Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2016; 7(6):746-757. doi:10.7150/jca.13289 This issue Cite
Research Paper
Silencing of hERG1 Gene Inhibits Proliferation and Invasion, and Induces Apoptosis in Human Osteosarcoma Cells by Targeting the NF-κB Pathway
Wenrong Zeng1*, Qingjun Liu1*, Zhida Chen1, Xinyu Wu2, Yuanfu Zhong3, Jin Wu1 ![]()
1. Department of Orthopaedics, the Affiliated Southeast Hospital of Xiamen University, Orthopaedic Center of People's Liberation Army, Zhangzhou, 363000, People's Republic of China.
2. Department of Neurology, the Affiliated Southeast Hospital of Xiamen University, Zhangzhou, 363000, People's Republic of China.
3. Department of central laboratory, the Affiliated Southeast Hospital of Xiamen University, Zhangzhou, 363000, People's Republic of China.
*Wenrong Zeng and Qingjun Liu contributed equally to this work.
Received 2015-7-20; Accepted 2016-2-11; Published 2016-4-10
Abstract
Recently, the human ether à go-go (eag) related gene 1 (hERG1) channel, a member of the voltage-dependent potassium channel (Kv) family, was determined to have a critical role in cancer cell proliferation, invasion, tumorigenesis and apoptosis. However, the expression levels and functions of hERG1 in osteosarcoma cells remain poorly characterized. In this study, hERG1 transcript and protein levels in osteosarcoma cells and tissues were measured using semi-quantitative real time PCR (RT-PCR), Western blot, and immunohistochemistry. The effects of hERG1 knockdown on osteosarcoma cell proliferation, apoptosis and invasion were examined using CCK-8, colony formation, flow cytometry, caspase-3 activity, wound healing and transwell based assays. Furthermore, semi-quantitative RT-PCR, Western blot and a luciferase reporter assay were used to assess the effects of hERG1 inhibition on the nuclear factor-κB (NF-κB) pathway. In addition, the effect of NF-κB p65-siRNA and NF-κB p65 expression on the survival of osteosarcoma cells was investigated. Through this work, a relationship for hERG1 with the NF-κB pathway was identified. Osteosarcoma cells and tissues were found to express high levels of hERG1. Knockdown of hERG1 significantly suppressed cellular proliferation and invasion, and induced apoptosis, while inhibition of hERG1 significantly decreased activation of NF-κB. Overall, hERG1 may stimulate nuclear translocation of p65, thus regulating the NF-κB pathway through the activation of the hERG1/beta1 integrin complex and PI3K/AKT signaling. Taken together, these results demonstrate that hERG1 is necessary for regulation of osteosarcoma cellular proliferation, apoptosis and migration. Furthermore, this regulation by hERG1 is, at least in part, through mediation of the NF-κB pathway.
Keywords: Human ether à go-go related gene 1, Osteosarcoma, Cell proliferation, Apoptosis, Invasion, NF-κB.
Introduction
Osteosarcomas are the most common types of malignant primary bone tumors and most frequently occur in children and adolescents [1, 2]. These malignant cells are derived from primitive bone-forming mesenchymal stem cells [3], and the majority of osteosarcomas originate from the metaphysis of long bones, particularly the distal femur and proximal tibia [4]. These tumors are typically rapid growers with high metastatic potential, especially to the lungs and bones [5]. Despite the availability of aggressive treatment options, such as high-dose chemotherapy and wide tumor resection, the ten year survival without recurrence of disease is only approximately 60% and 30% in patients with localized or metastatic disease upon diagnosis, respectively [6]. Furthermore, the application of high-dose chemotherapy is restricted due to its adverse side effects, including gastrointestinal problems, suppression of bone marrow, and renal and cardiac toxicity. Overall, the availability of efficacious treatment options for osteosarcomas is far from satisfactory. Therefore, the development of new treatments is an urgent clinical need. Recently, a significant genetic instability was identified that leads to the aberrant and uncoordinated expression of a number of gene products with a direct relationship with the development of osteosarcomas [7]. Therefore, the sources of this genetic instability are potential targets for osteosarcoma diagnosis and treatment, such as Kv channel families.
Kv channels play a number of necessary roles in cells, which can be categorized into electrically excitable and non-excitable. Recently, these functions of Kv channels and their relationships to tumor biology have become a focus of investigation [8]. Two Kv channels, heag1 (Kv10.1) and hERG1 (Kv11.1), are the most notable members of the Kv family due to their aberrant expression in many primary human cancers and their pleiotropic effects in cancer cells [9, 10]. Our previous studies have focused on characterizing heag1 and its role in osteosarcoma cells, and have yielded encouraging results [11, 12]. Interestingly, both heag1 and hERG1 are members of the same family of Kv channels and have 47% amino acid sequence homology [13]. However, the expression, functions and mechanisms of action of hERG1 in osteosarcoma cells are still poorly understood. In this study, the expression of hERG1 was quantified in human osteosarcoma cells and tissues firstly. Then, the functions of hERG1 in relation to proliferation, apoptosis and invasion of osteosarcoma cells were studied. Finally, possible mechanisms of action for these effects incurred upon hERG1 inhibition were investigated.
Materials and Methods
Cell culture
MG-63, SH-SY5Y and HEK293-wt cell lines (human osteosarcoma, neuroblastoma and wild-type embryonic kidney cell lines, respectively) were purchased from the American Type Culture Collection (ATCC). All cell lines were incubated at 37°C in a humidified atmosphere of 5% CO2 in media containing 10% fetal bovine serum (FBS) (Gibco, Rockville, MD, USA), 100 U/ml penicillin and 100 μg/ml streptomycin, and were split every 48-72 hours. MG-63 and SH-SY5Y cells were cultured in RPMI-1640 medium (Gibco), while HEK293-wt (HEK-293wt) and HEK-293 cells with stable expression of hERG1 (HEK293-hERG1) were cultured in DMEM medium (Gibco).
