Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2016; 7(8):900-909. doi:10.7150/jca.14506 This issue Cite
Research Paper
P53 Is Involved in a Three-Dimensional Architecture-Mediated Decrease in Chemosensitivity in Colon Cancer
Jianming He1 ![]() *, Xi Liang1,2*, Fen Luo2, Xuedan Chen3, Xueqing Xu2, Fengchao Wang4, Zhenping Zhang5
*, Xi Liang1,2*, Fen Luo2, Xuedan Chen3, Xueqing Xu2, Fengchao Wang4, Zhenping Zhang5
1. Department Of Oncology And Southwest Cancer Center, Southwest Hospital, Third Military Medical University, Chongqing, 400038 China;
2. Molecular Biology Center, State Key Laboratory of Trauma, Burn, and Combined Injury, Daping Hospital, Third Military Medical University, Chongqing, 400042 China;
3. Department Of Medical Genetics, Third Military Medical University, Chongqing, 400038 China;
4. Institute of Combined Injury, State Key Laboratory of Trauma, Burns and Combined Injury, Third Military Medical University, Chongqing, 400038 China;
5. Department Of Oncology, First Hospital of Shijiazhuang City, Shijiazhuang, Hebei Province, 050011 China.
*These authors contributed equally to this manuscript.
Received 2015-11-22; Accepted 2016-3-16; Published 2016-4-29
Citation:
He J, Liang X, Luo F, Chen X, Xu X, Wang F, Zhang Z. P53 Is Involved in a Three-Dimensional Architecture-Mediated Decrease in Chemosensitivity in Colon Cancer. J Cancer 2016; 7(8):900-909. doi:10.7150/jca.14506. https://www.jcancer.org/v07p0900.htm
Other stylesAbstract
Three-dimensional (3D) culture models represent a better approximation of solid tumor tissue architecture, especially cell adhesion, in vivo than two-dimensional (2D) cultures do. Here, we explored the role of architecture in chemosensitivity to platinum in colon cancer. Under the 3D culture condition, colon cancer cells formed multicellular spheroids, consisting of layers of cells. 3D cultures displayed significantly decreased sensitivity to platinum compared with 2D cultures. Platinum increased p53 in a dose-dependent and time-dependent manner. There was no detectable difference in basal p53 levels between 3D cultures and 2D cultures but cisplatin induced less p53 in both HCT116 3D cultures and LoVo 3D cultures. It was not due to cisplatin concentration because cisplatin induced similar γ-H2AX in 3D vs 2D. Knockdown of p53 significantly decreased sensitivity to platinum in 3D cultures. Knockdown of p53 decreased cleaved caspase 3 and apoptosis induced by cisplatin. These findings indicate that 3D architecture confers decreased chemosensitivity to platinum and p53 is involved in the mechanism. Knockdown of p53 decreased cisplatin's induction of c-Jun N-terminal kinase 1/2 (JNK1/2) activation, whereas inhibition of JNK1/2 activation increased chemosensitivity. Inhibition of p38 activation decreased cisplatin's induction of p53, but no difference in p38 activation by cisplatin was observed between 2D cultures and 3D cultures. Taken together, our results suggest that p53 is involved in a 3D architecture-mediated decrease in chemosensitivity to platinum in colon cancer. Mitogen-activated protein kinases (JNK1/2 and p38) do not play a dominant role in the mechanism.
Keywords: p53, platinum, three-dimensional culture, colorectal cancer cells.
Introduction
Colorectal cancer is the third most commonly diagnosed cancer in males and the second most in females worldwide, with an estimated 1.4 million cases occurring in 2012[1]. The incidence of colorectal cancer in developing countries is increasing, which is partly attributable to lipid metabolism [1-2]. Colorectal cancer is the fourth most lethal disease among all malignancies in males and the third most in females, with an estimated 693,900 deaths occurring in 2012[1]. Platinum (including cisplatin (CDDP), oxaliplatin (L-OHP), nedaplatin and so on) remains one of the most effective and most commonly used drugs to treat this disease [3]. Unfortunately, resistance to chemotherapy remains a critical barrier to the development of an effective cure [1, 3-4]. More than 35% of colorectal cancer patients die within 5 years after diagnosis, even in developed countries such as the United States [4].
Most in vitro experimental studies of anticancer drugs testing and resistance mechanisms are performed with monolayer cell cultures (two-dimensional (2D) cell cultures) [5-6]. 2D cell cultures are not an optimal experimental system for studies of anticancer drugs because these cultures do not well recapitulate solid tumor tissue architecture in vivo, and the architecture is important for tumor cells' biological behaviors, including chemosensitivity [7-10]. Three-dimensional (3D) cell cultures represent a better approximation of solid tumor tissue architecture than 2D cultures do [7-12].
A few researchers have reported that certain types of tumor cells in 3D cultures are more sensitive to specific anticancer drugs than in 2D cultures[13], but most researchers believe that most tumor cells in 3D cultures are more resistant to anticancer drugs[7, 9-11, 14]. The differences in sensitivities to most drugs are not attributed to drug concentrations because drug penetration and delivery in 3D cultures has been shown to be negligible for most anticancer drugs, including platinum, 5-fluorouracil, carmustine, lomustine, chlorambucil, bleomycin, and mitomycin, among others [9-10].
In the present study, we explored the influence of architecture on the response of human colorectal cancer cells to platinum using both 2D cultures and 3D cultures. Our data demonstrate that 3D architecture conferred decreased chemosensitivity to platinum, by or partly by weaker p53 induction.
Materials and Methods
Cell lines and cell culture
The human colorectal cancer cell lines HCT116, LoVo and the human embryonic kidney cell line 293T were obtained from the Cell Bank of the Chinese Academy of Science.
2D cultures were routinely grown and passaged as previously described [7]. In brief, cells were grown in McCoy's 5A (Gibco, Grand Island, NY, USA, Cat: 16600108) (HCT116), F12K (Gibco, Cat: 12500-062) (LoVo) or DMEM (Gibco, Cat: 12100-046) (293T) supplemented with 100 ml/L fetal bovine serum (Gibco, Cat: 10438018), 100,000 IU/L penicillin, and 100 μg/ml streptomycin (Gibco, Cat: 15140122) under a humidified atmosphere of 5% CO2 at 37°C.
