Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2016; 7(6):633-649. doi:10.7150/jca.12663 This issue Cite
Review
Understanding and Targeting MET Signaling in Solid Tumors - Are We There Yet?
Witthawat Ariyawutyakorn1,3, Siriwimon Saichaemchan2,3, Marileila Varella-Garcia3 ![]()
1. Faculty of Medicine, Chiang Mai University, 110 Intavarorod Rd., Muang, Chiang Mai, Thailand 50200.
2. Division of Oncology, Department of Medicine, Phramongkutklao Hospital and College of Medicine, 315 Phayathai Rd., Ratchathewi, Bangkok, Thailand 10400.
3. Department of Medicine, University of Colorado, Anschutz Medical Campus, 12801 East 17th Ave, RC1 South, L18-8118, Mail Stop 8117, Aurora, Colorado, USA 80045.
Received 2015-5-11; Accepted 2016-1-22; Published 2016-3-20
Abstract
The MET signaling pathway plays an important role in normal physiology and its deregulation has proved critical for development of numerous solid tumors. Different technologies have been used to investigate the genomic and proteomic status of MET in cancer patients and its association with disease prognosis. Moreover, with the development of targeted therapeutic drugs, there is an urgent need to identify potential biomarkers for selection of patients who are more likely to derive benefit from these agents. Unfortunately, the variety of technical platforms and analysis criteria for diagnosis has brought confusion to the field and a lack of agreement in the evaluation of MET status as a prognostic or predictive marker for targeted therapy agents. We review the molecular mechanisms involved in the deregulation of the MET signaling pathway in solid tumors, the different technologies used for diagnosis, and the main factors that affect the outcome, emphasizing the urge for completing analytical and clinical validation of these tests. We also review the current clinical studies with MET targeted agents, which mostly focus on lung cancer.
Keywords: MET, HGF, FISH, IHC, molecular testing, targeted therapy.
Introduction
With the continuum of research on the biology of human neoplasms, the therapeutic options in cancer have dramatically progressed over the last decade from cytotoxic and cytostatic regimens to molecular targeted agents, which overall provide better efficacy and less toxicity. The discovery of activation mechanisms in receptor tyrosine kinases (RTKs), such as the epidermal growth factor receptor (EGFR) mutations and the anaplastic lymphoma kinase (ALK) fusions, led to the development of successful targeted therapeutic agents, which are the proof of the ongoing revolution in cancer treatment [1-3]. Among the genes coding numerous human RTKs, the MET proto-oncogene (MET) is considered one of the most critical regulators of an extensive signaling pathway relevant in normal development and carcinogenesis [4-7].
The human gene MET is located on chromosome 7q31.2. It was identified as a proto-oncogene in a human osteogenic sarcoma cell line in 1984 and in 1987 found to encode an RTK called MET or c-Met [4,5,8]. The gene encoding its ligand protein, the hepatocyte growth factor (HGF), is located on chromosome 7q21.1 and was discovered in two independent approaches, as HGF in 1989 and as the scatter factor (SF) in 1989, which later were found to be the same protein [9].
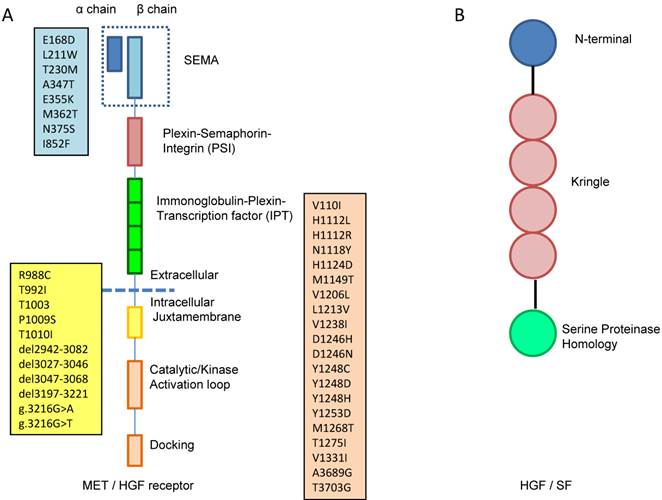
In general, receptor tyrosine kinases (RTKs) contain an N-terminal extracellular binding domain, a single transmembrane α helix, and a cytosolic C-terminal domain with tyrosine kinase activity. MET is a disulfide linked heteroimeric RTK consisting of an extracellular α chain, a β chain that encompasses the remainder of extracellular domain, the juxtamembrane and the kinase domains. As illustrated in Figure 1A, the extracellular component groups several domains including a large N-terminal semaphorin (Sema) domain (exon 2), a plexin-semaphorin-integrin (PSI) domain, and a stalk structure consisting of four immunoglobulin-plexin-transcription factor (IPT) domains. The intracellular component contains a juxtamembrane region responsible for signal downregulation and receptor degradation, a catalytic region with the enzyme activity, and a C-terminal region acting as a docking site for adaptor proteins such as GRB2 and GAB1, which leads to downstream signaling via PI3K, STAT and MAPK [10].
A. MET is a disulfide linked heteroimeric RTK consisting of an extracellular α chain, a β chain that encompasses the remainder of extracellular domain, the juxtamembrane and the kinase domains. The extracellular component groups several domains including a large N-terminal semaphorin (Sema) domain (exon 2), a plexin-semaphorin-integrin (PSI) domain, and a stalk structure consisting of four immunoglobulin-plexin-transcription factor (IPT) domains. The intracellular component contains a juxtamembrane region (exon 14), a catalytic region with the enzyme activity, and a C-terminal region (exon 15-21) acting as a docking site for adaptor proteins. The locations of gene mutation are shown in the boxes, the blue box lists the locations of mutation in sema domain (exon2), the yellow box lists the locations of mutation in juxtamembrane (exon 14) and the pink box lists the locations of mutation in tyrosine kinase domain (exon15-21). B. HGF consists of six domains including an N-terminal domain, four kringle domains and a C-terminal domain which is a serine proteinase homology (SPH) domain.
HGF, the only natural ligand of MET, is a large, disulfide, multidomain protein belonging to the plasminogen family. As shown in Figure 1B, HGF consists of six domains including an N-terminal domain, four kringle domains and a C-terminal domain which is a serine proteinase homology (SPH) domain [9,11]. HGF is secreted from mesenchymal cells as an inactive, single chain precursor and is converted to an active two-chain heterodimer by one of three serine proteinases: the soluble HGF activator, and the type II transmembrane enzymes matriptase and hepsin [12,13].
MET Signaling
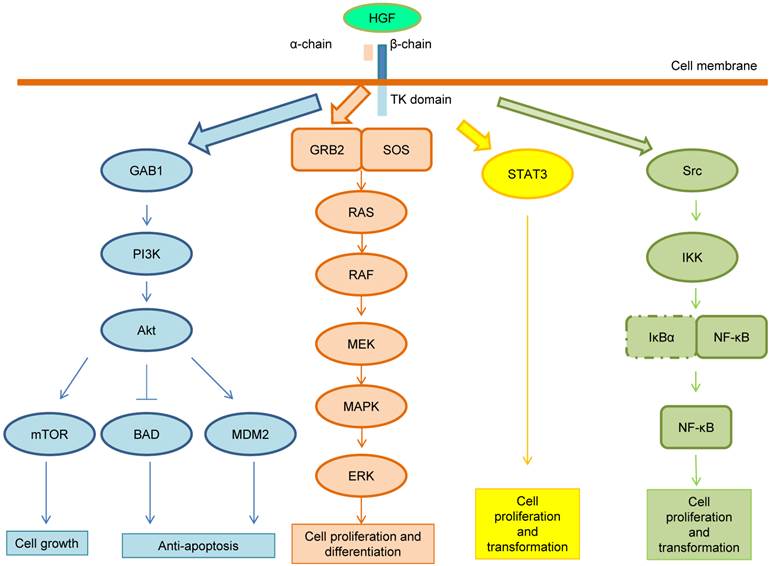
Following the HGF binding, the kinase activity of MET is switched on by receptor dimerization and trans-phosphorylation of two catalytic tyrosine residues (Tyr1234 and Tyr1235) within the kinase activation loop, then by phosphorylation of two additional docking tyrosine kinases in the C-terminal tail (Tyr1349 and Tyr1356). These tyrosines recruit signaling effectors including the adaptor proteins growth factor receptor-bound protein (GRB2), Src homology-2-containing (SHC), v-crk sarcoma virus CT10 oncogene homolog (CRK), and CRK-like (CRKL) [6,14]. Moreover, the adaptor protein GRB2-association binding protein 1 (GAB1) can bind either to MET directly or indirectly through GRB2 [6]. Phosphorylation of these different effectors initiate important downstream signaling pathways for cell proliferation, morphogenesis, survival, migration and angiogenesis, such as the cascades related to PI3K-Akt, RAS-MAP kinase, STAT3 and nuclear factor-κB (NF-κB) complex [6,14,15]. These pathways are summarized in Figure 2. After MET activation, PI3K can bind either to MET directly or indirectly with GAB1 and signals through the AKT/protein kinase B axis. AKT can inactivate the pro-apoptotic protein BCL-2 antagonist of cell death (BAD) and activate the E3 ubiquitin-protein ligase MDM2, which results in apoptosis suppression and cell survival promotion [16]. Moreover, AKT can also activate mammalian target of rapamycin (mTOR), which stimulates protein synthesis and cell growth. MET activation also signals through the RAS-MAPK pathway. The nucleotide exchanger protein Son of Sevenless (SOS) activates RAS by binding with SHC and GRB2 (GRB2-SOS complex). This leads to activation of the v-raf murine sarcoma viral oncogene homolog B1 (RAF) kinases, which subsequently stimulates the MAPK effector kinase (MEK) and results in MAPK activation. MAPK phosphorylates,the final effector of the cascade, ERK. The RAS-MAPK pathway is responsible for cell proliferation, cell motility and cell cycle progression [17]. A third major downstream axis of MET signaling is the Signal transducer and activator of transcription 3 (STAT3) pathway. STAT3 can bind to MET directly resulting in STAT3 phosphorylation, which regulates cell transformation, tubulogenesis and invasion [10]. Finally, the fourth major cascade of MET activation is through the IκBα-NF-κB complex. NF-κB is bound to IκBα forming an inactive form. The phosphorylation of either PI3K-AKT or SRC activates IκB kinase (IKK) and results in degradation of IκBs. NF-κB is then released and translocated to the nucleus to stimulate gene transcription [15].
MET signaling pathway. After MET activation, PI3K can bind either to MET directly or indirectly with GAB1 and signals through the AKT/protein kinase B axis. AKT can inactivate the pro-apoptotic protein BCL-2 antagonist of cell death (BAD) and activate the E3 ubiquitin-protein ligase MDM2, which results in apoptosis suppression and cell survival promotion. Moreover, AKT can also activate mammalian target of rapamycin (mTOR), which stimulates protein synthesis and cell growth. MET activation also signals through the RAS-MAPK pathway. The nucleotide exchanger protein Son of Sevenless (SOS) activates RAS by binding with SHC and GRB2 (GRB2-SOS complex). This leads to activation of the v-raf murine sarcoma viral oncogene homolog B1 (RAF) kinases, which subsequently stimulates the MAPK effector kinase (MEK) and results in MAPK activation. MAPK phosphorylates ERK, which is the final effector of the cascade. The RAS-MAPK pathway is responsible for cell proliferation, cell motility and cell cycle progression. A third major downstream axis of MET signaling is the Signal transducer and activator of transcription 3 (STAT3) pathway. STAT3 can bind to MET directly resulting in STAT3 phosphorylation, which regulates cell transformation, tubulogenesis and invasion. Finally, the fourth major cascade of MET activation is through the IκBα-NF-κB complex. NF-κB is bound to IκBα forming an inactive form. The phosphorylation of either PI3K-AKT or SRC activates IκB kinase (IKK) and results in degradation of IκBs. NF-κB is then released and translocated to the nucleus to stimulate gene transcription.