Drugs, siRNAs and plasmids
E-4031 (M5060), PD 118057 (p5624) and LY264002 (L9908) were purchased from Sigma (St. Louis, MO, USA), diluted into stock solutions (1mM) and then stored at -20°C until use. Control small interfering RNA (siRNA) (sc-37007) and anti-hERG1 (sc-42498) siRNA were purchased from Santa Cruz Biotechnology (CA, USA), while anti-NF-κB p65 (#6261) siRNA was purchased from Cell Signaling Technology (Danvers, MA, USA). Transient transfections of these siRNAs into 30-50% confluent cells in 6-well plates (Sigma) were performed using LipofectamineTM 2000 (Invitrogen, Rockville, MD, USA) according to the manufacturer's instructions. All these cells were harvested 48 h post-transfection
A pcDNA3.1-NF-κB p65 expression plasmid was constructed as previously described using PCR based cloning with the Promega PCR kit (Madison, WI) and successful construction was confirmed by DNA sequencing [14]. Overexpression of hERG1 was induced in HEK293 cells by transfecting in this plasmid using the technique described above.
Semi-quantitative RT-PCR
Total RNA from cultured cells was collected using Trizol (Invitrogen), and then a sample was run on a denaturing 1% agarose gel to confirm purity and integrity. This RNA was used as a cDNA template by combining 1 μg RNA, 200 U reverse transcriptase (Takara, Tokyo, Japan), 200 μM dNTPs and 2.5 μM oligo-dT primers in a 20 μl reaction. This solution was incubated at 30°C for 10 min, then at 42°C for 60 min and, finally, at 80°C for 5 min. The resulting cDNA was then amplified in a 25 μl PCR reaction consisting of 1 μl cDNA, 200 μM dNTPs and 2.5 U DNA polymerase. The primer sequences were as follows: hERG1 forward, 5'-GCT TTT GAG AAC GTG GAT GAG-3'; hERG1 reverse, 5'-CGA AGA TGG TGG CAT AGA GAA-3'; cIAP-1 forward, 5′-TTC CCA GGT CCC TCG TAT CAA AA-3′; cIAP-1 reverse, 5′-TGG AGA AAG GCT GGA GTA AGA ACC-3′; XIAP forward, 5′-TGG CAC GAG CAG GGT TTC TTT A-3′; XIAP reverse, 5′-TGG GGT TAG GTG AGC ATA GTC TGG-3′; Bcl-2 forward, 5′-ATG TGT GTG GAG AGC GTC AAC C-3′; Bcl-2 reverse, 5′-TGA GCA GAG TCT TCA GAG ACA GCC-3′; Survivin forward, 5′-GCC TGG CAG CCC TTT CTC AA-3′; Survivin reverse, 5′- CTC GAT GGC ACG GCG CAC TTT CT-3′; NF-κB p65 forward, 5′-AGC TCA AGA TCT GCC GAG TG-3′; NF-κB p65 reverse, 5′- ACA TCA GCT TGC GAA AAG GA-3′; MMP-2 forward, 5′-GCT ATG GAC CTT GGG AGA A-3'; MMP-2 reverse,5′-TGG AAG CGG AAT GGA AAC-3′; MMP-9 forward, 5′-TCC CTG GAG ACC TGA GAA CC-3′; MMP-9 reverse, 5′-CGG CAA GTC TTC CGA GTA GTT T-3'; β-actin forward, 5′-TCC ACC TTC CAG CAG ATG TG-3'; and β-actin reverse, 5′-GCA TTT GCG GTG GAC GAT-3'. An aliquot of PCR product from each reaction was run on a 2% agarose gel that was stained with ethidium bromide and then exposed to a UV transilluminator to visualize the DNA. To confirm specificity of the PCR and primers, several of the resulting PCR products were sequenced. Each experiment was performed three times total.
Western blot
To obtain protein, lysates were generated by incubating 5-6 × 107 cells for 30 min in ice-cold lysis buffer consisting of 150 mmol/L NaCl, 50 mmol/L Tris-Cl (pH 7.5), 1 mmol/L PMSF, 0.2 mmol/L EDTA, and 1% (v/v) Nonidet-P40. These lysates were centrifuged for 10 min at 13,200 rpm at 4°C to separate out the cellular debris from the proteins, and then the supernatants containing the proteins were collected and used for Western blotting. SDS-PAGE on 12% gels was used to resolve the proteins (25 μg) and then the proteins were transferred from the gels to nitrocellulose membranes (Bio-Rad, Richmond, CA). Blocking of the membranes was performed at room temperature for 1 h with 10% (w/v) nonfat milk powder, and then the membranes were incubated with anti-hERG1 (ab196301, Abcam, Cambridge, MA), pro-caspase 3 (ab32150, Abcam), active caspase-3 (ab2302, Abcam), PARP (ab6079, Abcam), cleaved PARP (ab32064, Abcam), cIAP-1 (ab2399, Abcam), XIAP (ab21278, Abcam), Bcl-2 (ab117115, Abcam ), Survivin (ab76424, Abcam), MMP-2 (sc-13595, Santa Cruz), MMP-9 (sc-21733, Santa Cruz), IκBα phosphorylation (#2859, Cell Signaling Technology) IκBα (#4812, Cell Signaling Technology), NF-κB p65 (#3034, Cell Signaling Technology), Akt (#9272, Cell Signaling Technology), p-Akt (#9271, Cell Signaling Technology), Integrin β1 antibody (12G10) (ab30394, Abcam) or GAPDH (ab9484, Abcam) antibodies overnight. The membranes were incubated with horseradish peroxidase-conjugated goat secondary antibodies against rabbit or mouse (Abcam), treated with a chemiluminescent detection kit (Zhongshan Biotechnology, Beijing, China) and then exposed to X-ray film. Experiments were performed at least three times and representative blots were shown. Densitometry was analyzed using NIH ImageJ software (http://rsb.info.nih.gov/ij).
Sample collection
A total of 34 formalin-fixed, paraffin-embedded specimens taken from 22 osteosarcomas and 12 osteosfibrous dysplasias present in 18 patients that had not been treated with neoadjuvant chemotherapy and 9 patients, respectively, were acquired along with clinicopathologic factors from the affiliated Southeast Hospital of Xiamen University between January 2010 and June 2014. For a positive control, a sample of a healthy human brain biopsy was used [15]. Prior to specimen collection, written informed consent was obtained from patients in accordance with a protocol approved by the Institutional Review Board of the affiliated Southeast Hospital of Xiamen University.