3D cultures were prepared by using the liquid overlay technique as previously described [7-8]. In brief, exponentially growing cancer cells were seeded into plates that had been previously coated with 2% agarose. The plates were then gently horizontally swirled for 10 minutes every 6 hours for the first 24 hours. The cells were incubated under a humidified atmosphere of 5% CO2 at 37°C. The medium was appropriately refreshed every day.
Hematoxylin and eosin (H&E) staining
HCT116 3D cultures were fixed in 4% paraformaldehyde, and OCT-embedded samples were sectioned at a thickness of 10 μm. Sample slides were routinely stained with H&E [12].
LoVo 3D cultures were fixed in 4% paraformaldehyde, and paraffin-embedded samples were sectioned at a thickness of 6 μm. Sample slides were routinely stained with H&E [15].
Slides preparation for transmission electron microscopy
Sample slides were routinely prepared as previously described [7, 11]. In brief, 3D cultures were fixed in 2.5% glutaraldehyde and then in 1% osmium tetroxide. Samples were dehydrated using graded alcohol, and ultrathin sections were generated. The sections were stained with uranium acetate and lead citrate and observed using a transmission electron microscope (TECNAI10, Philip, Holland).
Lentiviral delivery of shRNA
p53 was knocked down by lentiviral vector-mediated small hairpin (sh) RNA interference using The RNAi Consortium system (Open Biosystems, Inc., Huntsville, AL, USA) according to the manufacturer's instructions[6, 11]. In brief, a p53-targeting shRNA-pLKO.1 vector or a control shRNA-pLKO.1 vector, with the packaging plasmid pCMV-Dr8.91 and the enveloping plasmid pCMV-VSV-G, was co-transfected into 293T cells using Lipofectamine® 2000 (Invitrogen, Carlsbad, CA, USA, Cat: 11668-030) according to the manufacturer's instructions. Virus-containing medium was collected at 48 and 72 hours post-transfection and was filtered. Cells were infected with lentivirus encoding the shRNA targeting p53 or with control lentivirus and were then selected using puromycin (Sigma-Aldrich, St. Louis, MO, USA, Cat: P8833). The sense sequence of the shRNA targeting p53 is GACTCCAGTGGTAATCTACT[6]. Control shRNA (shcontrol) is targeted against green fluorescent protein, and the sense sequence of this shRNA is TACAACAGCCACAACGTCTAT[11]. Knockdown efficiency was confirmed by western blot.
Water-soluble tetrazolium salt (WST) assay
Cells were treated with 50 μg/ml CDDP or 20 μg/ml L-OHP for 24 hours. Control cultures received 10 μl PBS (Fig. 1C and 3F) or SP600125 (Fig. 4C) only. Cell viability was assayed using WST (2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt). The WST assay was performed using Cell Counting Kit 8 (Dojindo Laboratories, Kumamoto, Japan, Cat: CK04) according to the manufacturer's instructions, as previously described [7, 12]. In brief, 2D cultures or 3D cultures detached using accutase were incubated with WST/media for 2-4 hours, after which the absorbance at 450 nm was measured using a microplate reader with a reference wavelength of 650 nm. Cell viability was normalized to the control.
Clonogenic assay
Clonogenic assay in vitro was routinely grown and passaged as previously described [11-12]. In brief, the same numbers of cells were treated with 50 μg/ml CDDP for 24 hours. Then, the 3D cultures were detached using accutase, and the same ratio of single-cell suspensions was seeded into 24-well plates in duplicate. The cells were cultured at 37°C for 7 days and were subsequently stained with crystal violet.
Immunohistochemical staining
HCT116 3D cultures were fixed in 4% paraformaldehyde, and OCT-embedded samples were sectioned at a thickness of 10 μm. Immunohistochemistry was performed according to the protocol of the SPlink Detection Kits (ZSGB-BIO, Beijing, China, Cat: SP-9000), as previously described [12, 15].
Apoptosis assay
shcontrol HCT116 3D cultures and shp53 HCT116 3D cultures were treated with 20 μg/ml CDDP for 24 hours and then fixed in 4% paraformaldehyde. OCT-embedded samples were sectioned at a thickness of 10 μm. The terminal deoxynucleotidyl transferase-mediated nick end-labeling (TUNEL) stain was used to detect DNA fragmentation resulting from apoptosis via an In Situ Cell Death Detection Kit, Fluorescein (Roche, Germany, Cat: 11684795910), according to the manufacturer's instructions, as described previously [7].
Preparation of cell lysates
Cells were lysed in RIPA buffer (50 mM Tris base, 150 mM NaCl, 1% Nonidet P-40, 0.25% Na-deoxycholate, 1 mM EDTA) with protease inhibitors and phosphatase inhibitors (1 mM PMSF, 5 μg/ml leupeptin, 2 μg/ml pepstatin, 4 μg/ml aprotinin, 10 mM NaF, 1 mM Na3VO4, and 10 mM β-glycerophosphate disodium salt pentahydrate) for 30 minutes on ice. Following centrifugation (26,000 g, 16 minutes, 4°C), the supernatant was collected as total cell protein [11-12].
Western blot analysis
The protein was resolved by SDS/PAGE and blotted on nitrocellulose membranes (Bio-Rad, Richmond, CA, USA, Cat: 162-0115) as previously described [2, 7]. The nitrocellulose membranes were incubated with specific primary antibodies overnight. After incubation with secondary antibodies, immunoreactive proteins were visualized using the Enhanced Chemiluminescent Substrate (Thermo Scientific, Pittsburgh, PA, USA, Cat: 34095).
Primary antibodies against cleaved caspase 3 (Cat: 9664, 1:500), p53 (Cat: 2524, 1:1000), phospho-p53 (Cat: 9286, 1:500), β-catenin (Cat: 9582, 1:1000), γ-H2AX (Cat: 9718, 1:1000), α-tubulin (Cat: 3873, 1:1000), β-actin (Cat: 3700, 1:1000), Histone H3 (Cat: 3638, 1:1000) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Cat: 8884, 1:1000); the phospho-mitogen-activated protein kinase (MAPK) Family Antibody Sampler Kit (Cat: 9910, 1:1000); and HRP-linked secondary antibodies were from Cell Signaling Technology (Beverly, MA, USA).
