting CDK4/6 improves the response to paclitaxel in breast cancer patients with HR-positive/HER2-negative residual disease after neoadjuvant chemotherapy. Second, the fact that palbociclib did not re-sensitize T47DPAC cells to paclitaxel suggests that more caution should be exercised when the use of palbociclib in combination with taxanes is considered for cases that do not respond to neoadjuvant chemotherapy. Current ongoing clinical trials are investigating the potential benefits of palbociclib in combination with paclitaxel in patients with Rb-expressing metastatic breast cancer22.
Acknowledgements
We would like to acknowledge Nancy Azizian for technical support with the western blotting experiments and Stephanie Deming and Markeda Wade from the Department of Scientific Publications at The University of Texas MD Anderson Cancer Center for manuscript editing. The Characterized Cell Line Core Facility and the Flow Cytometry and Imaging Core Facility at The University of Texas MD Anderson Cancer Center are both funded by NCI Cancer Center Support Grant P30CA016672.
Source of funding
This work was supported in part by Komen for the Cure Catalytic Award KG090341 (AMG-A), Komen for the Cure Grant SAC 100004 (AMG-A), American Cancer Society Research Scholar Grant 121329-RSG-11-187-01-TBG (AMG-A), The Commonwealth Foundation for Cancer Research (AMG-A).
Abbreviations
ANOVA, analysis of variance; CDK, cyclin-dependent kinase; DMSO, dimethylsulfoxide; ER, estrogen receptor; FBS, fetal bovine serum; HER2, human epidermal growth factor receptor 2; HR, hormone receptor; pRb, retinoblastoma tumor suppressor protein; SDS, sodium dodecyl sulfate; p-NPM, phosphorylated nucleophosmin; PCNA, proliferating cell nuclear antigen.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Guarneri V, Broglio K, Kau SW. et al. Prognostic value of pathologic complete response after primary chemotherapy in relation to hormone receptor status and other factors. J Clin Oncol. 2006;24:1037-44
2. Symmans WF, Peintinger F, Hatzis C. et al. Measurement of residual breast cancer burden to predict survival after neoadjuvant chemotherapy. J Clin Oncol. 2007;25:4414-22
3. Sulpher J, Dent R, Dent S. Neoadjuvant chemotherapy in breast cancer: what questions remain? Curr Opin Support Palliat Care. 2014;8:59-63
4. Gonzalez-Angulo AM, Liu S, Chen H. et al. Functional proteomics characterization of residual breast cancer after neoadjuvant systemic chemotherapy. Ann Oncol. 2013;24:909-16
5. Arnold A, Papanikolaou A. Cyclin D1 in breast cancer pathogenesis. J Clin Oncol. 2005;23:4215-24
6. Schwartz GK, Shah MA. Targeting the cell cycle: a new approach to cancer therapy. J Clin Oncol. 2005;23:9408-2
7. Fry DW, Harvey PJ, Keller PR. et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther. 2004;3:1427-38
8. Thangavel C, Dean JL, Ertel A. et al. Therapeutically activating RB: reestablishing cell cycle control in endocrine therapy-resistant breast cancer. Endocr Relat Cancer. 2011;18:333-4
9. Finn RS, Dering J, Conklin D. et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009;11:R77
10. Finn RS, Crown JP, Lang I. et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. Lancet Oncol. 2015;16:25-35
11. Turner NC, Ro J, André F. et al. Palbociclib in hormone-receptor-positive advanced breast cancer. N Engl J Med. 2015;373:209-19
12. McClendon AK, Dean JL, Rivadeneira DB. et al. CDK4/6 inhibition antagonizes the cytotoxic response to anthracycline therapy. Cell Cycle. 2012;11:2747-55