Immunohistochemistry
To remove paraffin, slides and coverslips with adherent cells were baked on a rack in a dry oven for 2 h at 60°C. The slides and coverslips were then immersed in xylene (Zhongshan Biotechnology) for 3 min twice, hydrated with consecutive incubations in 100%, 95%, 70% and then 50% ethanol (Zhongshan Biotechnology) and rinsed for 5 min in cold tap water. The sections were dewaxed, treated to inhibit endogenous peroxidases, exposed to EDTA at 100°C (1 mM, pH 8.0) to retrieve antigens and then incubated with anti-hERG1 antibody (1:500, Abcam) at 4°C overnight. The next day, the samples were rinsed with PBS and then incubated at room temperature for 1 h with biotinylated goat anti-rabbit IgG (Abcam). The tissues were treated with avidin biotin complex (ABC) (Zhongshan Biotechnology) and diaminobenzamidine (DAB) (Zhongshan Biotechnology) according to manufacturer's instructions, and then counterstained with haematoxylin. Imaging was performed using a confocal microscope (Olympus, Japan).
Cellular proliferation assay
Quantification of cellular proliferation was performed using a Cell Counting Assay Kit-8 (CCK-8; Dojindo Molecular Technologies, Gaithersburg, MD) according to the manufacturer's instructions. Briefly, 1 × 105 cells were starved by incubating in serum-free medium for 12 h, transduced, harvested after 48h, treated CCK-8 solution (10 µl) and incubated for another 1 h. Then the absorbance (A) was measured by spectrophotometer at 490 nm (Bio-Rad). Experiments were performed at least three times and representative data were shown.
Colony formation assay
To perform colony formation assays, the inside bottoms of 60 mm dishes were coated with 0.5% agar (Sigma), and then another layer of treated osteosarcoma cells, 1 × 103 cells per dish, that had been mixed with 0.3% soft agar was added on top. These plates were incubated for up to 2 weeks, and the assays were stopped when the colonies were clearly visible by eye.
Flow cytometry
Cells that had been transfected with siRNA for 48 h were collected, washed twice with cold PBS, resuspended to 1 × 106 cells/ml and fixed in 70% cold ethanol at 4°C overnight. The frequency of apoptosis was determined by staining cells using an Annexin V-FITC/PI Apoptosis kit according to the manufacturer's instructions and measuring by a FACScan flow cytometer (Becton Dickinson, San Jose, CA). The percentage of cells in the early stages of apoptosis was calculated by counting the number of cells positive for annexin V and negative for propidium iodide (PI).
Caspase-3 Activity Assay
Caspase-3 activity was quantified with the Apo-ONE homogeneous caspase-3/7 assay (Promega) in accordance with the manufacturer's instructions. Briefly, 20 µl of a 1 × 105 cells/ml suspension was dispersed into the wells of 96-well plates and incubated at 37°C with 5% CO2 overnight. The cells were first exposed to the adenovirus constructs, then Apo-ONE Caspase-3/7-reagent was added and the samples were incubated for 1 h at room temperature. A plate-reader luminometer (Tecan, Switzerland) was used to measure the luminescence of each sample. Experiments were performed at least three times and representative data were shown.
Wound healing assay
5 × 105 cells were cultured in each well of 6-well plates (Sigma) in serum-free RPMI-1640 medium until 100% confluency was reached. The layer of cells were scratched with a sterile 10 μl Eppendorf tip (Sigma), washed 3 times with PBS, and then incubated in fresh serum-free RPMI-1640 medium for 24 h. An inverted microscope (Olympus) was used to examine and photograph random fields.
Cell invasion assay
Cell invasion assays were done in 24-well matrigel-coated 8-μm pore transwell chambers (Corning, NY, USA) according to the manufacturer's instructions. The upper transwell chambers were seeded with 2 ×104 cells that had been transfected with siRNAs for 24 h in media containing 0.1% FBS, while the bottom chamber contained media containing 5% FBS. After 24 h, the chambers were collected and cells that had moved into the bottom chamber (i.e. invasive cells) were stained with Giemsa and photographed under a microscope. The number of invading cells on each membrane was determined by counting the number of cells in ten high-power (400 ×) fields and then calculating the mean number of cells per field.
Luciferase Reporter Assay
Co-transfection of 200 ng each of a NF-κB p65 luciferase reporter and a Renilla luciferase construct (Promega) was performed on 1×106 osteosarcoma cells in each well of a 6-well plate (Sigma) using LipofectamineTM 2000 (Invitrogen) according to the manufacturer's instructions. These cells were washed with PBS twice, transfected with siRNA and then lysed using reporter lysis buffer (Promega). For the luciferase assay, a mixture was made at room temperature of 20 μl cell extract and 100 μl luciferase assay reagent. The firefly luciferase activity of this mixture was then quantified using a Dual-Luciferase Reporter Assay System (Promega).
Statistical analysis
All data are presented as mean ± standard error of mean (SEM). Student's t-test or analysis of variance (ANOVA) performed with SPSS 18.0 were used to determined statistical significance, where P <0.05 was considered significant.
Results
Expression of hERG1 in osteosarcoma cells
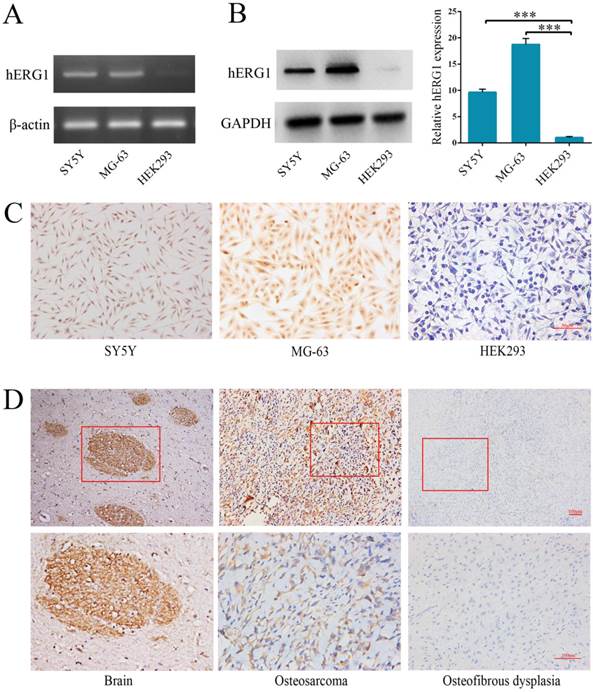
Semi-quantitative RT-PCR and Western blot were performed to quantify hERG1 transcript and protein levels in osteosarcoma cells. The human neuroblastoma SH-SY5Y [16] and HEK293 cell lines were used as positive and negative controls, respectively. As shown in Fig. 1A, high levels of hERG1 transcripts were noted in MG-63 cells, and hERG1 protein expression levels in MG-63 cells were confirmed by Western blot and immunohistochemistry (Fig. 1B and 1C). Next, immunohistochemistry was used to visualize the expression of hERG1 in osteosarcoma patient tissue samples. Positive hERG1 staining was noted in 16/22 (72.72%) of osteosarcoma samples studied, but 12/12 osteosfibrous dysplasia samples tested were negative for hERG1 staining. hERG1 staining primarily occurred in the cytoplasm, which is consistent with previous reports [17]. Representative images of hERG1 staining in the positive control (healthy human brain sample), osteosarcoma and osteosfibrous dysplasia tissue samples were presented in Fig. 1D. Overall, these results demonstrate high expression of hERG1 in osteosarcoma cells and tissue samples.