MAPK activation inhibitors
The c-Jun N-terminal kinase (JNK) inhibitor SP600125 [16] (Cat: S1460) and the p38 inhibitor SB202190 [17] (Cat: S1077) were from Selleck Chemicals (Houston, TX, USA).The MAPK/extracellular signal-regulated kinase (ERK) kinase 1/2 inhibitor U0126 [18] (Cat: 9910) was from Cell Signaling Technology.
Statistical analyses
The data shown represent the mean ± standard error. Statistical differences between groups were analyzed by Student's t-test or one-way ANOVA. p< 0.05 was considered statistically significant.
Results
3D architecture confers decreased sensitivity to platinum in colorectal cancer
The phenotypes of the colon cancer cell lines HCT116 and LoVo when cultured as 2D and 3D cultures were analyzed. Colon cancer cells cultured on plastic (2D) grew as flat monolayers (Fig. 1A and B). When maintained under the 3D culture condition, colon cancer cells formed multicellular spheroids (Fig. 1A and B). H&E staining showed that the multicellular spheroids consisted of layers of cells. Necrotic core was not found (Fig. 1A and B). By employing a transmission electron microscope, cell-cell junctions were found in most cells in HCT116 3D culture (Fig. S1).
Figure 1
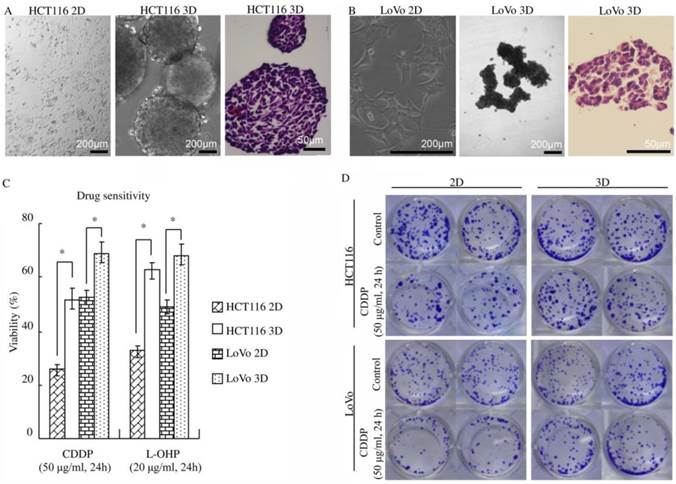
3D architecture confers decreased sensitivity to platinum in colorectal cancer. (A-B) HCT116 and LoVo cultured on plastic grew as flat monolayers (left) and formed multicellular spheroids (middle) using the liquid overlay technique. Multicellular spheroids sections were stained with H&E (right). (C) WST assay indicated that 3D cultures were less sensitive to platinum compared with 2D cultures. *:p<0.01 (D) After 3D or 2D cultures treated with or without CDDP, cells were detached and cultured as monolayer cells for 7 days and were subsequently stained with crystal violet. The clonogenicity of the 3D cultures treated with CDDP was much higher than that of the respective 2D cultures.
To explore the role of 3D architecture in chemosensitivity, cells were treated with platinum because it was well elucidated that platinum penetration and delivery in 3D cultures is negligible [9-10]. Cell viability was assayed using WST. 3D cultures displayed a significant decrease in chemosensitivity compared with 2D cultures (p<0.01) (Fig. 1C). The cell viability of HCT116 3D cultures treated with CDDP was 51.8±3.9%, whereas that of 2D cultures was 25.4±1.8% (p<0.01). The cell viability of HCT116 3D cultures treated with L-OHP was 62.7±3.0%, whereas that of 2D cultures was 32.3±2.7% (p<0.01). For LoVo cells, the viability of 3D cultures treated with CDDP was 69.1±3.7%, whereas that of 2D cultures was 52.4±2.7% (p<0.01). The cell viability of LoVo 3D cultures treated with L-OHP was 68.3±4.1%, whereas that of 2D cultures was 48.9±2.8% (p<0.01). The results of the clonogenic assay were consistent with those of the WST assay. The clonogenicity of the HCT116 3D cultures and LoVo 3D cultures was much higher than that of the respective 2D cultures (Fig. 1D).
Weaker p53 induction contributes to the 3D architecture-mediated decrease in chemosensitivity
Platinum kills cells by damaging DNA and p53 plays a key role in the DNA damage response [19-21]. Therefore, the role of p53 in the 3D architecture-mediated decrease in sensitivity to platinum was explored.
Platinum increased the p53 protein level in both HCT116 and LoVo cells (both of them contain stabilized wild-type p53 protein [22-23]), regardless of whether the cells were in 2D or 3D cultures (Fig. 2). p53 protein was increased by platinum in a dose-dependent and time-dependent manner (Fig. 2). There was no detectable difference in basal p53 levels between 3D cultures and 2D cultures but CDDP induced less p53 in both HCT116 3D cultures and LoVo 3D cultures. (Fig. 2C and D).
Although it was well elucidated that platinum penetration and delivery in 3D cultures is negligible [9-10], γ-H2A.X, a maker of DNA damage [24], was assayed by western blot to confirm this aspect. CDDP induced similar γ-H2AX protein in HCT116 3D cultures as in 2D cultures (Fig. 2E). This finding suggests that 3D architecture does not decrease the concentration of CDDP to cells in 3D cultures.