Independently of the HGF-dependent pathway, MET may be upregulated and become overexpressed by growth factors and cytokines such as PAX5, PAX8 and hypoxia-inducible factor (HIFα) [18-20]. Conditions such as hypoxia induce overexpression of both HGF and MET [20]. The downregulation of MET is initiated after ligand activation by internalization of MET receptor through endocytosis. The internalized receptor is ubiquitinized by the binding of casitas B-lineage lymphoma (CBL) at the juxtamembrane domain and is then degraded in the lysosome. The ubiquitin E3 ligase CBL also initiates early endocytosis by interaction with CBL-interacting protein 85, which enhances the adaptation of cell membrane for early endocytosis [21]. Another mechanism of downregulation of MET is a two-step proteolytic cleavage. The first step is cleavage of the extracellular domain by a disintegrin and metalloprotease (ADAM)-like receptor protease, which in turn creates a decoy extracellular domain MET fragment to block activation of other MET ligands; the second step is cleavage of intracellular domain by γ-secretase into fragment subsequently degraded by proteasome [22].
Overall, the physiologic function of MET pathway involves cell proliferation, cell motility, and morphogenesis and organ regeneration. During embryonal development, MET has a crucial role in proliferation of hepatocytes and placental trophoblasts. Ablation of the MET gene causes impaired development of liver and placenta leading to death in utero [4]. During development, MET induces migration of progenitor cell to form the hypaxial muscle and neurons [4]. MET and HGF also play an important role in healing process. MET and HGF are upregulated in the response of inflammation and injury. Overexpression of MET and HGF is observed in organ injuries such as liver, kidneys, heart and skin from toxins or chemicals and damage. HGF is secreted from mesenchyme after hepatectomy and induces MET downstream signaling in hepatocytes and result in liver regeneration and an increase size of liver. MET ablational mice had impaired liver regeneration. MET pathway plays a protective role against tubular necrosis of kidneys [4], myocardial injury after ischemic or reperfusion injury, and administration of recombinant HGF can reduce the area of myocardial infarction [23]. When wound occurs, HGF and MET are expressed in keratinocytes and stimulate the wound healing [24].
Over-activation of MET pathway induces cell overgrowth and invasion. Several preclinical studies have provided evidence of MET deregulation in carcinogenesis. Invasive activity of HGF was shown by Rong et al. [25], with MET- transformed NIH 3TC cells displaying motility activity in the absence of HGF while MET-transfected (MT) cells, transformed by a non-autocrine mechanism and used as control, did not show migration activity. When HGF was introduced, MT cells showed dramatic migration. Transgenic mice with MET cDNA developed hepatocellular carcinoma (HCC) which regressed after the transgene was inactivated [26]. Transduced MET oncogene introduced to mouse liver progenitor cells induced phenotypic changes, characterized by increased proliferation rate, loss of contact inhibition and formation of transformation foci. Transplant of the MET transduced cells into the spleen of immune-deficient mice led to colonization of spleen and liver with tumors similar to HCC [27].
MET pathway also promotes angiogenesis, which has an important role in wound healing and tumor development, by upregulation of vascular endothelial growth factor (VEGF) and downregulation of thrombospondin-1 (TSP1) [28]. Crosstalk of MET with other cell surface proteins (CD44, α6β4Integrin, SEMA4D and Plexin B1) has been proposed as mechanism to promote cell motility, invasion and metastasis [29], crosstalk with G protein receptors such as EGFR and HER2 to promote downstream signaling [7] and crosstalk with FAS ligand to promote anti-apoptosis [30]. These aforementioned interactions between MET and other proteins have a demonstrated contribution to carcinogenesis and drug resistance in in vitro studies [31].
Molecular Mechanisms of MET Activation in Carcinogenesis
Deregulation of the HGF-MET cellular axis in cancer can be detected at different molecular levels such as by changes in extent of protein expression, by variation in gene copy number and by presence of gene mutations. Each of these levels may be explored by different technologies, including immunohistochemistry staining (IHC) and enzyme-linked immunosorbent assays (ELISA) for evaluation of protein expression, and SNP arrays, fluorescence in situ hybridization (FISH), and PCR-based or sequencing-based techniques for evaluation of genomic status.
HGF and MET Overexpression
HGF is secreted either by primary and metastatic tumors as an autocrine mechanism or by mesenchymal cells as a paracrine or combined auto-para mechanisms [32]. Olivera et al reported a 10 to 100 fold increase in HGF and a 2 to 10 fold increase of MET in NSCLC compared to normal lung [33]. As shown in Table 1, levels of HGF expression have been sporadically accessed by either ELISA or IHC in bladder [34-36], breast [37-39], colorectal [40,41], head and neck [42,43], and lung cancers [31,32,44]. IHC was the common technique used to determine MET expression. Overexpression of the MET protein has been more commonly investigated in various solid tumors as seen in Table 2, with studies in bladder [45], breast [37-39, 46-51], colorectal [49,52-56], gastric [57], hepatobiliary [58], head and neck[42,43], lung [32,49,59-61], ovarian [49] and renal [49,62] cancers.
HGF protein expression in solid tumors.
| Tumor Type | Diagnostic Method | Antibody - Clone; Manufacturer | Dilution | Interpretation for Positive | Positive/ Informative Cases (%) | Overexpression Prognosis | Reference |
|---|---|---|---|---|---|---|---|
| Bladder | IHC | H4, Otsuka Pharmaceutical | 1:30 | >10% of cells with cytoplasmic staining | nodular - 5/27 (18%) | More frequent in papillary (more invasive) type (p<0.001) | Li [34] |
| papillary - 33/33 (100%) | |||||||
| ELISA | Not reported | Not reported | Continuous analysis | Not reported | Higher level in muscle-invasive tumor (p<0.001) | Wang [35] | |
| ELISA | Not reported | Not reported | >1820 pg/mg protein | 232/240 (97%) | Not reported | Eissa [36] | |
| Breast | ELISA | Not reported | Not reported | Continuous analysis | Not reported | Shorter disease relapse (p<0.001) and OS (p<0.001) | Nagy [37] |
| IHC | R & D Systems | 1:50 | ≥10% of both cancer and stromal cells/matrix - no intensity specified | 64/88 (73%) | Lower 10-year survival rate (p = 0.0096) | Edakuni [38] | |
| IHC | R & D Systems | Not reported | >10 % of tumor cells with moderate +2 and intense +3 staining | 147/323 (46%) | NS for OS | Kang [39] | |
| Colorectal | ELISA | Not reported | Not reported | Serum HGF > 0.37 ng/ml | 67/184 (36%) | HR for OS 3.1 (p=0.0001) | Toiyama [40] |
| ELISA | Not reported | Not reported | Serum HGF ≥1393.55 pg/ml | 52/103 (50%) | PFS (p<0.001), OS (p = 0.002) | Takahashi [41] | |
| HNSCC | IHC | H145, Santa Cruz Biotechnology | 1:50 | any tumor cell ≥+1 stain | 58/68 (85%) | Not reported | Seiwert [43] |
| Hypopharynx | IHC | R&D Systems | 50mcg/ml | >30% of cancer cells with moderate +2 and intense +3 staining | 31/40 (78%) | OS (p = 0.07), Recurrence rate (p=0.22), Advanced stage (p=0.016) | Kim [42] |
| Lung Adenocarcinoma | IHC | Immuno Biological Laboratories | 5mcg/ml | ≥50% of tumor cells positively stained | 5/16 (31%) | Not reported | Yano [31] |
| IHC | SC-7949, Santa Cruz Biotechnology | 1:100 | Allred score : (positive 3-8 score) | 104/183 (57%) | RR for OS 1.5 (p = 0.012) | Onitsuka [44] | |
| Lung Non-Small Cell | IHC | SC-7949, Santa Cruz Biotechnology | 1:100 | ≥50% of tumor cells positively stained | 14/88 (16%) | NS for OS | Masuya [32] |
ELISA; enzyme-linked immunosorbent assay, IHC; immunohistochemistry, NS; not significant, RR; risk ratio, OS; overall survival.
MET protein expression in solid tumors.
| Tumor Type | Diagnostic Method | MET antibody - Clone, Manufacturer | Dilution | Interpretation for Positive | Positive/ Informative Cases (%) | Overexpression Prognosis | Reference |
|---|---|---|---|---|---|---|---|
| Bladder | IHC | AC, Zymed Laboratories Inc | Not reported | > 50% of tumor cells with strong staining intensity | 37/133 (28%) | HR 3.76 (p=0.041) | Miyata [45] |
| Breast | Western blot | Not reported | Not reported | Any detectable protein | 26/73 (36%) | Disease relapse (p=0.012) | Nagy [37] |
| IHC | Santa Cruz Biotechenology Inc | 1:250 | >70-80% intense any reactivity | 20/91 (22%) | HR for survival 3.27 (p=0.02) | Ghoussoub [46] | |
| IHC | Santa Cruz Biotechenology Inc | 1:200 | ≥30% of area of slide | 64/88 (73%) | Lower 10-year survival rate 61.5% vs 97.9% (p= 0.0096) | Edakuni [38] | |
| IHC | 3D4; Zymed Laboratories | Not reported | moderate +2 and intense +3 staining | 91/320 (28%) | Relative Risk 1.862 (p=0.0064) | Kang [39] | |
| IHC | AC, Zymed Laboratories | 1:1 | +3 staining | 71/324 (22%) | Relative Risk 2.04 (p=0.0035) | Tolgay Ocal [47] | |
| IHC | Santa Cruz Biotechenology Inc | 1:100 | > 5% of tumor cells with intense cytoplasmic or membranous reactivity | Early stage - 23/51 (45%) | More frequent in metastasis (p=0.0002), Lower 10 yr-DFS rate 35.4% vs 73.7% (p = 0.0004) | Lee [48] | |
| Metastasis - 42/52 (81%) | |||||||
| IHC | Zymed-Invitrogen | Not reported | +2 and +3 staining | 6/37 (16%) | Not reported | Ma [49] | |
| IHC | Novocastra Laboratories | Not reported | IRS Remmele score : High (9-12 score) | 80/302 (27%) | Better prognosis (p=0.0089) | Gisterek [50] | |
| IHC | SP44, Novus Biological | 1:50 | > 25% positive tumor cells (no intensity specified) | 52/97 (54%) | More frequent in grade 3 tumor (p=0.004); OS (p=0.9) | Inanc [51] | |
| Colorectal | IHC | Zymed-Invitrogen | Not reported | +2 and +3 staining | 31/40 (78%) | Not reported | Ma [49] |
| IHC | Invitrogen | 1:300 | >200 based on H-score criteria | 274/339 (81%) | OS (p=0.889) | Uddin [52] | |
| IHC | Novocastra Laboratories | 1:30 | >10% of cells positive with ≥+1 (0 - 3 intensity) | 131/183 (72%) | More frequent in higher stage (p=0.02) | Garouniatis [53] | |
| IHC | C28, Santa Cruz Biotechenology | 1:100 | +3 (0 - 3 intensity) | Primary - 8/79 (10%) | Shorter survival HR 4.6 (p<0.0001) | Voutsina [54] | |
| Metastasis - 14/76 (18%) | |||||||
| IHC | SP44, Spring Biosciences | Not reported | 4 or greater score (intensity 0-3+ %of cells 1 (<25%) 2(25-50%) 3(>50%)) | 36/75 (48%) | Shorter PFS HR1.46 (p=0.018) | Kishiki [55] | |
| IHC | SP44 (CONFIRM), Ventana MedicalSystems | Not reported | +2 and +3 staining | 56/108 (52%) | Shorter RFS 9.7 vs 21.1 mo. (p = 0.013) | Shoji [56] | |
| Gastric | IHC | SP44 (CONFIRM), Ventana MedicalSystems | Prediluted | > 50% of tumor cells with moderate 2+ or strong 3+ intensity | 22/229 (10%) | Shorter PFS (p <0.001) and OS (p <0.001) | An [57] |
| Hepatocellular | IHC | 80/297 (28%) | Not significant for Survival | Lee [58] | |||
| HNSCC | IHC | CVD-12V | 1:100 | moderate +2 and intense +3 staining | 84/97 (85%) | Not reported | Seiwert [43] |
| Hypopharynx | IHC | R&D Systems Inc | 0.625 mcg/ml | > 30% of cells with moderate and strong intensity | 28/40 (70%) | More frequent in lymphnode met and advanced stage, OS (p>0.05) | Kim [42] |
| Lung | IHC | Zymed-Invitrogen | Not reported | +2 and +3 staining | 16/40 (40%) | Not reported | Ma [49] |
| Lung Adenocarcinoma | IHC | C28, Santa Cruz Biotechenology | 1:100 | > 50% of tumor cells with moderate 2+ or strong 3+ intensity | 33/110 (30%) | Not reported | Xia [59] |
| Lung Non-Small Cell | IHC | SC-10, Santa Cruz Biotechenology | 1:100 | > 50% of tumor cells with > grade 1 intratumoral staining | 36/88 (41%) | Relative Risk 2.64 (p=0.0029) for 5-yr survival | Masuya [32] |
| IHC | SP44 (CONFIRM), Ventana Med Systems | Not reported | > 50% of tumor cells with moderate 2+ or strong 3+ intensity | 210/1048 (20%) | HR for PFS 0.72 (p=0.014), HR for OS 0.70 (p=0.03) | Scagliotti [60] | |
| IHC | SP44 (CONFIRM), Ventana Med Systems | Not reported | > 50% of tumor cells with moderate 2+ or strong 3+ intensity | 66/128 (52%) | Good predictive factor for onartuzumab plus erlotinib, OS HR 0.42 (p=0.01), PFS (p = 0.69) | Spigel [61] | |
| Ovarian | IHC | Zymed-Invitrogen | Not reported | +2 and +3 staining | 12/40 (30%) | Not reported | Ma [49] |
| Renal Cell | IHC | AC, Zymed Laboratories | 1:350 | >50% of cancer cells with higher staining than normal kidney | 73/114 (64%) | OR for cause specific survival 2.94 (p=0.028) | Miyata [62] |
| IHC | Zymed-Invitrogen | Not reported | +2 and +3 staining | 28/40 (70%) | Not reported | Ma [49] |
IHC; immunohistochemistry, HR; hazard ratio, OS; overall survival, PFS; progression-free survival, RFS; relapse-free survival, OR; odd ratio.