Highly expression of hERG1 in osteosarcoma cells and tissues. (A) Semi-quantitative RT-PCR was performed to measure hERG1 mRNA levels in SH-SY5Y, MG-63 and HEK293 cells. β-actin was used as the internal standard. (B) Protein expression of hERG1 was measured in SH-SY5Y, MG-63, and HEK293 cells by Western blot. GAPDH was used as the internal standard. (C) Immunohistochemistry staining for hERG1 in MG-63 cells. Images were captured using an Olympus light microscope equipped with a CCD color camera. (D) Immunohistochemistry of hERG1 in human brain (as positive control), osteosarcoma and osteosfibrous dysplasia samples. *** P < 0.001.
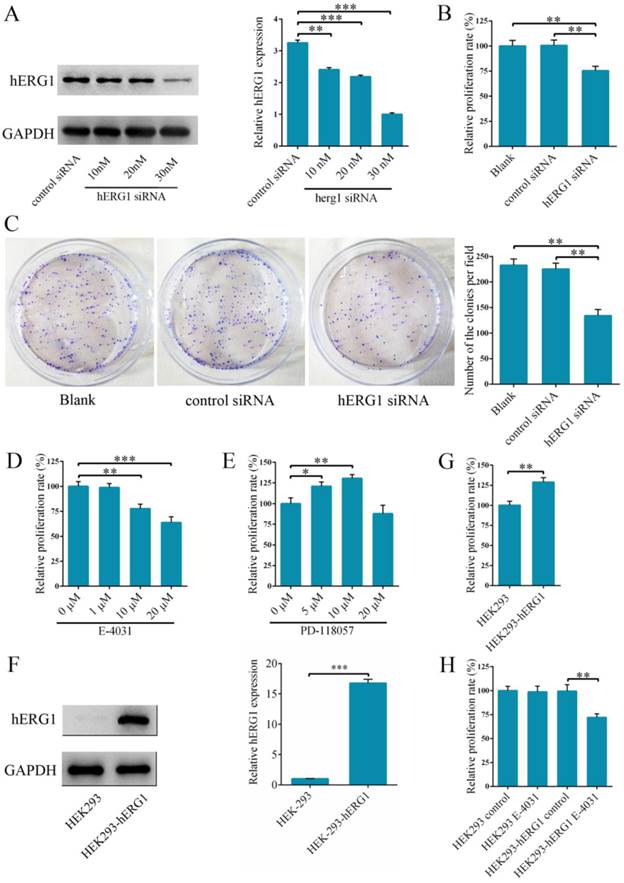
Effects of hERG1 inhibition on osteosarcoma cell proliferation
In order to delineate the role of hERG1 in osteosarcoma tumorigenesis, hERG1 knockdown was performed in MG-63 cells by transfecting in hERG1-siRNA. Following transfection, hERG1-siRNA treated cells had significantly lower hERG1 protein levels than cells given control-siRNA (Fig. 2A), indicating that hERG1-siRNA is an efficient means of hERG1 knockdown. Next, the effects of hERG1 knockdown on MG-63 cellular proliferation and growth were assessed using CCK-8 and colony formation assays following treatment with hERG1-siRNA (30 nM) for 48 h. As shown in Fig. 2B and 2C, hERG1-siRNA transfected cells had a remarkable decrease in proliferation and growth compared to untreated and control-siRNA treated cells. Similarly, proliferation was also attenuated in MG-63 cells incubated for 48 h with E-4031 (10 μM and 20 μM), a hERG1 specific inhibitor (Fig. 2D). Furthermore, incubation for 48 h with PD 118057 (5 μM and 10 μM), a hERG1 activator, resulted in an increase in MG-63 cell proliferation compared to untreated cells (Fig. 2E). Next, hERG1 protein levels in HEK293-hERG1 and HEK293-wt cells were measured by Western blot (Fig. 2F). There was an apparent increase in HEK293-hERG1 cell proliferation compared to the HEK-293wt cells after 48 h of incubation (Fig. 2G). When HEK293-wt and HEK293-hERG1 cells were treated with E-4031 (20 μM) for 48 h, a similar phenotype to MG-63 cells was observed, where the treated HEK293-hERG1 cellular proliferation was inhibited using E-4031 (Fig. 2H). These results indicated that hERG1 plays a critical role in the proliferation of osteosarcoma cells.
Knockdown of hERG1 reduces proliferation of osteosarcoma cells. (A) Efficiency of knockdown by hERG1-siRNA was measured by Western blot. (B-E) Proliferation of MG-63 cells transfected with hERG1-siRNA (30 nM) (B and C), or treated with hERG1 inhibitor E-4031 (D) or activator PD 118057 (E) was measured using CCK-8 or colony formation assay (n = 6). (F) Protein expression of hERG1 in HEK293-wt and HEK293-hERG1 cells was detected by Western blot. (G) 1 × 105 HEK293-wt and HEK293-hERG1 cells were cultured for 48 h and the CCK-8 assay was performed. (H) The effects of E-4031 on the proliferation of HEK293-wt and HEK293-hERG1 cells were determined by CCK-8 assay. * P < 0.05, ** P < 0.01, *** P < 0.001.