Following DNA damage, cells activate the DNA damage response, which either leads the cell to repair the lesion, promoting resistance to platinum, or causes cell death due to activation of the apoptotic response [19-20, 25]. p53 has been shown to play a key role in the DNA damage response in that p53 can initiate DNA repair; cell cycle arrest; senescence; and, notably, apoptosis[19-20, 25]. Hence, the role of p53 and apoptosis in 3D architecture-mediated decreased sensitivity to platinum was explored.
p53 was knocked down using The RNAi Consortium system. The efficiency and specificity of the sequence of the shRNA targeting p53 were previously verified [6]. Western blot showed that lentiviral delivery of the shRNA targeting p53 efficiently knocked down p53 in HCT116 3D cultures and that shcontrol did not detectably change the p53 protein level (Fig. 3A and B). Immunohistochemistry showed that p53 was expressed uniformly in cells in HCT116 3D cultures and that knockdown of p53 uniformly decreased its expression in cells (Fig. 3C). Western blot also showed that the system efficiently decreased CDDP's induction of p53 and p-p53. Knockdown of p53 significantly decreased cleaved caspase 3, which plays a central role in the execution phase of cell apoptosis, in 3D cultures (Fig. 3D and E) and 2D cultures (Fig. 3F)[26]. To confirm that knockdown of p53 decreased apoptosis induced by platinum treatment, TUNEL staining was used to detect apoptosis in HCT116 3D cultures. Treatment with 20 μg/ml CDDP for 24 hours clearly induced less apoptosis in HCT116 3D cultures with p53 knockdown than in shcontrol HCT116 3D cultures (Fig. 3G).
Inducing apoptosis is an important way of platinum killing cells by DNA damage [19-21]. Because knockdown of p53 decreased apoptosis induced by platinum, knockdown of p53 might decrease sensitivity to platinum. To validate this hypothesis, chemosensitivity was assayed using WST. Knockdown of p53 significantly decreased chemosensitivity (p<0.01) (Fig. 3H). The cell viability of shcontrol HCT116 3D cultures treated with CDDP was 48.6±3.7%, whereas that of treated HCT116 cells with p53 knockdown was 64.4±3.1% (p<0.01). The cell viability of shcontrol HCT116 3D cultures treated with L-OHP was 62.0±2.9%, whereas that of treated HCT116 cells with p53 knockdown was 73.0±2.1% (p<0.01).
Figure 2
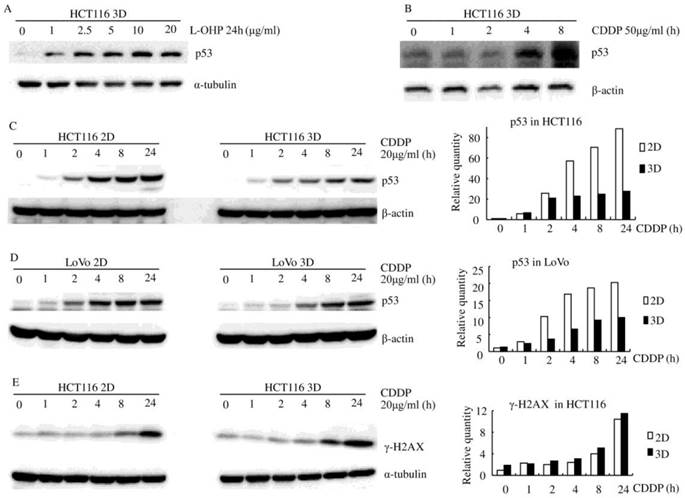
Platinum induces less p53 in 3D architecture (western blot). (A) L-OHP induced p53 in a dose-dependent manner. (B) CDDP induced p53 in a time-dependent manner. (C-D) CDDP induced less p53 in 3D cultures than in the respective 2D cultures. Relative quantities of p53 were normalized to β-actin and diagramed, respectively (right). (E) CDDP induced similar γ-H2A.X protein in HCT116 3D cultures as in 2D cultures. Relative quantities of p53 were normalized to α-tubulin and diagrammed (right).
Figure 3
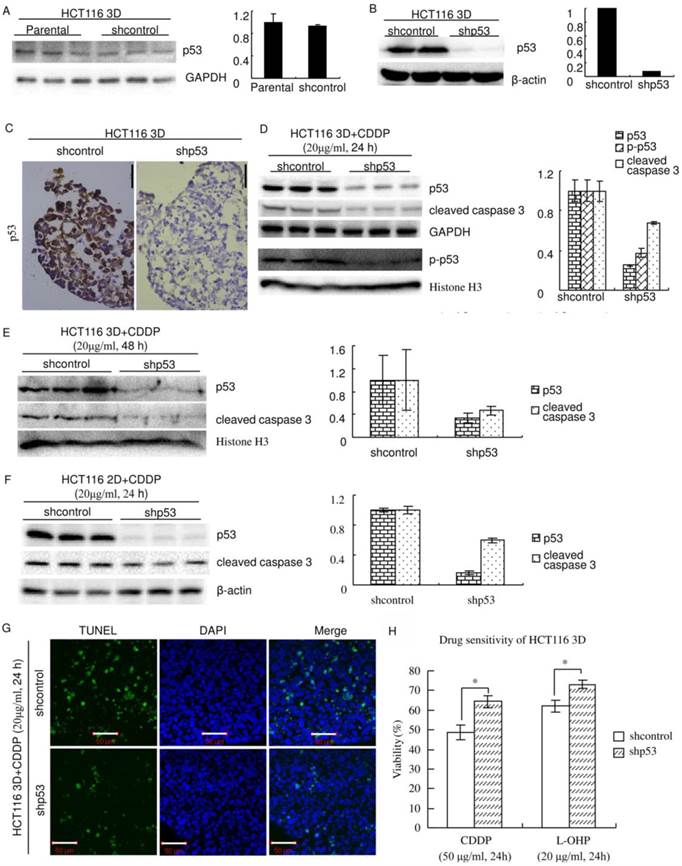
Knockdown of p53 decreases sensitivity to platinum in HCT116. (A) There was no detectable difference in the p53 protein level between Parental and shcontrol cells. Relative quantities of p53 were normalized to GAPDH and diagrammed (right). (western blot) (B) The protein p53 was knocked down by lentiviral delivery of shRNA (shp53), and the efficiency was assayed by western blot. Relative quantities of p53 were normalized to β-actin and diagrammed (right). (C) Immunohistochemical staining of p53 in HCT116 3D cultures. (D) After treatment of CDDP for 24 hours, p53, p-p53 and cleaved caspase 3 in 3D cultures were assayed by western blot. Relative quantities were normalized to their loading control and diagrammed, respectively (right). (E) After treatment of CDDP for 48 hours, p53 and cleaved caspase 3 in 3D cultures were assayed by western blot. Relative quantities were normalized to Histone H3 and diagrammed (right). (F) After treatment of CDDP for 24 hours, p53 and cleaved caspase 3 in 2D cultures were assayed by western blot. Relative quantities were normalized to β-actin and diagrammed (right). (G) After treatment of CDDP for 24 hours, apoptosis was detected with TUNEL stain. (H) Cell viabilities of HCT116 3D cultures were assayed by WST. *:p<0.01.
