Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2016; 7(5):555-568. doi:10.7150/jca.13614 This issue Cite
Research Paper
Tetrahydroanthraquinone Derivative (±)-4-Deoxyaustrocortilutein Induces Cell Cycle Arrest and Apoptosis in Melanoma Cells via Upregulation of p21 and p53 and Downregulation of NF-kappaB
Miroslav Genov1*, Birgit Kreiseder1*, Michael Nagl1*, Elisabeth Drucker1, Martina Wiederstein1, Barbara Muellauer1, Julia Krebs1, Teresa Grohmann1, Dagmar Pretsch1, Karl Baumann1, Markus Bacher2, Alexander Pretsch1, Christoph Wiesner1 ![]()
1. SeaLife Pharma GmbH, Technopark 1, A-3430 Tulln, Austria;
2. Division of Chemistry of Renewables, Department of Chemistry, University of Natural Resources and Life Sciences, Konrad Lorenz Straße 24, A-3430 Tulln, Austria.
*These authors contributed equally to this work.
Received 2015-8-20; Accepted 2016-1-22; Published 2016-3-12
Abstract
Background: Malignant melanoma is an aggressive type of skin cancer with high risk for metastasis and chemoresistance. Disruption of tightly regulated processes such as cell cycle, cell adhesion, cell differentiation and cell death are predominant in melanoma development. So far, conventional treatment options have been insufficient to treat metastatic melanoma and survival rates are poor. Anthraquinone compounds have been reported to have anti-tumorigenic potential by DNA-interaction, promotion of apoptosis and suppression of proliferation in various cancer cells.
Methods: In the current study, the racemic tetrahydroanthraquinone derivative (±)-4-deoxyaustrocortilutein (4-DACL) was synthesized and the cytotoxic activity against melanoma cells and melanoma spheroids determined by CellTiter-Blue viability Assay and phase contrast microscopy. Generation of reactive oxygen species (ROS) was determined with CellROX Green and Deep Red Reagent kit and microplate-based fluorometry. Luciferase reporter gene assays for nuclear factor kappa B (NF-κB) and p53 activities and western blotting analysis were carried out to detect the expression of anti-proliferative or pro-apoptotic (p53, p21, p27, MDM2, and GADD45M) and anti-apoptotic (p65, IκB-α, IKK) proteins. Cell cycle distribution and apoptosis rate were detected by flow cytometry, the morphological changes visualized by fluorescence microscopy and the activation of different caspase cascades distinguished by Caspase Glo 3/7, 8 and 9 Assays.
Results: We demonstrated that 4-DACL displayed high activity against different malignant melanoma cells and melanoma spheroids and only low toxicity to melanocytes and other primary cells. In particular, 4-DACL treatment induced mitochondrial ROS, reduced NF-κB signaling activity and increased up-regulation of the cell cycle inhibitors cyclin-dependent kinase inhibitor p21 (p21WAF1/Cip1) and the tumor suppressor protein p53 in a dose-dependent manner, which was accompanied by decreased cell proliferation and apoptosis via the intrinsic pathway.
Conclusion: According to these results, we suggest that 4-DACL may be a promising therapeutic agent for the treatment of malignant melanoma.
Keywords: 4-DACL, anthraquinone, melanoma, spheroids, NF-κB, ROS, cell cycle arrest, apoptosis.