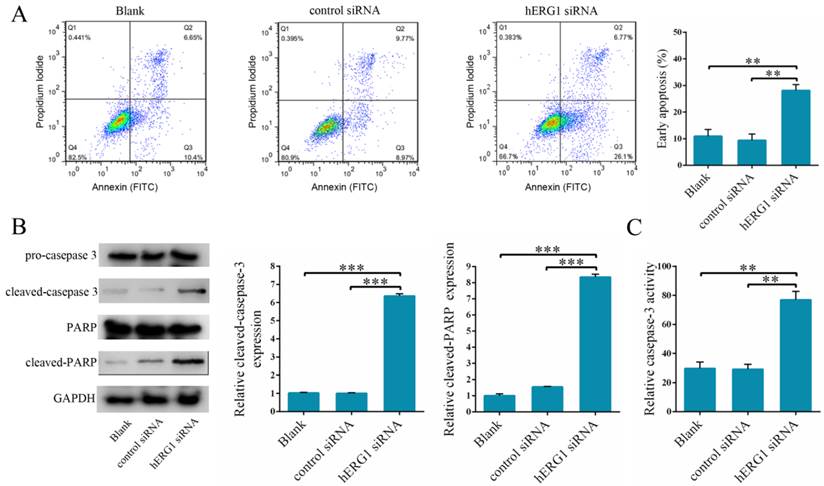
Effects of hERG1 inhibition on apoptosis of osteosarcoma cells
To delineate the mechanism of action behind hERG1 promotion of osteosarcoma cell proliferation, we examined whether inhibition of hERG1 triggers apoptosis. To this end, osteosarcoma cells were transfected with either control-siRNA or hERG1-siRNA, and then flow cytometry was used to measure the apoptotic cell population. Notably, there was a significant increase in the number of apoptotic cells in hERG1-siRNA treated cells compared to the control-siRNA and untreated cells (blank group) (Fig. 3A). Furthermore, measurement of caspase-3 cleavage and PARP expression, markers of apoptosis, was performed in these cells by Western blot. As shown in Fig. 3B, cells treated with hERG1-siRNA had significantly higher amounts of cleaved caspase-3 and PARP compared with control-siRNA or untreated (blank group) cells. Finally, caspase-3 became activated upon cleavage, and assays quantifying caspase-3 activity revealed a significant increase in caspase-3 cleavage when hERG1 was knocked down by siRNA compared to the controls (Fig. 3C).
The influence of hERG1 knockdown on osteosarcoma cell apoptosis. (A) MG-63 cells were transfected with hERG1-siRNA and then analyzed by flow cytometry. Cells untreated or transfected with control-siRNA were served as controls. Cells in the right lower quadrant are Annexin-positive, which are early apoptotic cells (n = 3). (B) Caspase-3 and PARP cleavage was determined by Western blot. (C) Cells transfected with hERG1-siRNA had a significant increase in caspase-3 activity compared to control-siRNA transfected and untreated cells. ** P < 0.01, *** P < 0.001.
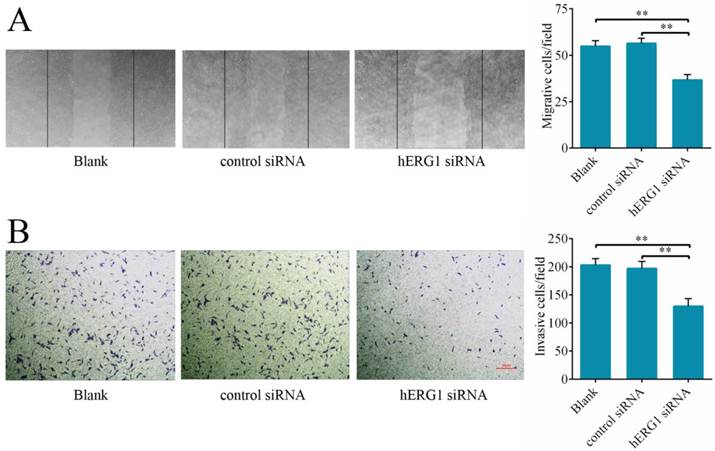
Effects of hERG1 inhibition on the migration and invasion of osteosarcoma cells
To delineate hERG1 regulation of cancer cell migration and invasion, the effects of hERG1 knockdown on osteosarcoma cells on wound healing and transwell assay translocation were examined. It was found that MG-63 cells transfected with hERG1-siRNA displayed reduced migration in wound healing assays (Fig. 4A) and invasion in transwell assays (Fig. 4B) compared to control-siRNA treated and untreated cells.
Knockdown of hERG1 suppresses migration of and invasion by osteosarcoma cells. (A) A scratch was made in a confluent, adherent layer of MG-63 cells that had undergone different treatments. After 24 h, cells that had migrated into the wound were counted (n = 3). Representative pictures of the wounds are shown. (B) MG-63 cells were transfected with the indicated siRNAs and the ability of the cells to invade was evaluated by transwell assay (n = 3). ** P < 0.01.
hERG1 regulation of proliferation, apoptosis and invasion of osteosarcoma cells
As shown in Fig. 2-4, hERG1 downregulation and/or inhibition prevented proliferation and invasion, and induced apoptosis of osteosarcoma cells. In order to detail the mechanism behind these phenotypes, we first focused on the NF-κB pathway. This pathway regulates the expression of a number of genes proposed to govern tumor survival, growth, apoptosis, invasion and angiogenesis [18-20]. As shown in Fig. 5A, transfection of cells with hERG1-siRNA resulted in the downregulation of NF-κB-regulated gene products, including cIAP-1, Bcl2, Survivin, XIAP, and MMP-2 and MMP-9. Western blot confirmed these results (Fig. 5B). Furthermore, a decrease in IκBα phosphorylation and an increase in total IκBα protein levels in osteosarcoma cells occured following hERG1-siRNA transfection (Fig. 5B). Likewise, translocation of NF-κB p65 protein into the nucleus, an indicator of NF-κB transcriptional activity, and NF-κB-luciferase reporter activity were also lower in osteosarcoma cells transfected with hERG1-siRNA than control cells (Fig. 5B-C). Moreover, overexpression of hERG1 in MG-63 cells also led to increased nuclear localization of NF-κB p65 (Fig. 5D).