Figure 4
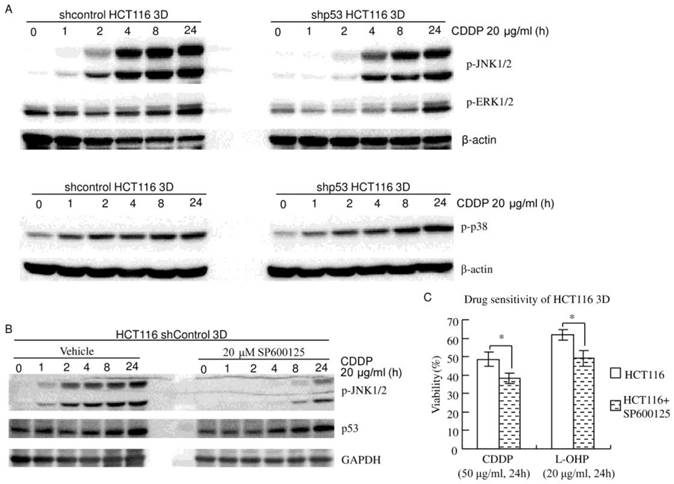
JNK1/2 do not play a dominant role in the 3D architecture-mediated p53-dependent decrease in chemosensitivity. (A) MAPKs (JNK1/2, p38 and ERK1/2) were activated by CDDP in a time-dependent manner. Knockdown of p53 decreased CDDP's activation of JNK1/2, but not p38 or ERK1/2 (western blot). (B) Cells were treated with vehicle or SP600125 1 hour prior to CDDP treatment. CDDP's activation of JNK1/2 was remarkably inhibited by SP600125. Inhibition of JNK1/2 activation did not detectably change CDDP's induction of p53 (western blot). (C) Cells were treated with vehicle or SP600125 1 hour prior to CDDP treatment. Cell viability was assayed using WST. Pretreatment with 20 μM SP600125 significantly increased chemosensitivity. *:p<0.05.
MAPKs (JNK1/2, p38 and ERK1/2) do not play a dominant role in the 3D architecture-mediated p53-dependent decrease in chemosensitivity
MAPKs play a remarkable role in apoptosis [5, 16, 18, 27-28]. JNK1/2, p38 and ERK1/2, three groups of MAPKs, have been reported to be related to platinum resistance and p53 [20-21, 29-30]. Here, the roles of JNK1/2, p38 and ERK1/2 in the 3D architecture-mediated p53-dependent decrease in chemosensitivity were explored.
HCT116 3D cultures were treated with 20 μg/ml CDDP for different amounts of time. Western blot showed that JNK1/2 was activated in a time-dependent manner (Fig. 4A and B). Knockdown of p53 decreased CDDP's activation of JNK1/2 in HCT116 3D cultures (Fig. 4A). CDDP's activation of JNK1/2 was remarkably inhibited by 20 μM SP600125 treatment 1 hour prior to CDDP treatment. Inhibition of JNK1/2 activation did not detectably change CDDP's induction of p53 in HCT116 3D cultures (Fig. 4B). These results indicate that p53 activates JNK1/2 in response to CDDP treatment, and this led to the hypothesis that JNK1/2 might play a role in the 3D architecture-mediated p53-dependent decrease in chemosensitivity. Then, chemosensitivities of HCT116 shcontrol 3D cultures were assayed. The result was opposite to our hypothesis: pretreatment with 20 μM SP600125 1 hour prior to platinum treatment significantly increased chemosensitivity (P<0.05) (Fig. 4C). The cell viability of shcontrol HCT116 3D cultures pretreated with SP600125 prior to CDDP was 38.4±2.8%, and that of shcontrol HCT116 3D cultures pretreated with SP600125 prior to L-OHP was 49.1±4.4%. These findings indicate that JNK1/2 do not play a dominant role in the 3D architecture-mediated p53-dependent decrease in chemosensitivity.
To explore the role of p38 in the 3D architecture-mediated p53-dependent decrease in sensitivity to platinum, both HCT116 3D cultures and LoVo 3D cultures were treated with CDDP for different amounts of time or were treated with a gradient of L-OHP for 24 hours. p-p38 protein was then analyzed by western blot. p38 was activated by platinum treatment in a dose-dependent and time-dependent manner (Fig. 4A, 5, 6A and 6B). Knockdown of p53 did not detectably change the p-p38 level induced by CDDP treatment (Fig. 4A). CDDP's activation of p38 was remarkably inhibited by 20 μM SB202190 treatment 1 hour prior to CDDP (Fig. 6A). Though inhibition of p38 activation decreased CDDP's induction of p53 (Fig. 6A), there was no detectable difference in CDDP's induction of p-p38 between HCT116 2D cultures and 3D cultures (Fig. 6B). These findings suggest that p38 does not play a role in the 3D architecture-mediated p53-dependent decrease in chemosensitivity.
The role of ERK1/2 in the 3D architecture-mediated p53-dependent decrease in chemosensitivity was explored in a similar way as for p38. ERK1/2 was activated in a dose-dependent and time-dependent manner by platinum treatment (Fig. 4A, 5 and 6C). Knockdown of p53 did not detectably change the p-ERK1/2 level induced by CDDP treatment (Fig. 4A). CDDP's induction of p-ERK1/2 was remarkably inhibited by 20 μM U0126 treatment 1 hour prior to CDDP (Fig. 6C). Inhibition of ERK1/2 activation did not detectably change CDDP's induction of p53 in HCT116 3D cultures (Fig. 6C). These findings suggest that ERK1/2 do not play a role in the 3D architecture-mediated p53-dependent decrease in chemosensitivity.