Introduction
In skin cancer, metastatic melanoma is the most aggressive type characterized by a bad prognosis for aggressive invasion and high drug resistance.4-7 In malignant melanoma important cellular processes such as cell cycle, cell adhesion, cell differentiation and cell death are disrupted via various cell signaling pathways, including nuclear factor kappa B (NF-κB) and tumor suppressor protein p53.7 The NF-κB signaling pathway is activated in response to numerous biological stimuli and stress signals and is a key player in diverse cellular functions. The transcription factor NF-κB is known to regulate the inflammatory response, cell growth, cell survival, and apoptosis and a constitutive activation of NF-κB is frequently observed in human cancers of diverse origins, including melanoma.8,9
The tumor suppressor protein p21WAF1/CIP1 is a universal cyclin-dependent kinase inhibitor (CDKI) that regulates the progression of cells through the cell cycle.10 Induction of p21 was first found to be directly mediated by p53 resulting in cell cycle arrest at the G1 phase.11,12 In melanoma cells, p21 expression was found mainly to be independent of wild-type p53 expression and that elevated expression of p21 is associated with melanoma differentiation, growth arrest and metastatic suppression.13,14 The tumor suppressor protein p53 controls several important physiologic functions such as DNA repair, translation, differentiation, cell cycle arrest, senescence and apoptosis.12,13 In comparison to other types of cancer, mutations in the p53 gene are rare in melanoma.17
The development of chemical compounds that show anti-proliferative or pro-apoptotic activity by interfering with specific cellular signaling pathways or transcription factors such as NF-κB, p21 or p53 are promising candidates for cancer therapy.
Anthraquinone compounds such as mitoxantrone, doxorubicin or epirubicin are known to be effective clinical anti-cancer drugs by interacting with DNA, inhibiting DNA and RNA synthesis and/or the DNA processing enzyme, topoisomerase II.18,19 Lijung Huang et al.20 reported that the anthraquinone compound G503, isolated from mangrove endophytic fungus, possesses anticancer potential by inducing apoptosis in gastric cancer cells through the mitochondrial apoptotic pathway.20 The marine anthraquinone SZ-685C suppresses the proliferation and promotes apoptosis by suppression of the Akt/FOXO pathway in various cancer cells.21,22 Anthraquinones, such as emodin, aloe-emodin and rhein, isolated from rhubarb show anti-tumorigenic potential in various cancer cells, including neuroblastoma, hepatocellular carcinoma, bladder cancer, lung adenocarcinoma and others.23 Kuma et al.24 clearly demonstrated that emodin inhibits NF-κB by suppressing NF-κB inhibitor, alpha (IκB) degradation.24 Kuo et al.25 showed that aloe-emodin induces G1/S arrest accompanied with upregulation of p53 and p21. They also demonstrated that aloe-emodin initiates apoptosis in p53-deficient Hep3B and p53 wild-type HepG2 cells suggesting that aloe-emodin triggers apoptosis via p53-independent p21 activation.25
The success of conventional chemotherapeutics such as dacarbazine or its derivative temozolomide but also in combinational therapy with other agents such as cisplatin in the treatment of malignant melanoma has been shown to be disappointing.26-28
As part of a MedChem-program, we synthesized more than 200 different anthraquinone derivatives and investigated their potential to be effective against melanoma cells. In this manuscript, we demonstrate that (±)-4-deoxyaustrocortilutein (4-DACL), a novel synthesized tetrahydroanthraquinone derivative, displays high antitumorigenic potential against different malignant melanoma cells and melanoma spheroids and low toxicity to melanocytes and other primary cells. 4-DACL was found to increase reactive oxygen species (ROS) generation, decrease specifically the activation of NF-κB signaling pathway also after tumor necrosis factor-alpha (TNF), lipopolysaccharide (LPS) and fetal calf serum (FCS) stimulation and cause upregulation of the cell cycle inhibitors p21 and p53 which was accompanied by reduced cell proliferation and enhanced apoptosis in melanoma cells.
Results
4-DACL, a tetrahydroanthraquinone derivative, decreases cell metabolism and cell survival in melanoma cells and melanoma spheroids
A substantial number of novel anthraquinone derivatives were synthesized and analyzed by means of different bioassays (data not shown). For melanoma screening, the anthraquinone derivatives were pre-screened for their cytotoxic potential in order to determine a therapeutic window between melanoma/cancer cells and melanocytes. From all screened anthraquinone derivatives, (±)-4-deoxyaustrocortilutein (4-DACL) showed the most promising potential (Figure 1). The enantiomerically pure 4-DACL was first isolated and characterized from Australian fungi of the genus Dermocube by Gill and coworkers and synthesized as described by Burns et al. (1991).1-3
4-DACL (racemic 2,5-dihydroxy-7-methoxy-2-methyl-1,2,3,4-tetrahydroanthracene-9,10-dione).