The involvement of NF-κB pathway in hERG1-regulated osteosarcoma cell survival. (A and B) Effect of hERG1 inhibition on NF-κB pathway-related gene expression. MG-63 cells were transfected hERG1-siRNA or control-siRNA, or left untreated, and the indicated mRNA and proteins were analyzed by semi-quantitative RT-PCR and Western blot. (C) Effect of hERG1 inhibition on NF-κB mediated transcription. (D) Nuclear localization of NF-κB p65 in MG-63 cells transfected with pcDNA3.1-hERG1 or pcDNA3.1. (E and F) CCK-8 assays were performed in MG-63 cells transiently transfected with NF-κB p65-siRNA or pCDNA3.1 NF-κB p65. (G) Effect of hERG1 knockdown on Akt phosphorylation. (H) Nuclear localization of NF-κB p65 (up panel) and relative cell proliferate rate (down panel) were determined. (I) Nuclear localization of NF-κB p65 in MG-63 cells treated with LY264002. * P < 0.05, ** P < 0.01.
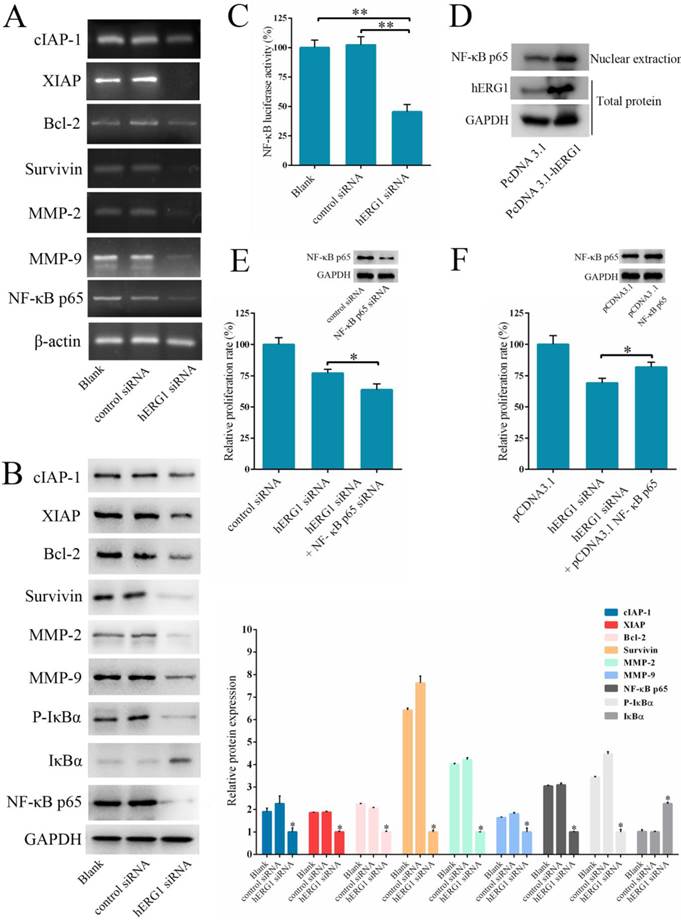
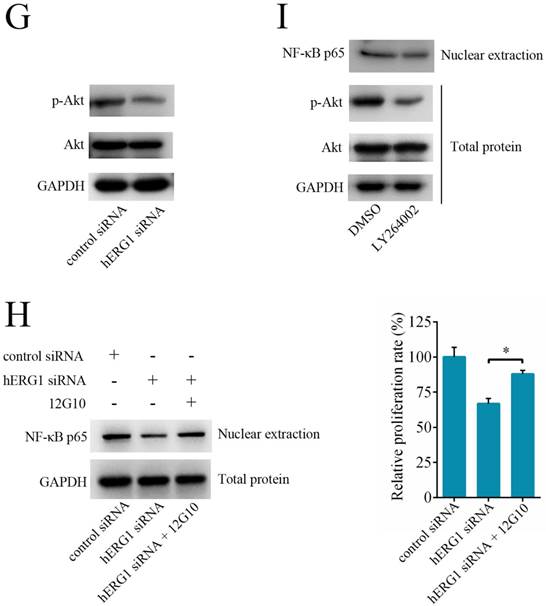
Next, we investigated the role of the NF-κB pathway in hERG1 mediated oncogenic properties of osteosarcoma cells. As shown in Fig. 5E, NF-κB downregulation by transfection with NF-κB p65-siRNA had an additive effect on the inhibition of osteosarcoma cell proliferation mediated by hERG1-siRNA. In addition, transfection with an NF-κB p65 expression plasmid partially reversed the hERG1-siRNA mediated decrease in cell viability (Fig. 5F). Furthermore, silencing of hERG1 resulted in a decrease in Akt phosphorylation (Fig. 5G). Importantly, the induction of osteosarcoma growth and inhibition of NF-κB by hERG1-siRNA could be reduced by activation of integrins using anti-integrin antibody 12G10 (Fig. 5H). Inhibition of Akt signaling using LY264002 led to reduced nuclear localization of NF-κB p65 (Fig. 5I), suggesting that hERG1 may induce nuclear translocation of p65, thus activating the NF-κB pathway through activation of PI3K/Akt signaling, which is consistent with a previous report [21]. Overall, these results strongly suggested that hERG1 regulates the NF-κB pathway in osteosarcoma cells.
Discussion
Characterization of phenotypic differences between healthy and tumor cells are an important method by which to study the mechanisms behind the malignant transformation of healthy cells. For example, ion channel encoding genes are differentially expressed in normal and tumor cells, and, because of this, these channels have become a strong research focus [22]. Researchers have connected potassium channels to known oncogenic properties, and, therefore, conclude that several potassium channels are directly related to the initiation and progression of cancers [9, 10]. The hERG1 gene encodes an inwardly rectifying potassium channel which is distinct from the majority of its fellow members of the evolutionarily conserved voltage-gated Eag family, which typically encode outwardly rectifying potassium channels [23]. hERG1 potassium channels are normally expressed in embryos, hearts and adult brains. They generate a current (Iherg) with inwardly rectifying properties and are indispensable for the maintenance of a normal heart rhythm, neuronal differentiation and other physiological functions [24]. A number of studies have noted an aberrant increase in expression of hERG1 in different human tumor cells compared to noncancerous matched tissues. Moreover, hERG1 has been linked to tumor cell proliferation, apoptosis, invasion and other malignant characteristics [9, 10, 25]. While work has found that erg K+ channels may play an anti-proliferative role in rat osteoblastic cells [26], until now, the relationship between hERG1 and osteosarcoma cells has received little attention. Along these lines, the expression levels and functions of hERG1 in osteosarcoma cells are still uncharacterized. In this study, semi-quantitative RT-PCR, Western blot and immunohistochemistry were employed to measure hERG1 expression in osteosarcoma cells. It was found that hERG1 expression was remarkably elevated in osteosarcoma cells compared to benign bone tumor cells, which is consistent with a previous report [27], providing support and laying the foundation for further research.