Figure 5
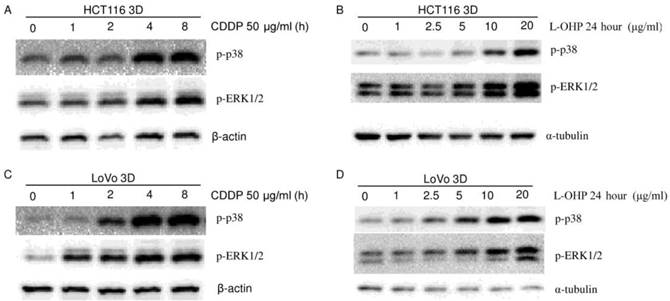
Platinum activates p38 and ERK1/2 (western blot). CDDP activated MAPKs (p38, ERK) in a time-dependent manner in HCT116 3D cultures (A) and LoVo 3D cultures (C). L-OHP activated MAPKs (p38, ERK) in a dose-dependent manner in HCT116 3D cultures (B) and LoVo 3D cultures (D).
Figure 6
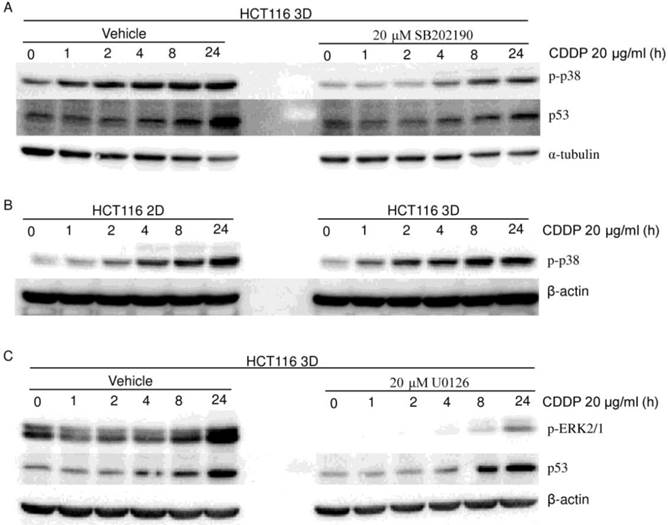
MAPKs (p38 and ERK1/2) do not contribute to the 3D architecture-mediated p53-dependent decrease in chemosensitivity. (western blot). (A) Cells were treated with vehicle or SB202190 1 hour prior to CDDP treatment. CDDP's activation of p38 was remarkably inhibited by SB202190. Inhibition of p38 activation decreased CDDP's induction of p53. (B) There was no detectable difference in p-p38 between 2D cultures and 3D cultures. (C) Cells were treated with vehicle or U0126 1 hour prior to CDDP treatment. CDDP's activation of ERK was remarkably inhibited by U0126. Inhibition of ERK activation did not detectably change CDDP's induction of p53.
In summary, although knockdown of p53 decreasing CDDP's induction of p-JNK1/2 and inhibition of p38 activation decreasing CDDP's induction of p53 were observed in HCT116 3D cultures, MAPKs (JNK1/2, p38 and ERK1/2) do not play a dominant role in the 3D architecture-mediated p53-dependent decrease in chemosensitivity.
Discussion
Solid tumor cells in vivo proliferate, survive and response to stimuli in a specific tissue architecture [9-11]. Most in vitro experiments on cancer are performed with 2D cultures [5-6], but 2D cultures do not well recapitulate tissue architecture [9-12]. In 3D cultures, cells adhered to each other within layers of cells, and cell-cell junctions were commonly found (Fig. 1A, B and Fig. S1). Their structures were more similar to those of tumors at avascular stage, avascular tumor regions or micrometastases than 2D cultures were [8-12]. Therefore, 3D cultures represent a better approximation of solid tumor tissue architecture in vivo [7-12].
Accumulating evidences demonstrate that the architecture of 3D cultures influences cell proliferation (3D cultures contain a core of cells which undergoes senescence), cell survival, nutrient supply, oxygen tension, pH and so on [7-10, 31-32]. These may play roles in chemosensitivity, more or less [7-10, 31-32]. 3D cultures displayed significantly decreased sensitivity to platinum compared with 2D cultures (Fig. 1C and D). Tumors in vivo were also reported to present the same characteristics [7-10, 31]. These data suggest that 3D cultures have the potential to provide predictable results regarding cancer chemotherapeutics [7, 9-12].
Platinum damages DNA by binding to and causing crosslink of DNA to kill cells [19-20]. Following DNA damage, cells respond by activating a DNA-damage response that either leads the cell to repair the lesion, promoting resistance to chemotherapy, or causes cell death by activation of the apoptotic response [19-20, 25]. Protein p53 plays a key role in the response to platinum-induced DNA damage [19-20]. In the current study, platinum treatment increased p53 protein level in a time-dependent and dose-dependent manner. 3D architecture clearly decreased p53 induced by CDDP (Fig. 2). Knockdown of wild-type p53 (Both HCT116 and LoVo contain a stabilized wild-type p53 protein [22-23].) decreased sensitivity to platinum (Fig. 3). These findings indicate that 3D architecture confers decreased chemosensitivity to platinum, by or partly by weaker p53 induction. This may contribute to explain the phenomenon that tumors in vivo display significantly a decreased chemosensitivity than 2D cultures do [7, 9-10, 31]. At the same time, loss of p53 or mutated p53 is frequently observed in human carcinomas [19, 33-34]. P53 mutations can also inactivate the protein's normal function [19, 34]. Our results may also contribute to explain that loss of p53 or mutated p53 often acquires oncogenic activities conferring drug resistance and that loss of p53 or mutated p53 is a negative predictor for survival of cancer patients, including colorectal cancer patients [33-36].
The role of apoptosis in the function of p53 in the DNA damage response is well established [19-20, 25]. p53 mediates the DNA-damage response in that p53 can initiate DNA repair; cell-cycle arrest; senescence; and, notably, apoptosis [19-20, 25]. If DNA damage is too great or repair is ineffective, cellular death pathways, such as apoptosis, are stimulated [19-20]. The fact that knockdown of wild-type p53 decreased apoptosis induced by platinum in the present study (Fig. 3) suggests that apoptosis may be involved in the 3D architecture-mediated p53-dependent decrease in sensitivity to platinum.