High levels of HGF and MET proteins have been reported in solid tumors, although in different frequencies in distinct tumor types. Tumor progression may be a differential factor in protein overexpression, but studies are not always concordant. Lee et al. [48] found MET overexpression in 45% of primary and in 81% of metastatic breast tumors while Voutsina et al. [54] found MET overexpression in 10% of primary and 18% of metastatic colorectal carcinomas (CRC).
However, there is also a large variation within specific tumor types that may be due to technical features such as reagents and experimental conditions used in the assay as well as to the qualitative nature of the criteria considered for diagnosis [32,38,42,43,48,53,55,61,62]. As seen in Tables 1 and 2, consensus for qualitative diagnosis of overexpression of HGF and MET has not been established yet. Several studies did not provide enough technical details for a comparison and even for studies in which data are available it is not easy to reconcile the different results. For instance, both Toiyama et al. [40] and Takahashi et al. [41] derived the cut-off value for quantification in high or low HGF expressors in the ELISA assays from the HGF level in the serum of their Japanese CRC patients usingROC curves to determine the cut-off for normal serum level. However, Toiyama et al. reached the cut-off of 0.37 ng/ml, which was surpassed by 36% of patients, while Takahashi obtained the cut-off of 1.39 ng/ml, which was surpassed by 50% of patients. Despite these discrepancies, both studies similarly concluded that high level of serum HGF conferred poorer survival to the patients.
The effects of using different assay reagents and specific criteria for determination of positivity for overexpression are obvious in the breast cancer studies, in which frequencies of MET overexpression varied from 22%46 to 73% [38]. Ghoussoub et al. [46] considered overexpressors (positive) the specimens displaying any reactivity in >70-80% of tumor cells whereas Edakuni et al. [38] considered any reactivity in ≥30% of the area. Therefore, it would be reasonable to expect that the frequency of cases with MET overexpression would be higher in the later (73% versus 22%). Similar effect occurred in CRC comparing Voutsina et al. [54], Ma et al. [49] and Shoji et al. [56] studies. In NSCLC, MET overexpression was reported by Spigel et al. [61] in 52% and by Masuya et al. [32] in 41% of the patients. These studies used different scoring criteria, however surprisingly the less stringent criteria (> grade 1 intensity in ≥50% of tumor cells) detected lower frequency of expressors than the more stringent (moderate 2+ or higher intensity in ≥50% of tumor cells). This could be due to the different reagents used, since some antibodies have higher level of efficacy for protein detection. Xia et al.59 using similar criteria to Spigel et al. [61] but a different antibody detected lower frequency of MET overexpression (30%) in Chinese patients with lung adenocarcinoma.
Few studies have applied semi-quantitative criteria to measure MET expression, such as the Allred score [63], the Immunoreactive-Remelle score (IRS) [64], or H-score [65], which are expected to perform better since they are less arbitrary in nature. Only one study [44] used Allred score to access HGF expression in lung adenocarcinoma and one study [50] has assessed MET expression level in breast cancer using IRS. The H-score has been widely employed to evaluate levels of hormone receptor in breast cancer [66] and EGFR in NSCLC [67] but has been applied to determine MET expression in only one study in CRC [52].
The variety of methodologies and classification criteria within the given methodologies used for definition of MET and HFG overexpression in the literature has created a set of circumstances that made it impossible to determine which molecular assay is best suitable to achieve this purpose. Consequently, the use of these variables as biomarkers for selecting cancer patients for novel MET targeted therapies has been significantly compromised as further discussed ahead.
MET Gene Mutation and Amplification
At least 30 distinct missense mutations in the MET gene have been found as somatic and germline events in cancer patients, and they are located mainly in three genomic areas: (a) the exon 2, which encodes the SEMA domain essential for dimerization [68], (b) the exon 14, which encodes the CBL-binding docking in the juxtamembrane domain that leads to ubiquitination and receptor degradation [69], and (c) the exons 15 to 21, which encompass the tyrosine kinase domain (Table 3, Figure 1A).
MET mutations in solid tumors.
| Types of cancer | Specimen | Technique | Positive/Case (%) | Reference |
|---|---|---|---|---|
| Breast | tissue | PCR | 1/30 (3%) | Lee [83] |
| tissue | targeted NGS/Sequenom Mass ARRAY/PCR-based extension assay | 8/88 (9%) | de Melo Gagliato [85] | |
| Colorectal | tissue | PCR | 0/20 (0%) | Schmidt [79] |
| tissue | qRT-PCR | 18/299 (6%) | Neklason [82] | |
| Gastric | tissue | PCR | 1/85 (1%) | Lee [83] |
| cell lines | PCR | 1/4 (25%) | Asaoka [69] | |
| HNSCC | tissue | RT-PCR | 4/15 (27%) | Di Renzo [71] |
| tissue | qRT-PCR | 9/66 (14%) | Seiwert [43] | |
| cell lines | 1/8(13%) | |||
| All | 10/74 (14%) | |||
| Lung adeno | tissue | WES | 16/230 (7%) | TCGA [76] |
| Lung non-small cell | tissue | PCR | 10/127 (8%) | Ma [74] |
| tissue | Multiplex PCR | 27/283 (10%) | Krisnaswamy [68] | |
| cell lines | 5/74 (7%) | |||
| All | 32/357 (9%) | |||
| tissue | RT-PCR | 7/262 (3%) | Onozato [75] | |
| Lung small cell | tissue | PCR | 4/32 (17%) | Ma [73] |
| Ovarian | tissue | targeted NGS/Sequenom Mass ARRAY/PCR-based extension assay | 9/122 (7%) | Tang [86] |
| Prostate | tissue | targeted NGS/Sequenom Mass ARRAY/PCR-based extension assay | 1/15 (7%) | Jardim [81] |
| Renal cell | tissue | targeted NGS/Sequenom Mass ARRAY/PCR-based extension assay | 2/10 (10%) | Jardim [81] |
| Renal cell, papillary, hereditary | tissue | PCR | 4/7 (57%) | Schmidt [70] |
| Renal cell, papillary, sporadic | tissue | PCR | 17/129 (13%) | Schmidt [79] |
WES; whole exome sequencing, PCR; polymerase chain reaction, RT-PCR; real time-polymerase chain reaction, qRT-PCR; quantitative real time-polymerase chain reaction.
MET mutations in solid tumors were first reported by Schmidt in hereditary papillary renal cell carcinoma (RCC) [70]. The surge of high resolution technologies for mutation detection such as massive parallel and target next generation sequencing so far has not impacted the knowledge in this field but is expect to contribute in the near future. Nowadays these mutations have been reported in HNSCC [43,71,72], lung [68,73-76], papillary RCC [77-81], CRC [82], gastric [69,83,84], breast [83,85], ovarian [86] and prostate [81] cancers (Table 3). Highest frequencies of MET mutations were found in hereditary and sporadic papillary RCC (57%) and HNSCC (27%). In HNSCC, missense mutations in the MET TK domain have been reported more frequently in metastases, increased from 2-6% in primary tumors to 50% in metastatic lymph nodes, supporting the hypothesis that MET mutations occur during the progression of tumorigenesis [71]. The recently identified mutations in exon 14 caused by nucleic acid deletions lead to splicing of RNA transcription and abnormal CBL-binding [43,73-75,82,87]. Kong-Beltren et al. [87] found that lung tumors harboring exon 14 splicing mutations had strong MET expression by IHC, decreased ubiquination and degradation and prolonged cell signaling. MET gene splice mutation was reported in 1% [74] to 2.7% [75] of lung cancer patients.
The MET pathway may also be activated by gain in the MET gene copy number per cell or gene amplification. Amplification of the MET gene has been investigated, albeit scarcely, in tumor types such as adrenocortical, biliary tract, breast, colorectal, gastric, genitourinary and HNSCC, and much more extensively in NSCLC, as summarized in Table 4. The frequencies of MET amplification in the listed studies are largely variable and the main factors accounting for this variation are both biological and technical in nature. The relevant biological variables include patient ethnicity and cancer treatment received pre-molecular testing, and enrichment of the selected cohort for clinical, demographic and histological characteristics. The technical variables mainly include differences in the sensitivity and specificity of the technical platforms used and in the selected diagnostic criteria.
MET gene amplification in solid tumors.