The association between hERG1 and proliferation of several types of cancer cells has long been known. Specifically, hERG1 is a player in neuroblastoma cell cycle regulation, and restriction of Iherg in these cells results in a decrease in proliferation [16]. Pillozz et al. reported that treatment with an Iherg specific inhibitor E-4031 lead to the growth of and G1 phase arrest in leukemia cell lines [28]. In addition, hERG1-siRNA has been found to not only be a suppressor of cancer cell proliferation in vitro, but also the growth of tumors in vivo [29]. The data from this present study determined the following: 1. HEK293-hERG1 cells had higher levels of proliferation compared to HEK293-wt cells, which lack endogenous expression of hERG1. 2. Inhibition of hERG1 with either E-4031 (10 μM and 20 μM) or knockdown with hERG1-siRNA reduced proliferation of osteosarcoma cells. Furthermore, there was a reduction in the proliferation of HEK293-hERG1 cells, not HEK293-wt, upon exposure to E-4031 (20 μM). 3. Activation of hERG1 after 48 h of PD 118057 treatment (5 μM and 10 μM) increased proliferation of osteosarcoma cells.
Research has also noted the involvement of hERG1 in tumor cell apoptosis. Wang et al. reported that the tumor necrosis factor α (TNF-α) is a regulator of tumor cell apoptosis in a hERG1 expression associated manner [30]. Also, Staudacher et al. found that the hERG1 small molecule ligand doxazosin induces the apoptosis of glioblastoma cells [31]. In this study, it was found that hERG1 silencing induced apoptosis in osteosarcoma cells.
Although data has been previously published suggesting that hERG1 plays a prominent role in the control of tumor cell proliferation and apoptosis, there has been a lack of evidence for a role for hERG1 in the regulation of tumor invasion. Lastraioli et al. found that hERG1 is involved in the invasive phenotype of colorectal cancer cells both in vitro and in vivo [32]. Other reports have demonstrated a functional association for hERG1 with integrin receptors [33] and vascular endothelial growth factor (VEGF) [16]. In this present study, we examined the role of hERG1 in osteosarcoma cell migration and invasion, and found that hERG1 is involved in regulation of the migration and invasion of osteosarcoma cells.
When characterizing the mechanisms by which hERG1 regulates osteosarcoma cell malignancy, we focused on the NF-κB pathway. The NF-κB family consists of NF-κB1 (p105), NF-κB2 (p100), Rel A (p65), Rel-B and Rel-C, which function as transcription factors and consist of special DNA binding peptides that participate in cellular physiological processes and regulation [34, 35]. Recent studies have found that aberrant activation of the NF-κB pathway, especially the Rel A (p65) subunit, leads to the abnormal expression of several cancer-associated genes, accelerates cellular proliferation, inhibits apoptosis and promotes the formation and metastasis of tumor vessels, thus directly affecting the occurrence and survival of tumors [36, 37]. In this study, we measured the expression of NF-κB-regulated gene products, including cIAP-1, XIAP, Bcl-2, Survivin, and MMP-2 and MMP-9, after siRNA knockdown of hERG1, and then investigated the role of the NF-κB pathway in hERG1 mediated oncogenic effects in osteosarcoma cells. Based on a previously published paper [21], we determined how hERG1 was related to the NF-κB pathway based on three phenotypes: 1. a decrease in p-AKT levels upon hERG1 knockdown; 2. rescue of hREG1-siRNA induced inhibition of cellular growth and reduction of NF-κB signaling by integrin/AKT activation; and 3. the induction of a phenotype similar to hERG1 knockdown upon inhibition of AKT signaling using a chemical inhibitor, such as LY264002, especially in terms of NF-κB signaling regulation. Overall, this work indicates hERG1 regulates the NF-κB pathway in osteosarcoma cells.
Taken together, the results of this study lead to the conclusion that hERG1 is highly expressed in osteosarcoma cells, and is involved in the regulation of malignant characteristics, such as proliferation, apoptosis and migration of osteosarcoma cells. Moreover, hERG1 likely participates in these cellular processes through the NF-κB pathway. However, there were limitations to this study that should be noted. First, all work was performed in vitro. In vivo experiments are needed to verify these data. Second, the mechanism behind hERG1 regulation of the NF-κB pathway remains uncharacterized. Finally, there is still the possibility that other pathways are involved in the regulation of malignancy that may also be affected by hERG1-siRNA. Delineating the pathways besides the NF-κB pathway affected by hERG1 is the focus of our future studies.
Acknowledgements
This study was supported by Natural Science Foundation of China (No. 81402217).
Competing Interests
The authors declare that they have no competing interests.