13. Dean JL, McClendon AK, Knudsen ES. Modification of the DNA damage response by therapeutic CDK4/6 inhibition. J Biol Chem. 2012;287:29075-87
14. Parinyanitikul N, Lei X, Chavez-MacGregor M. et al. Receptor status change from primary to residual breast cancer after neoadjuvant chemotherapy and analysis of survival outcomes. Clin Breast Cancer. 2015;15:153-60
15. Bosco EE, Wang Y, Xu H. et al. The retinoblastoma tumor suppressor modifies the therapeutic response of breast cancer. J Clin Invest. 2007;117:218-28
16. Witkiewicz AK, Ertel A, McFalls J. et al. RB-pathway disruption is associated with improved response to neoadjuvant chemotherapy in breast cancer. Clin Cancer Res. 2012;18:5110-22
17. Cadoo KA, Gucalp A, Traina TA. Palbociclib: an evidence-based review of its potential in the treatment of breast cancer. Dove Medical Press. 2014;6:123-33
18. Wang J, Kuiatse I, Lee AV. et al. Sustained c-Jun-NH2-kinase activity promotes epithelial-mesenchymal transition, invasion, and survival of breast cancer cells by regulating extracellular signal-regulated kinase activation. Mol Cancer Res. 2010;8:266-77
19. Mingo-Sion AM, Marietta PM, Koller E. et al. Inhibition of JNK reduces G2/M transit independent of p53, leading to endoreduplication, decreased proliferation, and apoptosis in breast cancer cells. Oncogene. 2004;23:596-604
20. Wang L, Gallo KA, Conrad SE. Targeting mixed lineage kinases in ER-positive breast cancer cells leads to G2/M cell cycle arrest and apoptosis. Oncotarget. 2013;4:1158-71
21. The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61-70
22. Vidula N, Rugo HS. Cyclin-Dependent Kinase 4/6 Inhibitors for the Treatment of Breast Cancer: A Review of Preclinical and Clinical Data. Clin Breast Cancer. 2016;16:8-17
Author contact
![]() Corresponding author: Naoto T. Ueno, Department of Breast Medical Oncology, Unit 1354, The University of Texas MD Anderson Cancer Center, 1515 Holcombe Blvd, Houston, TX 77030. Phone: 713-792-8754 Fax: 713-794-4385 E-mail: nuenoorg.
Corresponding author: Naoto T. Ueno, Department of Breast Medical Oncology, Unit 1354, The University of Texas MD Anderson Cancer Center, 1515 Holcombe Blvd, Houston, TX 77030. Phone: 713-792-8754 Fax: 713-794-4385 E-mail: nuenoorg.
Citation styles
APA
Trapé, A.P., Liu, S., Cortes, A.C., Ueno, N.T., Gonzalez-Angulo, A.M. (2016). Effects of CDK4/6 Inhibition in Hormone Receptor-Positive/Human Epidermal Growth Factor Receptor 2-Negative Breast Cancer Cells with Acquired Resistance to Paclitaxel. Journal of Cancer, 7(8), 947-956. https://doi.org/10.7150/jca.14441.
ACS
Trapé, A.P.; Liu, S.; Cortes, A.C.; Ueno, N.T.; Gonzalez-Angulo, A.M. Effects of CDK4/6 Inhibition in Hormone Receptor-Positive/Human Epidermal Growth Factor Receptor 2-Negative Breast Cancer Cells with Acquired Resistance to Paclitaxel. J. Cancer 2016, 7 (8), 947-956. DOI: 10.7150/jca.14441.
NLM
Trapé AP, Liu S, Cortes AC, Ueno NT, Gonzalez-Angulo AM. Effects of CDK4/6 Inhibition in Hormone Receptor-Positive/Human Epidermal Growth Factor Receptor 2-Negative Breast Cancer Cells with Acquired Resistance to Paclitaxel. J Cancer 2016; 7(8):947-956. doi:10.7150/jca.14441. https://www.jcancer.org/v07p0947.htm
CSE
Trapé AP, Liu S, Cortes AC, Ueno NT, Gonzalez-Angulo AM. 2016. Effects of CDK4/6 Inhibition in Hormone Receptor-Positive/Human Epidermal Growth Factor Receptor 2-Negative Breast Cancer Cells with Acquired Resistance to Paclitaxel. J Cancer. 7(8):947-956.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.