| Tumor Type | Cohort Type | Diagnostic Method | Reagent | Interpretation for Positive | Positive/Informative Cases (%) | Amplification Prognosis | Reference |
|---|---|---|---|---|---|---|---|
| Adrenocortical | untreated and previously treated (with chemotherapy and/or TKI) | FISH | Not reported | MET/CEP7 >2 or >20 copies of MET in >10% of tumor cells | 2/13 (15%) | NS in OS | Jardim [88] |
| Biliary tract | untreated and previously treated (with chemotherapy and/or TKI) | FISH | Not reported | MET/CEP7 >2 or >20 copies of MET in >10% of tumor cells | 2/21 (10%) | NS in OS | Jardim [88] |
| Bladder | untreated and previously treated (with chemotherapy and/or TKI) | FISH | Not reported | MET/CEP7 >2 or >20 copies of MET in >10% of tumor cells | 1/10 (10%) | NS in OS | Jardim [88] |
| Breast | untreated and previously treated (with chemotherapy and/or TKI) | FISH | Not reported | MET/CEP7 >2 or >20 copies of MET in >10% of tumor cells | 3/63 (5%) | NS in OS | de Melo Gagliato [85] |
| untreated and previously treated (with chemotherapy and/or TKI) | FISH | Not reported | MET/CEP7 >2 or >20 copies of MET in >10% of tumor cells | 3/64 (5%) | NS in OS | Jardim [88] | |
| Colorectal | Primary | PCR | > mean + 2SD of normal tissue | 21/67 (31%) | Not reported | Voutsina [54] | |
| Metastasis | 21/62 (34%) | ||||||
| untreated and previously treated (with chemotherapy and/or TKI) | FISH | Not reported | MET/CEP7 >2 or >20 copies of MET in >10% of tumor cells | 4/208 (2%) | NS in OS | Jardim [88] | |
| Gastric | Cell lines | Southern blot | ≥3 fold increase of signal intensit than non-neoplastic mucosa | 6/11 (55%) | Not reported | Kuniyasu [103] | |
| Tumor - advanced stage | 15/64 (23%) | ||||||
| Tumor - early stage | 0/11 (0%) | ||||||
| Treatment naïve | CGH | Not reported | MET/MTHFR >1.5 | 6/58 (10%) | Not reported | Sakakura [104] | |
| Pre chemo | FISH | MET/Cen 7, Kreatech Diagnostics | MET/CEP7 >2 or >20 copies of MET in >10% of tumor cells | 19/227 (8%) | shorter PFS and OS (p <0.001) | An [57] | |
| untreated and previously treated (with chemotherapy and/or TKI) | FISH | Not reported | MET/CEP7 >2 or >20 copies of MET in >10% of tumor cells | 5/77 (6%) | NS in OS | Jardim [88] | |
| Genitourinary | untreated and previously treated (with chemotherapy and/or TKI) | FISH | Not reported | MET/CEP7 >2 or >20 copies of MET in >10% of tumor cells | 7/97 (7%) | HR for OS 2.8 (p = 0.034) | Jardim [81] |
| HNSCC | Cell lines | FISH | LTD, RP11-163C9/LTD, RP11-144B2 | copy number > 4 | 4/14 (29%) | Not reported | Seiwert [43] |
| Recurrent/Metastasis | PCR | copy number ≥10 | 18/23 (78%) | Not reported | |||
| Lung Adenocarcinoma | Pre EGFR TKI | aCGH | LTD, RP11-163C9/CEP7 Abbott Molecular | MET/MTHFR >1.5 | 2/62 (3%) | Not reported | Bean [99] |
| Acquired resistance to EGFR TKI | 9/43 (21%) | ||||||
| EGFR TKI resistant cell lines | PCR | Not reported | 4/18 (22%) | Not reported | Engelman [100] | ||
| Pre EGFR TKI | PCR | > mean + 2SD of normal tissue | 10/49 (20%) | NS in event-free survival (p=0.09) | Beau-Faller [89] | ||
| Acquired resistance to EGFR TKI | FISH | LSI D7S522 /CEP7 Abbott Molecular | MET/CEP7 > 2 or tight MET gene cluster or > 15 copies of MET/cell in > 10% of cells | 1/6 (17%) | Not reported | Jiang [93] | |
| Pre EGFR TKI | PCR | Not reported | 0/11 | Not reported | Yano [31] | ||
| Acquired resistance to EGFR TKI | 0/5 | ||||||
| Pre EGFR TKI | qRT-PCR | > mean + 2SD of normal tissue | 2/53 (4%) | Not reported | Chen [94] | ||
| Acquired resistance to EGFR TKI | 5/29 (17%) | ||||||
| Pre EGFR TKI | qPCR | copy number > 3 | 0/8 (0%) | Not reported | Onitsuka [91] | ||
| Acquired resistance to EGFR TKI | 0/10 (0%) | ||||||
| Treatment naïve | qRT-PCR | > mean + 2SD of normal tissue | 8/183 (4%) | NS in OS | Onitsuka [44] | ||
| Acquired resistance to EGFR TKI | FISH | LDT, RP11-163C9; Kreatech MET probe/CEP7 Abbot Molecular | MET/CEP7 > 2 | 4/37 (11%) | Not reported | Arcila [95] | |
| Pre EGFR TKI | qPCR | MET/MTHFR >1.5 | 12/139 (9%) | shorter OS (p=0.042) | Chen [101] | ||
| Acquired resistance to EGFR TKI | FISH | LDT, CTB12N12 and EGFR probe (CTD-2113A18) | ≥4 copies in ≥40% of cells or presence of MET cluster and ratio of MET/CEP7 ≥ 2 or ≥ 15 copies of MET/cell in ≥ 10% of analyzed cells | 2/37 (5%) | Not reported | Sequist [96] | |
| Pre EGFR TKI | qPCR | > mean + 2SD of normal tissue | 6/110 (4%) | Not reported | Xia [59] | ||
| Acquired resistance to EGFR TKI | FISH | LDT, RP11 163C9; Kreatech MET probe/CEP7 Abbott Molecular | MET/CEP7 > 2 | 4/75 (5%) | Not reported | Yu [97] | |
| untreated and previously treated (with chemotherapy and/or TKI) | FISH | Not reported | MET/CEP7 >2 or >20 copies of MET in >10% of tumor cells | 1/18 (6%) | NS in OS | Jardim [88] | |
| untreated and previously treated (with chemotherapy and/or TKI) | FISH | Not reported | MET/CEP7 > 2.2 | 13/733 (1.7%) | Not reported | Kris [98] | |
| Lung Non-Small Cell | Treatment naïve | FISH | LDT, RP11-95I20 and CEP7 (Abbott Molecular) | copy number ≥5 | 48/435 (11%) | higher grade (p=0.016), advanced stage (p=0.01) and shorter survival (p=0.005) | Cappuzzo [90] |
| Pre EGFR TKI | qPCR | copy number ≥5 | 2/100 (2%) | Not reported | Kubo [100] | ||
| untreated and previously treated (with chemotherapy and/or TKI) | FISH | LTD, CTB12N13 and CEP7 (Abott Molecular) | ≥4 copies in ≥40% of cells or presence of MET cluster and ratio of MET/CEP7 ≥ 2 or ≥ 15 copies of MET/cell in ≥ 10% of analyzed cells | 37/167 (26%) | NS in PFS with treatment of Tivantinib and Erlotinib | Sequist [92] | |
| untreated and previously treated (with chemotherapy and/or TKI) | FISH | Not reported | MET/CEP7 ≥1.8 | not provided | improved ORR with Crizotinib in patients with intermediate and high MET level (ratio >2.2 - < 5.0 and ≥ 5) | Camidge [105] | |
| Lung Squamous Cell | Pre EGFR TKI | PCR | > mean + 2SD of normal tissue | 12/57 (21%) | NS in event-free survival | Beau-Faller [89] | |
| Pre EGFR TKI | qPCR | MET/MTHFR >1.5 | 7/59 (12%) | shorter OS p=0.042 | Chen [102] | ||
| Melanoma | untreated and previously treated (with chemotherapy and/or TKI) | FISH | Not reported | MET/CEP7 >2 or >20 copies of MET in >10% of tumor cells | 2/61 (3%) | NS in OS | Jardim [88] |
| Ovarian | untreated and previously treated (with chemotherapy and/or TKI) | FISH | Not reported | MET/CEP7 ≥ 2 | 4/13 (4%) | NS in OS | Tang [86] |
| untreated and previously treated (with chemotherapy and/or TKI) | FISH | Not reported | MET/CEP7 >2 or >20 copies of MET in >10% of tumor cells | 4/110 (4%) | NS in OS | Jardim [88] | |
| Renal | untreated and previously treated (with chemotherapy and/or TKI) | FISH | Not reported | MET/CEP7 >2 or >20 copies of MET in >10% of tumor cells | 4/28 (14%) | NS in OS | Jardim [88] |
| Salivary gland | untreated and previously treated (with chemotherapy and/or TKI) | FISH | Not reported | MET/CEP7 >2 or >20 copies of MET in >10% of tumor cells | 1/30 (3%) | NS in OS | Jardim [88] |
aCGH; array comparative genomic hybridization, CGH; comparative genomic hybridization, FISH; fluorescent in situ hybridization, HR; hazard ratio, ORR; objective response rate, OS; overall survival, PCR; polymerase chain reaction, PFS; progression-free survival, qRT-PCR; quantitative real time-polymerase chain reaction, RT-PCR; real time-polymerase chain reaction.
In non-lung tumors, frequency of MET amplification ranged from ~5% in breast cancer [85,88] to ~78% in HNSCC [43]. In lung tumors, MET amplification ranged from 0% to 26% of patients, in non-comparative studies, there is no clear difference in patients never-treated with EGFR inhibitors from 0% to 26% [44,59,89-92] and EGFR TKI resistant patients from 1.7% to 21% [88,93-98]. Nevertheless, the difference became obvious in comparative studies of EGFR TKI naïve and resistant patients, with 3%-4% of MET amplification in EGFR TKI naïve patients and 17%-21% in EGFR TKI resistant patients [94,99]. These results corroborated the findings of Engelman et al. [100] that MET amplification is a mechanism of resistance to EGFR TKIs. Moreover, Chen et al. [101] also found that MET amplification was more frequent in chemotherapy-treated than chemotherapy-naïve lung adenocarcinoma patients (36% vs 7%, respectively) in a subgroup analysis. In EGFR TKI naïve patients, MET amplification rate was higher in Caucasian, ranging from 11%-21%[89,90,99], than in Asian patients, ranging from 2%-17% [59,94,102]. However, lung tumor histology, adenocarcinoma or squamous cell carcinoma, did not show association with MET amplification [89,101].
MET amplification has been detected in a variety of technical platforms. Few early studies have used southern blotting [103] or comparative genomic hybridization (aCGH) [104], while more recent investigations have used PCR-based or FISH techniques. The PCR-based and aCGH assays evaluate extracted DNA, thus the high proportion of non-tumor cells in the tissue sample analyzed may compromise the measurement. Conversely, FISH is an in situ assay and is not impacted by presence of non-tumor cells. FISH measures gene amplification usually in relation to an internal control; in the instance of MET amplification the internal control is the number of copies of chromosome 7 centromere. Both laboratory developed and commercially available DNA FISH probes have been used in the MET FISH assays, as shown in Table 4, but since these reagents should have been validated they would not influence the study conclusions. However, the criteria to classify a specimen as MET amplification positive by FISH were also highly variable. MET amplification has been defined by different cut-offs of the ratio MET/centromere 7 such as ≥1.8 [105], ≥2 [81,82]81,88 or ≥ 2.2 [105]. Other studies have classified tumors as MET positive not based on ratio against an internal control but based strictly on the mean MET copy number per cell. In that case, copy number gain has been defined as ≥4 copies in ≥40% of cells [106], ≥5 copies on average per tumor cell [90], ≥ 15 copies of MET in ≥ 10% of cells analyzed [106] or >20 copies of MET in >10% of cells analyzed [88]. Obviously, this variability may be responsible for a large proportion of the discordant results among studies. As noticeable from the above descriptions, MET copy number gain usually occurs at lower level than gene amplification. In the study conducted by Cappuzzo et al. [90], it was found that post-operative NSCLC had MET copy number gain rate of 11.1% but the true MET gene amplification rate was only 4.1%.
Prognostic and Predictive Value of the Aberrant MET Pathway
As summarized in Table 1, HGF overexpression was found in more invasive [34,35] or advanced stage [42] cancers and conferred poor prognosis [37,38,40,41]. Nagy et al. [37] measured serum HGF level in breast cancer patients using ELISA and found that high level of HGF was significantly related to shorter disease relapse time and shorter overall survival (OS). Li et al. [34] and Wang et al. [35] measured serum and urine HGF levels in bladder cancer patients and reported occurrence of highly invasive tumors in patients with higher HGF levels compared to those with lower HGF levels. Patients with high HGF overexpression detected by IHC also had significantly shorter progression-free survival (PFS) and OS in breast, CRC and hypopharynx cancers [38,40-42].
Similarly, high expression of MET was associated with advanced stage and poor prognosis in breast, bladder, colorectal , gastric, lung and RCC tumors [32,48,53,55] as listed in Table 2. Studies have shown that MET pathway was more often aberrant in advanced stage tumors and metastases rather than in early stage tumors [42,48,51,54,90] and aberrant MET pathway consistently associated with poorer patients' prognosis [37,40-42,47,54,62]. In breast cancer, most investigations concluded that MET overexpression was associated to lower survival rate. Lee et al. [48] found higher rate of MET overexpression in breast cancer patients with advanced stage and patients with MET overexpressed tumors also had shorter 10-year disease free survival (DFS) compared to non-MET overexpressors (35.4% versus 73.7%; p = 0.0004). Similar findings were reported by Voutsina et al. [54] in CRC, MET overexpression was found at two fold higher rate in metastatic lesions compared to primary lesions (18% vs 10%) and conferred shorter survival (HR 4.6 p<0.0001).
The knowledge of prognostic impact of MET mutation is limited and its predictive impact remains undetermined. For MET amplification, despite the numerous studies suggesting trends toward poor prognosis this outcome was only confirmed in two lung cancer studies, as shown in Table 4. In resected NSCLC, MET copy number gain/amplification was associated with higher tumor grade (p=0.016), advanced clinical stage (p=0.01) and shorter OS (p=0.05) [90,101]. MET amplification was also associated with shorter PFS and OS in gastric cancer (p<0.001 for PFS and OS) [57] and with shorter OS in genitourinary cancer (HR for OS 2.8 p=0.034) [81].
In summary, tumors harboring MET pathway deregulation detected either as overexpression of HGF and MET or as amplification of MET were associated with higher pathological grade, advanced clinical stage and more invasive features [34,35,51,90] These findings significantly support the conclusion that increase in the MET signaling is one of the most common poor prognostic markers in lung carcinomas and maybe also in other carcinomas.