References
1. Damron TA, Ward WG, Stewart A. Osteosarcoma, chondrosarcoma, and Ewing's sarcoma: National Cancer Data Base report. Clin Orthop Relat Res. 2007;459:40-47
2. Sluga M, Windhager R, Pfeiffer M. et al. Osteosarcoma and Ewing's sarcoma-The most frequent malignant bone tumors in children-therapy and outcome. Z Orthop Ihre Grenzgeb. 2002;140(6):652-655
3. Benayahu D, Shur I, Marom R. et al. Cellular and molecular properties associated with osteosarcoma cells. J Cell Biochem. 2001;84(1):108-114
4. Klein MJ, Siegal GP. Osteosarcoma: anatomic and histologic variants. Am J Clin Pathol. 2006;125(4):555-581
5. Panizo-Santos A, Sola I, Lozano M. et al. Metastatic osteosarcoma presenting as a small bowel polyp: a case report and review of the literature. Arch Pathol Lab Med. 2000;124(11):1682-1684
6. Longhi A, Errani C, De Paolis M. et al. Primary bone osteosarcoma in the pediatric age: State of the art. Cancer Treat Rev. 2006;32(6):423-436
7. Tan ML, Choong PF, Dass CR. Osteosarcoma: Conventional treatment vs. gene therapy. Cancer Biol Ther. 2009;8(2):106-117
8. Pardo LA, Contreras-Jurado C, Zientkowska M. et al. Role of voltage-gated potassium channels in cancer. J Membr Biol. 2005;205(3):115-124
9. Pardo L A, Stühmer W. The roles of K+ channels in cancer. Nat Rev Cancer. 2014;14(1):39-48
10. Camacho J. Ether a go-go potassium channels and cancer. Cancer lett. 2006;233(1):1-9
11. Wu X, Zhong D, Lin B. et al. p38 MAPK regulates the expression of ether a go-go potassium channel in human osteosarcoma cells. Radiol Oncol. 2013;47(1):42-49
12. Wu X, Zhong D, Gao Q. et al. MicroRNA-34a inhibits human osteosarcoma proliferation by downregulating ether a go-go 1 expression. Int J Med Sci. 2013;10(6):676-682
13. Warmke J W, Ganetzky B. A family of potassium channel genes related to eag in Drosophila and mammals. Proc Natl Acad Sci U S A. 1994;91(8):3438-3442
14. Nishikawa T, Hagihara K, Serada S. et al. Transcriptional complex formation of c-Fos, STAT3, and hepatocyte NF-1α is essential for cytokine-driven C-reactive protein gene expression. J Immunol. 2008;180(5):3492-3501
15. Rampe D, Murawsky M K, Grau J. et al. The antipsychotic agent sertindole is a high affinity antagonist of the human cardiac potassium channel HERG. J Pharmacol Exp Ther. 1998;286(2):788-793
16. Arcangeli A, Rosati B, Crociani O. et al. Modulation of HERG current and herg gene expression during retinoic acid treatment of human neuroblastoma cells: potentiating effects of BDNF. J Neurobiol. 1999;40(2):214-225
17. Masi A, Becchetti A, Restano-Cassulini R. et al. hERG1 channels are overexpressed in glioblastoma multiforme and modulate VEGF secretion in glioblastoma cell lines. Br J Cancer. 2005;93(7):781-792
18. Lu Y, Zhao X, Luo G. et al. Thioredoxin-like protein 2b facilitates colon cancer cell proliferation and inhibits apoptosis via NF-κB pathway. Cancer lett. 2015;363(2):119-126
19. Hoesel B, Schmid J A. The complexity of NF-κB signaling in inflammation and cancer. Mol Cancer. 2013;12(86):1-15
20. Huang S, Pettaway C A, Uehara H. et al. Blockade of NF-kappaB activity in human prostate cancer cells is associated with suppression of angiogenesis, invasion, and metastasis. Oncogene. 2001;20(31):4188-4197
21. Crociani O, Zanieri F, Pillozzi S. et al. hERG1 channels modulate integrin signaling to trigger angiogenesis and tumor progression in colorectal cancer. Sci Rep. 2013;3:3308
22. Litan A, Langhans S A. Cancer as a channelopathy: ion channels and pumps in tumor development and progression. Front Cell Neurosci. 2015;9:86
23. Trudeau M C, Warmke J W, Ganetzky B. et al. HERG, a human inward rectifier in the voltage-gated potassium channel family. Science. 1995;269(5220):92-95
24. Cayabyab F S, Schlichter L C. Regulation of an ERG K+ current by Src tyrosine kinase. J Biol Chem. 2002;277(16):13673-13681
25. Jehle J, Schweizer P A, Katus H A. et al. Novel roles for hERG K+ channels in cell proliferation and apoptosis. Cell Death Dis. 2011;2:e193
26. Hernandez L, Park K H, Cai S Q. et al. The antiproliferative role of ERG K+ channels in rat osteoblastic cells. Cell Biochem Biophys. 2007;47(2):199-208
27. Cherubini A, Taddei G L, Crociani O. et al. HERG potassium channels are more frequently expressed in human endometrial cancer as compared to non-cancerous endometrium. Br J Cancer. 2000;83(12):1722-1729
28. Pillozzi S, Brizzi M F, Balzi M. et al. HERG potassium channels are constitutively expressed in primary human acute myeloid leukemias and regulate cell proliferation of normal and leukemic hemopoietic progenitors. Leukemia. 2002;16(9):1791-1798
29. Zhao J, Wei X L, Jia Y S. et al. Silencing of herg gene by shRNA inhibits SH-SY5Y cell growth in vitro and in vivo. Eur J Pharmacol. 2008;579(1-3):50-57
30. Wang H, Zhang Y, Cao L. et al. HERG K+ channel, a regulator of tumor cell apoptosis and proliferation. Cancer Res. 2002;62(17):4843-4848
31. Staudacher I, Jehle J, Staudacher K. et al. HERG K+ channel-dependent apoptosis and cell cycle arrest in human glioblastoma cells. PloS One. 2014;9(2):e88164
32. Lastraioli E, Guasti L, Crociani O. et al. hERG1 gene and HERG1 protein are overexpressed in colorectal cancers and regulate cell invasion of tumor cells. Cancer Res. 2004;64(2):606-611
33. Cherubini A, Hofmann G, Pillozzi S. et al. Human ether-a-go-go-related gene 1 channels are physically linked to β1 integrins and modulate adhesion-dependent signaling. Mol Biol Cell. 2005;16(6):2972-2983
34. Baldwin AS Jr. Series introduction: the transcription factor NF-κB and human disease. J Clin Invest. 2001;107(1):3-6
35. Siebenlist U, Franzoso G, Brown K. Structure, regulation and function of NF-kappaB. Annu Rev Cell Biol. 1994;10(1):405-455
36. Sasaki N, Morisaki T, Hashizume K. et al. Nuclear factor-κB p65 (RelA) transcription factor is constitutively activated in human gastric carcinoma tissue. Clin Cancer Res. 2001;7(12):4136-4142
37. Viatour P, Merville M P, Bours V. et al. Phosphorylation of NF-κB and IκB proteins: implications in cancer and inflammation. Trends Biochem Sci. 2005;30(1):43-52
Author contact
![]() Corresponding author: Jin Wu, Department of Orthopaedics, the Affiliated Southeast Hospital of Xiamen University, Orthopaedic Center of People's Liberation Army, Zhangzhou, 363000, People's Republic of China. Tel: +86 596 2931538; Fax: +86 596 2931538; Emails: wuxinyu0102com.
Corresponding author: Jin Wu, Department of Orthopaedics, the Affiliated Southeast Hospital of Xiamen University, Orthopaedic Center of People's Liberation Army, Zhangzhou, 363000, People's Republic of China. Tel: +86 596 2931538; Fax: +86 596 2931538; Emails: wuxinyu0102com.