The MAPK cascade is a critical pathway for human cancer cell apoptosis and resistance to chemotherapy [5, 16, 18, 27-29]. There are 4 independent MAPK pathways composed of 4 signaling families: JNK, p38 signaling families, the MAPK/ERK family and the Big MAP kinase-1[28]. In the current study, it was clearly observed that platinum activated JNK1/2, p38 and ERK1/2 in a dose-dependent and time-dependent manner in colon cancer cell 3D cultures (Fig. 4-6). These results are consistent with other reports using 2D cultures or in vivo models [5, 27, 29]. In HCT116 3D cultures, knockdown of p53 decreased CDDP's induction of p-JNK1/2, and inhibition of p38 activation decreased CDDP's induction of p53. However, inhibition of JNK1/2 activation enhanced HCT116 3D cultures' chemosensitivity, and no difference was observed in CDDP's activation of p38 between HCT116 2D cultures and 3D cultures (Fig. 6B). These findings indicate that MAPKs (JNK1/2, p38 and ERK1/2) do not play a dominant role in the 3D architecture-mediated p53-dependent decrease in chemosensitivity. Further experiments are needed to clarify the complicated mechanism.
Conclusions
Our results suggest that colorectal cancer cell 3D cultures represent a better approximation of solid tumor tissue architecture, especially cell-cell adhesion, in vivo than 2D cultures do and confer decreased sensitivity to platinum. Weaker p53 induction contributes to the control of colon cancer cells' sensitivity to platinum by 3D architecture. Although p53 regulating JNK1/2 and p38 regulating p53 under the condition of CDDP treatment were observed in HCT116 3D cultures, MAPKs (JNK1/2, p38 and ERK1/2) do not play a dominant role in the mechanism of the 3D architecture-mediated p53-dependent decrease in chemosensitivity.
Supplementary Material
Figure S1.
Abbreviations
CDDP: cisplatin; L-OHP: oxaliplatin; 2D: two-dimensional; 3D: three-dimensional; H&E: Hematoxylin and eosin; sh: small hairpin; shcontrol: control shRNA; WST: Water-soluble tetrazolium salt; TUNEL: the terminal deoxynucleotidyl transferase-mediated nick end-labeling; GAPDH: glyceraldehyde 3-phosphate dehydrogenase; MAPK: mitogen-activated protein kinase; JNK: c-Jun N-terminal kinase; ERK: extracellular signal-regulated kinase.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant no. 81000990), the Natural Science Foundation Project of CQ CSTC (grant no. 2009BB5339) and the Science Foundation of Third Military Medical University for the Young Scholar (grant no. 2009XQN32).
Conflict of interest
The authors disclose no potential conflicts of interest.
References
1. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87-108
2. He J, Shin H, Wei X, Kadegowda AK, Chen R, Xie SK. NPC1L1 knockout protects against colitis-associated tumorigenesis in mice. BMC Cancer. 2015;15:189
3. Schwartzberg LS, Rivera F, Karthaus M, Fasola G, Canon JL, Hecht JR. et al. PEAK: a randomized, multicenter phase II study of panitumumab plus modified fluorouracil, leucovorin, and oxaliplatin (mFOLFOX6) or bevacizumab plus mFOLFOX6 in patients with previously untreated, unresectable, wild-type KRAS exon 2 metastatic colorectal cancer. J Clin Oncol. 2014;32:2240-7
4. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5-29
5. Park GB, Choi Y, Kim YS, Lee HK, Kim D, Hur DY. Silencing of PKCeta induces cycle arrest of EBV(+) B lymphoma cells by upregulating expression of p38-MAPK/TAp73/GADD45alpha and increases susceptibility to chemotherapeutic agents. Cancer Lett. 2014;350:5-14
6. Godar S, Ince TA, Bell GW, Feldser D, Donaher JL, Bergh J. et al. Growth-inhibitory and tumor- suppressive functions of p53 depend on its repression of CD44 expression. Cell. 2008;134:62-73
7. He JM, Wang FC, Qi HB, Li Y, Liang HJ. Down-regulation of alphav integrin by retroviral delivery of small interfering RNA reduces multicellular resistance of HT29. Cancer Lett. 2009;284:182-8
8. Leithner K, Hrzenjak A, Trotzmuller M, Moustafa T, Kofeler HC, Wohlkoenig C. et al. PCK2 activation mediates an adaptive response to glucose depletion in lung cancer. Oncogene. 2015;34:1044-50
9. Durand RE. Chemosensitivity testing in V79 spheroids: drug delivery and cellular microenvironment. J Natl Cancer Inst. 1986;77:247-52
10. Durand RE. Distribution and activity of antineoplastic drugs in a tumor model. J Natl Cancer Inst. 1989;81:146-52
11. Liang X, Xu X, Wang F, Chen X, Li N, Wang C. et al. E-cadherin knockdown increases beta-catenin reducing colorectal cancer chemosensitivity only in three-dimensional cultures. Int J Oncol. 2015;47:1517-27
12. Liang X, Xu X, Wang F, Li N, He J. E-cadherin increasing multidrug resistance protein 1 via hypoxia-inducible factor-1alpha contributes to multicellular resistance in colorectal cancer. Tumour Biol. 2015
13. Gomes LR, Vessoni AT, Menck CF. Three-dimensional microenvironment confers enhanced sensitivity to doxorubicin by reducing p53-dependent induction of autophagy. Oncogene. 2015;34:5329-40
14. Casagrande N, Celegato M, Borghese C, Mongiat M, Colombatti A, Aldinucci D. Preclinical activity of the liposomal cisplatin lipoplatin in ovarian cancer. Clin Cancer Res. 2014;20:5496-506
15. Li Y, He J, Zhong D, Li J, Liang H. High-mobility group box 1 protein activating nuclear factor-kappaB to upregulate vascular endothelial growth factor C is involved in lymphangiogenesis and lymphatic node metastasis in colon cancer. J Int Med Res. 2015;43:494-505