The predictive value of MET pathway has only been explored in lung cancer and in two major settings, in relation to resistance to EGFR-TKI inhibitors and sensitivity to MET inhibitors. With the discovery of the activating mutations in the exons 19 and 21 of EGFR gene, EGFR-TKIs have become a successful therapeutic option for advanced NSCLC patients harboring these mutations. The response rate of EGFR mutant patients to EGFR TKIs is very high at 65% to 82% [1,2,107], but these patients commonly became resistant to therapy over a short period of time. MET amplification is among the principal mechanisms driving this acquired resistance, found in 15%-20% of EGFR TKI resistant patients [94,99,100]. Interestingly, MET deregulation may have a less prominent role in primary resistance to TKIs. In a small cohort, Chen et al. [94] found no impact of MET copy number on EFGR-TKI sensitivity of TKI-naïve lung adenocarcinoma patients, that is the high MET gene copy number (MET≥3 by qRT-PCR) in the diagnostic specimen has not affected the initial response to EGFR TKI treatment.
The efficiency of MET deregulation as biomarker for MET targeted therapies is less conclusive. Spigel et al. [61] conducted a phase II study in chemo-treated, EGFR-TKI naïve advanced NSCLC patients with the MET monoclonal antibody onartuzumab (METMab) in combination with erlotinib. The overexpression of MET was verified retrospectively by IHC. No difference in PFS and OS in the intention-to-treat (ITT) populations was found but addition of onartuzumab to erlotinib significantly prolonged PFS and OS in the MET IHC positive patients (moderate or strong intensity in ≥50% of tumor cells). Based on these results, MET high expression was assumed to be a predictive marker for response to the combined onartuzumab/erlotinib therapy and a randomized, placebo-controlled trial in previously treated stage IIIb or IV NSCLC with MET-positive (METLung) was conducted. Recent interim analysis in this phase 3 trial was disappointing and failed to show benefit of onartuzumab in OS. Final results are pending since the study is still in the follow up period [108].
Another relevant trial (A8081001) has been ongoing in NSCLC for several years with crizotinib, a small molecule kinase inhibitor of ALK, ROS1 and MET. This study was initiated as a phase I, expanded to a phase II for patients with MET amplification diagnosed by FISH, and more recently expanded to other patients such as with protein overexpression and exon 14 skipping mutation. By FISH, the patients are classified into three categories of amplification: high level (MET/CEP7 ≥5); intermediate level (MET/CEP7 >2.2 to <5); and low level (MET/CEP7 ≥1.8 to ≤2.2). In interim analyses, patients with high level MET amplification showed response rate of 67% (4 of 6 cases, including one complete response), while intermediate level of MET amplification showed response rate of 20% (1 of 5 cases) and no response was observed among the 2 low amplification cases [105]. As described, the METLung and A8001001 studies used different assays to classify patients' MET status. The METLung study measured MET protein expression by IHC using a low stringency cut-off that classified 52% of patients as positive. Conversely, the A8001001 study measured MET copy number by FISH with stringent cut-offs that only classified as positive about 3% of patients tested [105]. The difference between these two assays was also detected in direct comparison in lung cancer by Arriola et al. [109], who found MET IHC positive (moderate and strong intensity in ≥50% of tumor cells) in 48% of patients while MET amplification by FISH (gene copy number ≥5) was only found in 3% of patients. From the clinical trial findings, expression of MET by IHC with the current cut-off does not seem to be a good test to identify patients who will derive benefits from MET-targeted therapy. The FISH assay looks more promising in terms of clinical outcome, but the frequency of eligible patients according to this technology is very low. This limitation is reducing significantly the fraction of patients who may benefit from MET inhibitors and even compromising the conclusion of the ongoing trial with crizotinib. In an attempt to address this limitation, the A8001001 trial has been recently amended to include patients with MET mutations and copy number gain detected by Next-Generation Sequencing (NGS).
In lung cancer, it is critical to continue exploring different platforms to evaluate the molecular profile of MET as a marker in a more comprehensive and standardized way to identify the best algorithms for identification of patients sensitive to MET inhibitors. Moreover, any successful test identified in lung cancer will need to be prospectively validated in other tumors in which MET deregulation occur in substantial frequency.
Clinical Applications of Deregulation in the MET Pathway
With the better understanding of the role of the MET signaling in carcinogenesis, its pathway has been targeted by newly developed therapeutic agents included into three major categories based on their specific targets (HGF and MET) and mechanisms of action (monoclonal antibodies and TKIs). These targeted agents have been clinically investigated in numerous solid tumors, but mostly in lung cancer as summarized in Table 5.
Therapeutical agents targeting MET in lung cancer clinical trials.
| HGF antagonist | |||||||
|---|---|---|---|---|---|---|---|
| Drug | Chemical Name | Study Phase | Treatment Arms | Patient Population | Trial No. | Status | Primary outcome |
| AV-299 | Ficlatuzumab | I / II | Ficlatuzumab plus Gefitinib | Advanced untreated Asian lung adenocarcinoma | NCT01039948 | Active, not recruiting | Safety |
| II | Arm A : Ficlatuzumab plus Erlotinib Arm B : Placebo plus Erlotinib | Previously untreated metastatic EGFR-mutated NSCLC and BDX004 positive | NCT02318368 | Recruiting | PFS | ||
| AMG-102 | Rilotumamab | I / II | Rilotumamab plus Erlotinib | Advanced pre-treated NSCLC | NCT01233687 | Recruiting | Safety |
| I / II | Rilotumamab plus Chemotherapy of choice | Extensive SCLC | NCT00791154 | Completed | Safety | ||
| Anti-MET monoclonal antibody | |||||||
| MetMab | Onartuzumab | II | Onartuzumab plus Erlotinib | Previously treated NSCLC | NCT00854308 | Completed | PFS in ITT and MET-positive patients |
| II | Arm A : Onartuzumab plus Bevacizumab/Platinum/Paclitaxel or platinum/Pemetrexed Arm B : placebo plus Bevacizumab/Platinum/Paclitaxel or platinum/Pemetrexed | Previously untreated non-squamous NSCLC | NCT01496742 | Active, not recruiting | PFS in ITT and MET-positive patients | ||
| II | Arm A : Onartuzumab plus Platinum + paclitaxel Arm B : Placebo plus Platinum + paclitaxel | Previously untreated squamous NSCLC | NCT01519804 | Active, not recruiting | PFS in ITT and MET-positive patients | ||
| III (METLung) | Arm A : Onartuzumab plus Erlotinib Arm B : placebo plus Erlotinib | Advanced MET-positive NSCLC | NCT01456325 | Active, not recruiting | OS | ||
| III | Arm A : Onartuzumab plus Erlotinib Arm B : placebo plus Erlotinib | Advanced previously treated MET-positive NSCLC | NCT02031744 | Recruiting | OS | ||
| III | Arm A : Onartuzumab plus Erlotinib Arm B : placebo plus Erlotinib | Advanced previously untreated MET-positive NSCLC carrying activation EGFR mutation | NCT01887886 | Recruiting | PFS | ||
| MET tyrosine kinase inhibitor | |||||||
| MP-470 | Amuvatinib | II | Amuvatinib with Chemotherapy (Platinum and Etoposide) | Previously treated SCLC | NCT01357395 | Active, not recruiting | ORR |
| XL 184 | Cabozantinib | I/II | Arm A :Cabozatinib plus Erlotinib Arm B : placebo plus Erlotinib | Acquired resistant to Erlotinib | NCT00596648 | Completed | Safety |
| II | Cabozatinib | Previously untreated NSCLC positive for RET, ROS1, or NTRK fusion or increased MET activity | NCT01639508 | Recruiting | ORR | ||
| II | Arm A : Erlotinib Arm B : Cabozatinib Arm C : Combination | Previously treated NSCLC with wt-EGFR | NCT01708954 | Active, not recruiting | PFS | ||
| INC 280 | Capmatinib | Ib/II | Capmatinib plus Gefitinib vs Gefitinib alone | EGFR-TKI resistant EGR-mt, c-MET amplified NSCLC | NCT01610336 | Recruiting | Safety |
| I | Capmatinib plus Erlotinib vs Erlotinib alone | Erlotinib-treated NSCLC | NCT01911507 | Recruiting | Safety | ||
| PF 02341066 | Crizotinib | I | Crizotinib (A8081001 study) | Previously treated advanced malignancies that are known to be sensitive to PF-03241066 inhibition, e.g. ALK, c-MET and ROS | NCT00585195 | Recruiting | Safety |
| I/II | Arm A : Crizotinib plus Erlotinib Arm B : placebo plus Erlotinib | Previously treated lung adenocarcinoma | NCT00965731 | Completed Phase I; Phase II withdrawn | Safety | ||
| I | PF 00299804 (Sequential vs combination) | Previously treated NSCLC | NCT01121575 | Completed | Safety | ||
| I | Crizotinib plus PF 00299804 | Previously treated NSCLC | NCT01441128 | Completed | Safety | ||
| GSK 1363089 | Foretinib | I/II | Foretinib plus Cabozatinib | Previously treated NSCLC | NCT01068587 | Active, not recruiting | Safety |
| ARQ 197 | Tivantinib | I | Tivantinib plus Erlotinib | Previously treated NSCLC | NCT01069757 | Completed | Safety |
| II | Arm A : Tivantinib plus Erlotinib Arm B : placebo plus Erlotinib | Previously treated NSCLC | NCT00777309 | Completed | PFS | ||
| III | Arm A : Tivantinib plus Erlotinib Arm B : placebo plus Erlotinib (ATTENTION study) | Previously treated Asian advanced non-squamous NSCLC with wild-type EGFR | NCT01377376 | Discontinued due to high frequency of interstitial lung disease | OS | ||
| III | Arm A : Tivantinib plus Erlotinib Arm B : placebo plus Erlotinib (MARQUEE study) | Previously treated advanced non-squamous NSCLC | NCT01244191 | Discontinued due to failure to meet primary endpoint at planned intereim analysis | OS in ITT | ||
| I/II | Tivantinib plus Carbolatin + Pemetrexed | Previously untreated non-squamous NSCLC | NCT02049060 | Recruiting | Safety | ||
| II | Arm A : Tivantinib plus Erlotinib Arm B : Single chemotherapy (Docetaxel, Pemetrexed, Gemcitabine) | Previously chemotherapy-treated, K-Ras mutant NSCLC | NCT01395758 | Active, not recruiting | PFS | ||
ITT; intentiion-to-treat, OS; overall survival, ORR; objective response rate, PFS; progression-free survival.
Anti-HGF Antibodies
Two antibodies against HGF, ficlatuzumab and rilotumumab, are being tested in lung cancer clinical trials. Ficlatuzumab (AV-299) is a humanized HGF IgG1 monoclonal antibody under investigation in Asian patients with previously untreated lung adenocarcinoma likely to harbor activating EGFR mutations, e.g. never smoker with adenocarcinoma histology (NCT01039948). Patients were classified into two arms, one received ficlatuzumab 20mg/kg every 2 weeks plus gefitinib 250 mg/day and another received gefitinib 250mg/day alone. In preliminary analysis, there was no difference in PFS and OS in the ITT population. However, patients with high HGF expression who received the drug combination had better PFS (6.5 vs 2.7 mo, p=0.44) and OS (not reached vs 16 mo, p=0.03) in the subgroup analysis [110]. An ongoing phase II study is recruiting to compare ficlatuzumab plus erlotinib with erlotinib alone in treatment naïve, mutant EGFR, advanced stage IV NSCLC (NCT02318368). For this trial, mutant EGFR patients are selected based on the positive proteomic signature in the VeriStrat assay (Biodesix).
Rilotumumab (AMG 102) is a fully human monoclonal antibody against HGF, neutralizing HGF and preventing downstream METsignaling. Rilotumumab has been studied in a phase II trial of advanced gastric or esophagogastric cancer and has demonstrated efficacy in patients with METhigh tumors, defined as harboring >50% of cells with intermediate and strong MET staining, addition of rilotumumab to chemotherapy increased median PFS from 4.4 to 6.9 months (HR 0.31; p=0.007) when compared with chemotherapy alone [111]. More data are anticipated soon from a IB/II trial of rilotumumab plus chemotherapy in extensive SCLC patients (NCT00791154) that was completed in February 2015 and from a phase l/ll trial of combination of rilotumumab and erlotinib in NSCLC patients (NCT01233687) that is still actively recruiting participants.
Anti-MET Antibody
The anti-MET antibody onartuzumab (MetMab) has been extensively studied in lung cancer. This humanized, monoclonal antibody binds to the Sema domain of MET receptor inhibiting the docking of the ligand HGF. As discussed previously, a phase II study of onartuzumab plus erlotinib in EGFR-TKI naïve showed benefit of adding onartuzumab in MET positive patients in terms of PFS which was not confirmed in the interim analysis of the subsequent phase III METLung study [108]. Another phase III study of onartuzumab plus erlotinib as second- or third-line treatment is recruiting patients with MET IHC-positive NSCLC patients (NCT02031744). Final results of the first trial after longer follow up and preliminary results of the second trial are keenly awaited.
MET Tyrosine Kinase Inhibitors
There are six oral MET small molecule TKIs in clinical trials, namely amuvatinib, cabozantinib, capmatinib, crizotinib, foretinib and tivantinib. Four of them (amuvatinib, cabozantinib, crizotinib and foretinib) are multi-kinase inhibitors whereas the other two (capmatinib and tivantinib) are selective MET inhibitors.
Amuvatinib (MP-470) acts on MET, KIT and platelet-derived growth factor receptor (PDGFR). In a phase IB trial in adult solid tumors, amuvatinib was well tolerated and showed 12% PR when combined with chemotherapy [112]. There is an ongoing phase II study of amuvatinib with chemotherapy in previously treated SCLC (NCT01357395).
Cabozantinib (XL184) inhibits MET, vascular endothelial growth factor receptor2 (VEGFR2), and RET.The agent is approved by the US FDA for treatment of aggressive, unresectable, locally advanced or metastasis medullary thyroid carcinoma [113]. Cabozantinib was also tested in phase ll randomized discontinuation cohort of different solid tumors e.g. melanoma, NSCLC, HCC and breast cancer, in which it reached objective tumor regression respectively in 60%, 64%, 5% and 64% of patients [114]. Cabozantinib is being tested as single agent in previously untreated NSCLC patients positive for RET, ROS1 or NTRK fusion or with increased MET activity (NTC01639508) or in combination with erlotinib in previously treated NSCLC patients with wild-type EGFR (NTC01708954). In these studies, MET activity was defined as overexpression, mutation or amplification. Moreover, there are several trials investigating the efficacy of cabozantinib in other conditions such as CRC and prostate cancer.
Capmatinib (INC 280) has effectively induced apoptosis in in vitro conditions and has shown significant negative effect in tumor cell proliferation and migration [115]. There are two phase I studies with this drug in NSCLC patients, one with capmatinib in combination with gefitinib in EGFR-TKI resistant EGFR-mutant, MET amplified (NTC01610336) and another in combination with erlotinib in NSCLC patients with progressive disease after erlotinib (NTC01911507). There are also ongoing phase I and II studies of Capmatinib in other solid tumors such as HCC, HNSCC, CRC and papillary RCC.
Crizotinib (PF02341066) inhibits ALK, ROS1 and MET and is a FDA approved TKI for ALK-positive NSCLC patients. Moreover, crizotinib showed a high efficacy in NSCLC patients with de novo MET amplification [116]. As discussed previously, crizotinib is under an expanded phase I/II trial (A80081001) in NSCLC patients with MET amplification detected by FISH, which have shown promising results. More recently, efficacy of crizotinib was also detected in advanced NSCLC harboring splicing mutations in the MET exon 14 [117], which is relevant since these mutations have been found with relatively high frequency in NSCLC. Several other phasel/ll trials of crizotinib alone or in combination with other TKIs are ongoing in various types of cancers.
Foretinib (GSK1363089, XL880) inhibits MET, VEGF, RON, AXL, TIE-2 receptor and potentially also VEGFR2. Efficacy of foretinib was shown in a phase II study of papillary RCC with ORR of 13.5% [118]. Foretinib is under evaluation in phase I/II trial of previously treated NSCLC patients (NTC01068587).
Tivantinib (ARQ197) blocks both ligand-dependent and ligand-independent MET downstream signaling by stabilizing the inactive conformation. A randomized, phase ll study of tivantinib plus erlotinib versus erlotinib alone in previously treated, EGFR TKI-naïve NSCLC patients showed improvement of median PFS in the drug combination arm (3.8 versus 2.3 months; adjusted HR 0.68; p=0.04) [92]. An exploratory molecular analyses in a subset of 50 patients with tissue available for molecular testing has not found significant difference in PFS and OS for MET-positive tumors between the combination arm and the erlotinib alone and no correlation was found between MET expression by IHC and MET copy number by FISH (r= 0.14, p = 0.35) [119]. A phase lll study in previously treated, EGFR- and MET-TKI naïve, non-squamous lung cancer patients (MARQUEE trial) was discontinued prematurely because the interim analysis failed to show an improvement of OS in the ITT population. PFS improvement after addition of tivantinib to erlotinib was significant (HR 0.74; p<0.01) but this benefit was not carried on to OS (HR 0.89; p=0.81). However, OS was significantly improved in the combination treatment arm in patients with METhigh tumors (HR 0.7; p=0.03) [60]. Another phase III study in previously treated, EGFR- and MET-TKI naïve, wild type EGFR, non-squamous cell lung cancer Asian patients (ATTENTION trial) was discontinued prematurely because of the higher rate of interstitial lung disease (0.8% in combination with erlotinib group vs 0.2% in erlotinib alone group). ITT analysis showed non-statistically significant improvement of OS (12.9 months vs 11.2 months HR 0.88 p=0.4) [120].
The adverse effects of these multi-kinase and selective MET inhibitors are quite similar. Fatigue, skin rash and gastrointestinal adverse effects including diarrhea, anorexia, nausea, vomiting, and constipation were commonly seen in both groups. Dysguesia was more common in cabozantinib and crizotinib which are multi-kinase inhibitors. Cabozantinib was reported to cause hypertension and mucositis and crizotinib was reported to cause visual disorder and edema [3,92,113]. Tivantinib had interstitial lung disease as serious adverse event which led to discontinuation of ATTENTION trial.
As noticeable, there are far more small molecule TKIs as MET inhibitors in clinical trials than antibodies, but favorable results have been observed in both drug categories. However, one of the major limiting factors for the success of these clinical studies is the lack of molecular markers validated to predict which patients are more likely to benefit from the novel targeted agents.
Conclusion
Extensive studies on the MET pathway have uncovered its important physiologic function in embryogenesis, organ development and tissue regeneration. Deregulation of MET pathway, including overexpression of HGF or MET, MET gene mutations and amplification, has shown to play an essential role in carcinogenesis progression and metastasis and, specifically in NSCLC, was also found to be important player in acquired resistance to targeted therapeutic agents such as the EGFR-TKIs.
This cellular pathway may be blocked by numerous targeted therapy agents, including HGF-antagonists and anti-MET antibodies and TKIs, which provide new venues for clinical benefit to patients whose tumors are significantly driven by this pathway. Many of those agents are currently in clinical investigation and some such as onartuzumab, tivantinib and crizotinib have shown promising preliminary results. However, molecular biomarkers for the MET pathway are not well defined. There is no proved technical platform for detection of activation, and no standardized methodology or criteria for diagnosis. Among the two more promising assays, MET expression by IHC failed to confirm its validation in randomized clinical trial and MET amplification by FISH still lacks standardization. Moreover, following the selection of the optimal biomarkers, prospective studies with larger number of patients are needed for their validation at the clinical level.
Acknowledgements
The authors acknowledge the Thai Society of Clinical Oncology (TSCO) for a fellowship to Witthawat Ariyawutyakorn, the Phramongkutklao Hospital for a fellowship to Siriwimon Saichamchan, and the support from NCI grants Colorado Lung Cancer SPORE (P50CA058187) and CCSG (P30CA046934) to Marileila Varella-Garcia.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Mok TS, Wu Y-L, Thongprasert S. et al. Gefitinib or Carboplatin-Paclitaxel in Pulmonary Adenocarcinoma. N Engl J Med. 2009;361(10):947-957
2. Rosell R, Carcereny E, Gervais R. et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012;13(3):239-246
3. Solomon BJ, Mok T, Kim D-W. et al. First-Line Crizotinib versus Chemotherapy in ALK-Positive Lung Cancer. N Engl J Med. 2014;371(23):2167-2177
4. Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF. Met, metastasis, motility and more. Nat Rev Mol Cell Biol. 2003;4(12):915-925
5. Ma PC, Maulik G, Christensen J, Salgia R. c-Met: Structure, functions and potential for therapeutic inhibition. Cancer Metastasis Rev. 2003;22(4):309-325
6. Trusolino L, Bertotti A, Comoglio PM. MET signalling: principles and functions in development, organ regeneration and cancer. Nat Rev Mol Cell Biol. 2010;11(12):834-848
7. Scagliotti GV, Novello S, von Pawel J. The emerging role of MET/HGF inhibitors in oncology. Cancer Treat Rev. 2013;39(7):793-801
8. Park M, Dean M, Kaul K, Braun MJ, Gonda MA, Vande Woude G. Sequence of MET protooncogene cDNA has features characteristic of the tyrosine kinase family of growth-factor receptors. Proc Natl Acad Sci USA. 1987;84(18):6379-6383
9. Bottaro DP, Rubin JS, Faletto DL. et al. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science. 1991;251(4995):802-804
10. Syed ZA, Yin W, Hughes K, Gill JN, Shi R, Clifford JL. HGF/c-met/Stat3 signaling during skin tumor cell invasion: indications for a positive feedback loop. BMC Cancer. 2011;11:180-190
11. Lokker NA, Mark MR, Luis EA. et al. Structure-function analysis of hepatocyte growth factor: identification of variants that lack mitogenic activity yet retain high affinity receptor binding. EMBO J. 1992;11(7):2503-2510
12. Owen KA, Qiu D, Alves J. et al. Pericellular activation of hepatocyte growth factor by the transmembrane serine proteases matriptase and hepsin, but not by the membrane-associated protease uPA. Biochem J. 2010;426(2):219-228
13. Lee S-L, Dickson RB, Lin C-Y. Activation of Hepatocyte Growth Factor and Urokinase/Plasminogen Activator by Matriptase, an Epithelial Membrane Serine Protease. J Biol Chem. 2000;275(47):36720-36725
14. Furge KA, Zhang Y-W, Woude GF. Met receptor tyrosine kinase: enhanced signaling through adapter proteins. Oncogene. 2000;19(49):5582-5589
15. Müller M, Morotti A Ponzetto C. Activation of NF-κB Is Essential for Hepatocyte Growth Factor-Mediated Proliferation and Tubulogenesis. Mol Cell Biol. 2002;22(4):1060-1072