16. Bennett BL, Sasaki DT, Murray BW, O'Leary EC, Sakata ST, Xu W. et al. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc Natl Acad Sci U S A. 2001;98:13681-6
17. Fabian MA, Biggs WH 3rd, Treiber DK, Atteridge CE, Azimioara MD, Benedetti MG. et al. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat Biotechnol. 2005;23:329-36
18. Zheng B, Fiumara P, Li YV, Georgakis G, Snell V, Younes M. et al. MEK/ERK pathway is aberrantly active in Hodgkin disease: a signaling pathway shared by CD30, CD40, and RANK that regulates cell proliferation and survival. Blood. 2003;102:1019-27
19. Cavallo F, Feldman DR, Barchi M. Revisiting DNA damage repair, p53-mediated apoptosis and cisplatin sensitivity in germ cell tumors. Int J Dev Biol. 2013;57:273-80
20. Zhu Y, Regunath K, Jacq X, Prives C. Cisplatin causes cell death via TAB1 regulation of p53/MDM2/MDMX circuitry. Genes Dev. 2013;27:1739-51
21. Wu ZZ, Sun NK, Chien KY, Chao CC. Silencing of the SNARE protein NAPA sensitizes cancer cells to cisplatin by inducing ERK1/2 signaling, synoviolin ubiquitination and p53 accumulation. Biochem Pharmacol. 2011;82:1630-40
22. Furuwatari C, Yagi A, Yamagami O, Ishikawa M, Hidaka E, Ueno I. et al. A comprehensive system to explore p53 mutations. Am J Clin Pathol. 1998;110:368-73
23. Landers JE, Cassel SL, George DL. Translational enhancement of mdm2 oncogene expression in human tumor cells containing a stabilized wild-type p53 protein. Cancer Res. 1997;57:3562-8
24. Ivashkevich A, Redon CE, Nakamura AJ, Martin RF, Martin OA. Use of the gamma-H2AX assay to monitor DNA damage and repair in translational cancer research. Cancer Lett. 2012;327:123-33
25. Pappano WN, Zhang Q, Tucker LA, Tse C, Wang J. Genetic inhibition of the atypical kinase Wee1 selectively drives apoptosis of p53 inactive tumor cells. BMC Cancer. 2014;14:430
26. Yu Z, Zhang C, Wang H, Xing J, Gong H, Yu E. et al. Multidrug resistance-associated protein 3 confers resistance to chemoradiotherapy for rectal cancer by regulating reactive oxygen species and caspase-3-dependent apoptotic pathway. Cancer Lett. 2014;353:182-93
27. Sui X, Kong N, Ye L, Han W, Zhou J, Zhang Q. et al. p38 and JNK MAPK pathways control the balance of apoptosis and autophagy in response to chemotherapeutic agents. Cancer Lett. 2014;344:174-9
28. Burotto M, Chiou VL, Lee JM, Kohn EC. The MAPK pathway across different malignancies: a new perspective. Cancer. 2014;120:3446-56
29. Koti M, Gooding RJ, Nuin P, Haslehurst A, Crane C, Weberpals J. et al. Identification of the IGF1/PI3K/NF kappaB/ERK gene signalling networks associated with chemotherapy resistance and treatment response in high-grade serous epithelial ovarian cancer. BMC Cancer. 2013;13:549
30. El Fajoui Z, Toscano F, Jacquemin G, Abello J, Scoazec JY, Micheau O. et al. Oxaliplatin sensitizes human colon cancer cells to TRAIL through JNK-dependent phosphorylation of Bcl-xL. Gastroenterology. 2011;141:663-73
31. Kolosenko I, Fryknas M, Forsberg S, Johnsson P, Cheon H, Holvey-Bates EG. et al. Cell crowding induces interferon regulatory factor 9, which confers resistance to chemotherapeutic drugs. Int J Cancer. 2015;136:E51-61
32. Mellor HR, Ferguson DJ, Callaghan R. A model of quiescent tumour microregions for evaluating multicellular resistance to chemotherapeutic drugs. Br J Cancer. 2005;93:302-9
33. Sarasqueta AF, Forte G, Corver WE, de Miranda NF, Ruano D, van Eijk R. et al. Integral analysis of p53 and its value as prognostic factor in sporadic colon cancer. BMC Cancer. 2013;13:277
34. Wei S, Wang H, Lu C, Malmut S, Zhang J, Ren S. et al. Activating Transcription Factor 3 Suppresses the Oncogenic Function of Mutant p53. J Biol Chem. 2014;289:8947-59
35. Stefancikova L, Moulis M, Fabian P, Vasova I, Zedek F, Ravcukova B. et al. Prognostic impact of p53 aberrations for R-CHOP-treated patients with diffuse large B-cell lymphoma. Int J Oncol. 2011;39:1413-20
36. Liu BW, Liu Y, Liu JR, Feng ZX, Liu T. Prognostic effect of p53 expression in patients with completely resected colorectal cancer. Tumour Biol. 2014;35:9893-6
Author contact
![]() Corresponding author: Tel: +86 23 68773057; Email: hejmedu.cn (J. He).
Corresponding author: Tel: +86 23 68773057; Email: hejmedu.cn (J. He).
Citation styles
APA
He, J., Liang, X., Luo, F., Chen, X., Xu, X., Wang, F., Zhang, Z. (2016). P53 Is Involved in a Three-Dimensional Architecture-Mediated Decrease in Chemosensitivity in Colon Cancer. Journal of Cancer, 7(8), 900-909. https://doi.org/10.7150/jca.14506.
ACS
He, J.; Liang, X.; Luo, F.; Chen, X.; Xu, X.; Wang, F.; Zhang, Z. P53 Is Involved in a Three-Dimensional Architecture-Mediated Decrease in Chemosensitivity in Colon Cancer. J. Cancer 2016, 7 (8), 900-909. DOI: 10.7150/jca.14506.
NLM
He J, Liang X, Luo F, Chen X, Xu X, Wang F, Zhang Z. P53 Is Involved in a Three-Dimensional Architecture-Mediated Decrease in Chemosensitivity in Colon Cancer. J Cancer 2016; 7(8):900-909. doi:10.7150/jca.14506. https://www.jcancer.org/v07p0900.htm
CSE
He J, Liang X, Luo F, Chen X, Xu X, Wang F, Zhang Z. 2016. P53 Is Involved in a Three-Dimensional Architecture-Mediated Decrease in Chemosensitivity in Colon Cancer. J Cancer. 7(8):900-909.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.