16. Xiao G-H, Jeffers M, Bellacosa A, Mitsuuchi Y, Vande Woude GF, Testa JR. Anti-apoptotic signaling by hepatocyte growth factor/Met via the phosphatidylinositol 3-kinase/Akt and mitogen-activated protein kinase pathways. Proc Natl Acad Sci USA. 2001;98(1):247-252
17. Paumelle R, Tulasne D, Kherrouche Z. et al. Hepatocyte growth factor/scatter factor activates the ETS1 transcription factor by a RAS-RAF-MEK-ERK signaling pathway. Oncogene. 2002;21(15):2309-2319
18. Kanteti R, Nallasura V, Loganathan S. et al. PAX5 is expressed in small cell lung cancer and positively regulates c-Met transcription. Lab Invest. 2009;89(3):301-314
19. Kanteti R, El-Hashani E, Dhanasingh I. et al. Role of PAX8 in the regulation of MET and RON receptor tyrosine kinases in non-small cell lung cancer. BMC Cancer. 2014;14:185-196
20. Pennacchietti S, Michieli P, Galluzzo M, Mazzone M, Giordano S, Comoglio PM. Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell. 2003;3(4):347-361
21. Petrelli A, Gilestro GF, Lanzardo S, Comoglio PM, Migone N, Giordano S. The endophilin-CIN85-Cbl complex mediates ligand-dependent downregulation of c-Met. Nature. 2002;416(6877):187-190
22. Foveau B, Ancot F, Leroy C. et al. Down-Regulation of the Met Receptor Tyrosine Kinase by Presenilin-dependent Regulated Intramembrane Proteolysis. Mol Biol Cell. 2009;20(9):2495-2507
23. Nakamura T, Mizuno S, Matsumoto K, Sawa Y, Matsuda H, Nakamura T. Myocardial protection from ischemia/reperfusion injury by endogenous and exogenous HGF. J Clin Invest. 2000;106(12):1511-1519
24. Chmielowiec J, Borowiak M, Morkel M. et al. c-Met is essential for wound healing in the skin. J Cell Biol. 2007;177(1):151-162
25. Rong S, Segal S, Anver M, Resau JH, Vande Woude GF. Invasiveness and metastasis of NIH 3T3 cells induced by Met-hepatocyte growth factor/scatter factor autocrine stimulation. Proc Natl Acad Sci U S A. 1994;91(11):4731-4735
26. Wang R, Ferrell LD, Faouzi S, Maher JJ, Bishop JM. Activation of the Met Receptor by Cell Attachment Induces and Sustains Hepatocellular Carcinomas in Transgenic Mice. J Cell Biol. 2001;153(5):1023-1034
27. Boccaccio C, Sabatino G, Medico E. et al. The MET oncogene drives a genetic programme linking cancer to haemostasis. Nature. 2005;434(7031):396-400
28. Zhang Y-W, Su Y, Volpert OV, Woude GFV. Hepatocyte growth factor/scatter factor mediates angiogenesis through positive VEGF and negative thrombospondin 1 regulation. Proc Natl Acad Sci USA. 2003;100(22):12718-12723
29. Conrotto P, Corso S, Gamberini S, Comoglio PM, Giordano S. Interplay between scatter factor receptors and B plexins controls invasive growth. Oncogene. 2004;23(30):5131-5137
30. Bivona TG, Hieronymus H, Parker J. et al. FAS and NF-κB signalling modulate dependence of lung cancers on mutant EGFR. Nature. 2011;471(7339):523-526
31. Yano S, Wang W, Li Q. et al. Hepatocyte Growth Factor Induces Gefitinib Resistance of Lung Adenocarcinoma with Epidermal Growth Factor Receptor-Activating Mutations. Cancer Res. 2008;68(22):9479-9487
32. Masuya D, Huang C, Liu D. et al. The tumour-stromal interaction between intratumoral c-Met and stromal hepatocyte growth factor associated with tumour growth and prognosis in non-small-cell lung cancer patients. Br J Cancer. 2004;90(8):1555-1562
33. Olivero M, Rizzo M, Madeddu R. et al. Overexpression and activation of hepatocyte growth factor/scatter factor in human non-small-cell lung carcinomas. Br J Cancer. 1996;74(12):1862-1868
34. Li B, Kanamaru H, Noriki S, Fukuda M, Okada K. Differential Expression of Hepatocyte Growth Factor in Papillary and Nodular Tumors of the Bladder. Int J Urol. 1998;5(5):436-440
35. Wang P, Nishitani M-A, Tanimoto S. et al. Bladder Cancer Cell Invasion Is Enhanced by Cross-Talk with Fibroblasts Through Hepatocyte Growth Factor. Urology. 2007;69(4):780-784
36. Eissa S, Swellam M, Labib RA, El-Zayat T, El Ahmady O. A Panel of Angiogenic Factors for Early Bladder Cancer Detection: Enzyme Immunoassay and Western Blot. J Urol. 2009;181(3):1353-1360
37. Nagy J, Curry GW, Hillan KJ. et al. Hepatocyte growth factor/scatter factor expression and c-met in primary breast cancer. Surg Oncol. 1996;5(1):15-21
38. Edakuni G, Sasatomi E, Satoh T, Tokunaga O, Miyazaki K. Expression of the hepatocyte growth factor/c-Met pathway is increased at the cancer front in breast carcinoma. Pathol Int. 2001;51(3):172-178
39. Kang JY, Dolled-Filhart M, Ocal IT. et al. Tissue Microarray Analysis of Hepatocyte Growth Factor/Met Pathway Components Reveals a Role for Met, Matriptase, and Hepatocyte Growth Factor Activator Inhibitor 1 in the Progression of Node-negative Breast Cancer. Cancer Res. 2003;63(5):1101-1105
40. Toiyama Y, Miki C, Inoue Y, Okugawa Y, Tanaka K, Kusunoki M. Serum hepatocyte growth factor as a prognostic marker for stage II or III colorectal cancer patients. Int J Cancer. 2009;125(7):1657-1662
41. Takahashi N, Yamada Y, Furuta K. et al. Serum levels of hepatocyte growth factor and epiregulin are associated with the prognosis on anti-EGFR antibody treatment in KRAS wild-type metastatic colorectal cancer. Br J Cancer. 2014;110(11):2716-2727
42. Kim C-H, Moon S-K, Bae J-H. et al. Expression of hepatocyte growth factor and c-Met in hypopharyngeal squamous cell carcinoma. Acta Otolaryngol. 2006;126(1):88-94
43. Seiwert TY, Jagadeeswaran R, Faoro L. et al. The MET Receptor Tyrosine Kinase is a potential novel Therapeutic Target for Head and Neck Squamous Cell Carcinoma. Cancer Res. 2009;69(7):3021-3031
44. Onitsuka T, Uramoto H, Ono K. et al. Comprehensive Molecular Analyses of Lung Adenocarcinoma with Regard to the Epidermal Growth Factor Receptor, K-ras, MET, and Hepatocyte Growth Factor Status. J Thorac Oncol. 2010;5:591-596
45. Miyata Y, Sagara Y, Kanda S, Hayashi T, Kanetake H. Phosphorylated hepatocyte growth factor receptor/c-Met is associated with tumor growth and prognosis in patients with bladder cancer: correlation with matrix metalloproteinase-2 and -7 and E-cadherin. Hum Pathol. 2009;40(4):496-504
46. Ghoussoub RAD, Dillon DA, D'Aquila T, Rimm EB, Fearon ER, Rimm DL. Expression of c-met is a strong independent prognostic factor in breast carcinoma. Cancer. 1998;82(8):1513-1520
47. Tolgay Ocal I, Dolled-Filhart M, D'Aquila TG, Camp RL, Rimm DL. Tissue microarray-based studies of patients with lymph node negative breast carcinoma show that met expression is associated with worse outcome but is not correlated with epidermal growth factor family receptors. Cancer. 2003;97(8):1841-1848
48. Lee W-Y, Chen HHW, Chow N-H, Su W-C, Lin P-W, Guo H-R. Prognostic Significance of Co-expression of RON and MET Receptors in Node-Negative Breast Cancer Patients. Clin Cancer Res. 2005;11(6):2222-2228
49. Ma PC, Tretiakova MS, MacKinnon AC. et al. Expression and Mutational Analysis of MET in Human Solid Cancers. Genes Chromosomes Cancer. 2008;47(12):1025-1037
50. Gisterek I, Lata E, Halon A. et al. Prognostic role of c-met expression in breast cancer patients. Rep Pract Oncol Radiother. 2011;16(5):173-177
51. Inanc M, Ozkan M, Karaca H. et al. Cytokeratin 5/6, c-Met expressions, and PTEN loss prognostic indicators in triple-negative breast cancer. Med Oncol. 2014;31(1):801-808
52. Uddin S, Hussain AR, Ahmed M. et al. Coexpression of Activated c-Met and Death Receptor 5 Predicts Better Survival in Colorectal Carcinoma. Am J Pathol. 2011;179(6):3032-3044
53. Garouniatis A, Zizi-Sermpetzoglou A, Rizos S, Kostakis A, Nikiteas N, Papavassiliou AG. FAK, CD44v6, c-Met and EGFR in colorectal cancer parameters: tumour progression, metastasis, patient survival and receptor crosstalk. Int J Colorectal Dis. 2013;28(1):9-18
54. Voutsina A, Tzardi M, Kalikaki A. et al. Combined analysis of KRAS and PIK3CA mutations, MET and PTEN expression in primary tumors and corresponding metastases in colorectal cancer. Mod Pathol. 2013;26(2):302-313
55. Kishiki T, Ohnishi H, Masaki T. et al. Overexpression of MET is a new predictive marker for anti-EGFR therapy in metastatic colorectal cancer with wild-type KRAS. Cancer Chemother Pharmacol. 2014;73(4):749-757
56. Shoji H, Yamada Y, Taniguchi H. et al. Clinical impact of c-MET expression and genetic mutational status in colorectal cancer patients after liver resection. Cancer Sci. 2014;105(8):1002-1007
57. An X, Wang F, Shao Q. et al. MET amplification is not rare and predicts unfavorable clinical outcomes in patients with recurrent/metastatic gastric cancer after chemotherapy. Cancer. 2014;120(5):675-682
58. Lee SJ, Lee J, Sohn I. et al. A survey of c-MET expression and amplification in 287 patients with hepatocellular carcinoma. Anticancer Res. 2013;33(11):5179-5186
59. Xia N, An J, Jiang Q, Li M, Tan J, Hu C. Analysis of EGFR, EML4-ALK, KRAS, and c-MET mutations in Chinese lung adenocarcinoma patients. Exp Lung Res. 2013;39(8):328-335
60. Scagliotti G, Pawel J von, Novello S, Ramlau R, Favaretto A, Barlesi F. et al. Phase III Multinational, Randomized, Double-Blind, Placebo-Controlled Study of Tivantinib (ARQ 197) Plus Erlotinib Versus Erlotinib Alone in Previously Treated Patients With Locally Advanced or Metastatic Nonsquamous Non-Small-Cell Lung Cancer. J Clin Oncol. 2015;33(24):2667-74
61. Spigel DR, Ervin TJ, Ramlau RA. et al. Randomized Phase II Trial of Onartuzumab in Combination With Erlotinib in Patients With Advanced Non-Small-Cell Lung Cancer. J Clin Oncol. 2013;31(32):4105-4114
62. Miyata Y, Kanetake H, Kanda S. Presence of Phosphorylated Hepatocyte Growth Factor Receptor/c-Met Is Associated with Tumor Progression and Survival in Patients with Conventional Renal Cell Carcinoma. Clin Cancer Res. 2006;12(16):4876-4881
63. Allred DC, Harvey JM, Berardo M, Clark GM. Prognostic and predictive factors in breast cancer by immunohistochemical analysis. Mod Pathol. 1998;11(2):155-168
64. Kaemmerer D, Peter L, Lupp A. et al. Comparing of IRS and Her2 as immunohistochemical scoring schemes in gastroenteropancreatic neuroendocrine tumors. Int J Clin Exp Pathol. 2012;5(3):187-194
65. Dowsett M, Allred C, Knox J. et al. Relationship Between Quantitative Estrogen and Progesterone Receptor Expression and Human Epidermal Growth Factor Receptor 2 (HER-2) Status With Recurrence in the Arimidex, Tamoxifen, Alone or in Combination Trial. J Clin Oncol. 2008;26(7):1059-1065
66. Hammond MEH, Hayes DF, Dowsett M. et al. American Society of Clinical Oncology/College of American Pathologists Guideline Recommendations for Immunohistochemical Testing of Estrogen and Progesterone Receptors in Breast Cancer. Arch Pathol Lab Med. 2010;134(6):907-922
67. Pirker R, Pereira JR, von Pawel J. et al. EGFR expression as a predictor of survival for first-line chemotherapy plus cetuximab in patients with advanced non-small-cell lung cancer: analysis of data from the phase 3 FLEX study. Lancet Oncol. 2012;13(1):33-42
68. Krishnaswamy S, Kanteti R, Duke-Cohan JS. et al. Ethnic Differences and Functional Analysis of MET Mutations in Lung Cancer. Clin Cancer Res. 2009;15(18):5714-5723
69. Asaoka Y, Tada M, Ikenoue T. et al. Gastric cancer cell line Hs746T harbors a splice site mutation of c-Met causing juxtamembrane domain deletion. Biochem Biophys Res Commun. 2010;394(4):1042-1046
70. Schmidt L, Duh F-M, Chen F. et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat Genet. 1997;16(1):68-73
71. Di Renzo M, Olivero M, Martone T. et al. Somatic mutations of the MET oncogene are selected during metastatic spread of human HNSC carcinomas. Oncogene. 2000;19(12):1547-1555
72. Lorenzato A, Olivero M, Patanè S. et al. Novel Somatic Mutations of the MET Oncogene in Human Carcinoma Metastases Activating Cell Motility and Invasion. Cancer Res. 2002;62(23):7025-7030
73. Ma PC, Kijima T, Maulik G. et al. c-MET Mutational Analysis in Small Cell Lung Cancer Novel Juxtamembrane Domain Mutations Regulating Cytoskeletal Functions. Cancer Res. 2003;63(19):6272-6281
74. Ma PC, Jagadeeswaran R, Jagadeesh S. et al. Functional Expression and Mutations of c-Met and Its Therapeutic Inhibition with SU11274 and Small Interfering RNA in Non-Small Cell Lung Cancer. Cancer Res. 2005;65(4):1479-1488
75. Onozato R, Kosaka T, Kuwano H, Sekido Y, Yatabe Y, Mitsudomi T. Activation of MET by gene amplification or by splice mutations deleting the juxtamembrane domain in primary resected lung cancers. J Thorac Oncol. 2009;4(1):5-11
76. The Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511(7511):543-550
77. Jeffers M, Schmidt L, Nakaigawa N. et al. Activating mutations for the Met tyrosine kinase receptor in human cancer. Proc Natl Acad Sci USA. 1997;94(21):11445-11450
78. Lubensky IA, Schmidt L, Zhuang Z. et al. Hereditary and Sporadic Papillary Renal Carcinomas with c-met Mutations Share a Distinct Morphological Phenotype. Am J Pathol. 1999;155(2):517-526
79. Schmidt L, Junker K, Nakaigawa N. et al. Novel mutations of the MET proto-oncogene in papillary renal carcinomas. Oncogene. 1999;18(14):2343-2350
80. Medová M, Pochon B, Streit B. et al. The Novel ATP-Competitive Inhibitor of the MET Hepatocyte Growth Factor Receptor EMD1214063 Displays Inhibitory Activity against Selected MET-Mutated Variants. Mol Cancer Ther. 2013;12(11):2415-2424
81. Jardim DLF, de Melo Gagliato D, Falchook G. et al. MET Abnormalities in Patients With Genitourinary Malignancies and Outcomes With c-MET Inhibitors. Clin Genitourin Cancer. 2015;13(1):e19-e26
82. Neklason DW, Done MW, Sargent NR. et al. Activating mutation in MET oncogene in familial colorectal cancer. BMC Cancer. 2011;11:424-430
83. Lee J-H, Han S-U, Cho H, Jennings B, Gerrard B. A novel germ line juxtamembrane Met mutation in human gastric cancer. Oncogene. 2000;19(43):4947-4953
84. Kim I-J, Park J-H, Kang HC. et al. A novel germline mutation in the MET extracellular domain in a Korean patient with the diffuse type of familial gastric cancer. J Med Genet. 2003;40(8):e97-e97
85. De Melo Gagliato D, Jardim DLF, Falchook G. et al. Analysis of MET Genetic Aberrations in Patients With Breast Cancer at MD Anderson Phase I Unit. Clin Breast Cancer. 2014;14(6):468-474
86. Tang C, Fontes Jardim DL, Falchook GS. et al. MET nucleotide variations and amplification in advanced ovarian cancer: characteristics and outcomes with c-Met inhibitors. Oncoscience. 2013;1(1):5-13
87. Kong-Beltran M, Seshagiri S, Zha J. et al. Somatic Mutations Lead to an Oncogenic Deletion of Met in Lung Cancer. Cancer Res. 2006;66(1):283-289
88. Jardim DLF, Tang C, Gagliato DDM. et al. Analysis of 1,115 Patients Tested for MET Amplification and Therapy Response in the MD Anderson Phase I Clinic. Clin Cancer Res. 2014;20(24):6336-6345
89. Beau-Faller M, Ruppert A-M, Voegeli A-C. et al. MET gene copy number in non-small cell lung cancer: molecular analysis in a targeted tyrosine kinase inhibitor naïve cohort. J Thorac Oncol. 2008;3(4):331-339
90. Cappuzzo F, Marchetti A, Skokan M. et al. Increased MET Gene Copy Number Negatively Affects Survival of Surgically Resected Non-Small-Cell Lung Cancer Patients. J Clin Oncol. 2009;27(10):1667-1674
91. Onitsuka T, Uramoto H, Nose N. et al. Acquired resistance to gefitinib: The contribution of mechanisms other than the T790M, MET, and HGF status. Lung Cancer. 2010;68(2):198-203
92. Sequist LV, Pawel J von, Garmey EG. et al. Randomized Phase II Study of Erlotinib Plus Tivantinib Versus Erlotinib Plus Placebo in Previously Treated Non-Small-Cell Lung Cancer. J Clin Oncol. 2011;29(24):3307-3315
93. Jiang S-X, Yamashita K, Yamamoto M. et al. EGFR genetic heterogeneity of nonsmall cell lung cancers contributing to acquired gefitinib resistance. Int J Cancer. 2008;123(11):2480-2486
94. Chen H-J, Mok TS, Chen Z-H. et al. Clinicopathologic and molecular features of epidermal growth factor receptor T790M mutation and c-MET amplification in tyrosine kinase inhibitor-resistant Chinese non-small cell lung cancer. Pathol Oncol Res. 2009;15(4):651-658
95. Arcila ME, Oxnard GR, Nafa K. et al. Rebiopsy of Lung Cancer Patients with Acquired Resistance to EGFR Inhibitors and Enhanced Detection of the T790M Mutation Using a Locked Nucleic Acid-Based Assay. Clin Cancer Res. 2011;17(5):1169-1180
96. Sequist LV, Waltman BA, Dias-Santagata D. et al. Genotypic and Histological Evolution of Lung Cancers Acquiring Resistance to EGFR Inhibitors. Sci Transl Med. 2011;3(75):75ra26
97. Yu HA, Arcila ME, Rekhtman N. et al. Analysis of Tumor Specimens at the Time of Acquired Resistance to EGFR TKI therapy in 155 patients with EGFR mutant Lung Cancers. Clin Cancer Res. 2013;19(8):2240-2247
98. Kris MG, Johnson BE, Berry LD, Kwiatkowski DJ. Using Multiplexed Assays of Oncogenic Drivers in Lung Cancers to Select Targeted Drugs. JAMA. 2014;311(19):1998-2006
99. Bean J, Brennan C, Shih J-Y. et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci USA. 2007;104(52):20932-20937
100. Engelman JA, Zejnullahu K, Mitsudomi T. et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316(5827):1039-1043
101. Chen Y-T, Chang JW-C, Liu H-P. et al. Clinical implications of high MET gene dosage in non-small cell lung cancer patients without previous tyrosine kinase inhibitor treatment. J Thorac Oncol. 2011;6(12):2027-2035
102. Kubo T, Yamamoto H, Lockwood WW. et al. MET gene amplification or EGFR mutation activate MET in lung cancers untreated with EGFR tyrosine kinase inhibitors. Int J Cancer. 2009;124:1778-1784
103. Kuniyasu H, Yasui W, Kitadai Y, Yokozaki H, Ito H, Tahara E. Frequent amplification of the c-met gene in scirrhous type stomach cancer. Biochem Biophys Res Commun. 1992;189(1):227-232
104. Sakakura C, Mori T, Sakabe T. et al. Gains, losses, and amplifications of genomic materials in primary gastric cancers analyzed by comparative genomic hybridization. Genes Chromosomes Cancer. 1999;24(4):299-305
105. Camidge DR, Ou S-HI, Shapiro G. et al. Efficacy and safety of crizotinib in patients with advanced c-MET-amplified non-small cell lung cancer (NSCLC). ASCO Meet Abstr. 2014;32(15_suppl):8001
106. Cappuzzo F, Hirsch FR, Rossi E. et al. Epidermal Growth Factor Receptor Gene and Protein and Gefitinib Sensitivity in Non-Small-Cell Lung Cancer. J Natl Cancer Inst. 2005;97(9):643-655
107. Zhou C, Wu Y-L, Chen G. et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011;12(8):735-742
108. Spigel DR, Edelman MJ, O'Byrne K. et al. Onartuzumab plus erlotinib versus erlotinib in previously treated stage IIIb or IV NSCLC: Results from the pivotal phase III randomized, multicenter, placebo-controlled METLung (OAM4971g) global trial. J Clin Oncol. 2014;32:5s (suppl; abstr 8000)
109. Arriola E, Casadevall D, Gimeno J. et al. Met heterogeneity evaluation by immunohistochemistry (IHC) and fluorescence in situ hybridization (FISH) in nonsquamous non-small cell lung cancer (nsNSCLC). J Clin Oncol. 2014;32:5s (suppl; abstr 11005)
110. Mok TS, Park K, Geater SL, Agarwal S. A randomized phase (PH) 2 study with exploratory biomarker analysis of ficlatuzumab (F) a humanized hepatocyte growth factor (HGF) inhibitory mAb in combination with gefitinib (G) versus G in Asian patients (pts) with lung adenocarcinoma (LA). Abstr Book 37th ESMO Congr. 2012
111. Iveson T, Donehower RC, Davidenko I. et al. Rilotumumab in combination with epirubicin, cisplatin, and capecitabine as first-line treatment for gastric or oesophagogastric junction adenocarcinoma: an open-label, dose de-escalation phase 1b study and a double-blind, randomised phase 2 study. Lancet Oncol. 2014;15(9):1007-1018
112. Mita M, Gordon M, Rosen L. et al. Phase 1B study of amuvatinib in combination with five standard cancer therapies in adults with advanced solid tumors. Cancer Chemother Pharmacol. 2014;74(1):195-204
113. Elisei R, Schlumberger MJ, Müller SP. et al. Cabozantinib in Progressive Medullary Thyroid Cancer. J Clin Oncol. 2013;31(29):3639-3646
114. Gordon MS, Vogelzang NJ, Schoffski P. et al. Activity of cabozantinib (XL184) in soft tissue and bone: Results of a phase II randomized discontinuation trial (RDT) in patients (pts) with advanced solid tumors. J Clin Oncol. 2011:29 (suppl; abstr 3010)
115. Bang Y-J, Su W-C, Nam D-H. et al. Phase I study of the safety and efficacy of INC280 in patients with advanced MET-dependent solid tumors. J Clin Oncol. 2014;32:5s (suppl; abstr 2520)
116. Ou S-HI, Kwak EL, Siwak-Tapp C. et al. Activity of Crizotinib (PF02341066), a dual mesenchymal-epithelial transition (MET) and anaplastic lymphoma kinase (ALK) inhibitor, in a non-small cell lung cancer patient with de novo MET amplification. J Thorac Oncol. 2011;6(5):942-946
117. Drilon A, Yu HA, Krug L. Response to MET tyrosine kinase inhibition in advanced lung adenocarcinomas harbor MET exon 14 splice site mutations. Proc 15th Annu Target Ther Treat Lung Cancer St Monica Feb. 2015
118. Choueiri TK, Vaishampayan U, Rosenberg JE. et al. Phase II and biomarker study of the dual MET/VEGFR2 inhibitor foretinib in patients with papillary renal cell carcinoma. J Clin Oncol. 2013;31(2):181-186
119. Rodig S, Sequist LV, Schiller JH. et al. An exploratory biomarker analysis evaluating the effect of the c-MET inhibitor tivantinib (ARQ 197) and erlotinib in NSCLC patients in a randomized, double-blinded phase 2 study. Present Annu Meet Am Assoc Cancer Res March 31-April 4 2012 Chic IL Abstr. 1729
120. Arque/Daiichi-Sankyo world-wide news release: ArQule provides updates on clinical trials in hepatocellular carcinoma and non-small cell lung cancer with tivantinib. http://investors.arqule.com/releasedetail.cfm?ReleaseID=819847
Author contact
![]() Corresponding author: Marileila Varella-Garcia, PhD. University of Colorado Anschutz Medical Campus, Department of Medicine/ Medical Oncology Division, 12801 East 17th Ave, RC1 South, L18-8118. Mail Stop 8117, Aurora CO 80045. Telephone number 303-724-3147 Fax number 303-724-3889 Email address marileila.garciaedu.
Corresponding author: Marileila Varella-Garcia, PhD. University of Colorado Anschutz Medical Campus, Department of Medicine/ Medical Oncology Division, 12801 East 17th Ave, RC1 South, L18-8118. Mail Stop 8117, Aurora CO 80045. Telephone number 303-724-3147 Fax number 303-724-3889 Email address marileila.garciaedu.