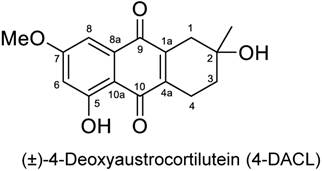
To examine the effect of 4-DACL on cell metabolism and survival in more detail we compared human melanoma cells with primary cells. Therefore, human melanoma cell lines (MCM1, MCM1G, IGR37, IGR39, A375, Mel.7, Mel.17, Mel.15), primary cells (normal human primary melanocytes (NHM), human keratinocytes (hKER)) and other tumor cell lines (HaCaT, CaCo-2, MCF7) were treated with different concentrations of 4-DACL. Cell metabolism and survival was determined 24 h later normalized to untreated control cells and the IC-50 calculated (Figure 2 A and B). Interestingly, all human melanoma cells were 8-fold more susceptible to 4-DACL than normal human primary melanocytes (NHMs). In particular, in melanoma cells the IC-50 values were between 2.5 µg/ml (IGR39) and 26 µg/ml (A375) and between 76 µg/ml and 101 µg/ml in human melanocytes after 24 h treatment. Also other primary cells such as human keratinocytes showed higher resistance (IC-50: 62 µg/ml) to 4-DACL than melanoma cells. HaCaT, CaCo-2 and MCF7 showed IC-50 values of 10 µg/ml, 20 µg/ml and 35 µg/ml, respectively (Figure 2A and B). To study the cellular morphology, NHM and melanoma cells (IGR37 and A375) were grown on 96 well plates and treated with 5 µg/ml 4-DACL for 16 h, fixed, permeabilised and immunostained with phalloidin-568 and Hoechst and subsequently investigated using phase contrast and fluorescence microscopy (Figure 2C). Microscopic analysis confirmed that NHM showed robust growth and barely visible morphological changes after 4-DACL treatment while melanoma cells changed their morphology, resulting in a rounding of cells, suggestive of further progression towards apoptosis.
4-DACL promotes cell toxicity in melanoma cells and melanoma spheroids in a dose dependent manner. (A) Cells were plated at 2×105 cells/ml in 96 well plates for 6 h and stimulated with 200 µg/ml-0.04 µg/ml 4-DACL for 24 h before CellTiter blue was added (10%) and fluorescence intensity measured. (B) Primary cells (NHM_1, NHM_2, hKER) and tumor cells (melanoma, HaCaT, CaCo-2, MCF7) were treated as in (A) and the IC50 determined. (C) The actin and nuclear of NHM, IGR37 and A375 were stained with Alexa Fluor 568 phalloidin and Hoechst 33342, respectively and viewed by fluorescence and phase contrast microscopy; bar represents 100 μm (D-F) NHM, IGR37 and A375 were cultured in matrigel for spheroid formation and spheroids treated with 5 µg/ml 4-DACL for 48 h or left untreated (n.c.). (D) Microscopic images. (E) Diameters of spheroids were determined before and after treatment and statistically evaluated. (F) Cell metabolism and viability of spheroids were analyzed using CellTiter blue assay.
In order to confirm these observations in a more physiological assay spheroids consisting of NHM, IGR37 and A375 cells were fabricated (see Material and Methods), treated with 5 µg/ml 4-DACL for 48 hours, visualized and the size of the spheroids determined before and after treatment (Figure 2D and E). Interestingly, the melanoma spheroids shrank to a considerable smaller size, whereas the spheroids consisting of melanocytes indicated no significant effect after treatment (Figure 2D and E). To examine the effect of 4-DACL on cell metabolism and survival spheroids were treated with 5 µg/ml 4-DACL or left untreated and cell metabolism and survival was determined 48 h later normalized to untreated control cells. As depicted in figure 2F, metabolic decreases were between 55-60% in melanoma spheroids and only 30% in melanocyte spheroids.
4-DACL is no DNA intercalator but induces mitochondrial ROS
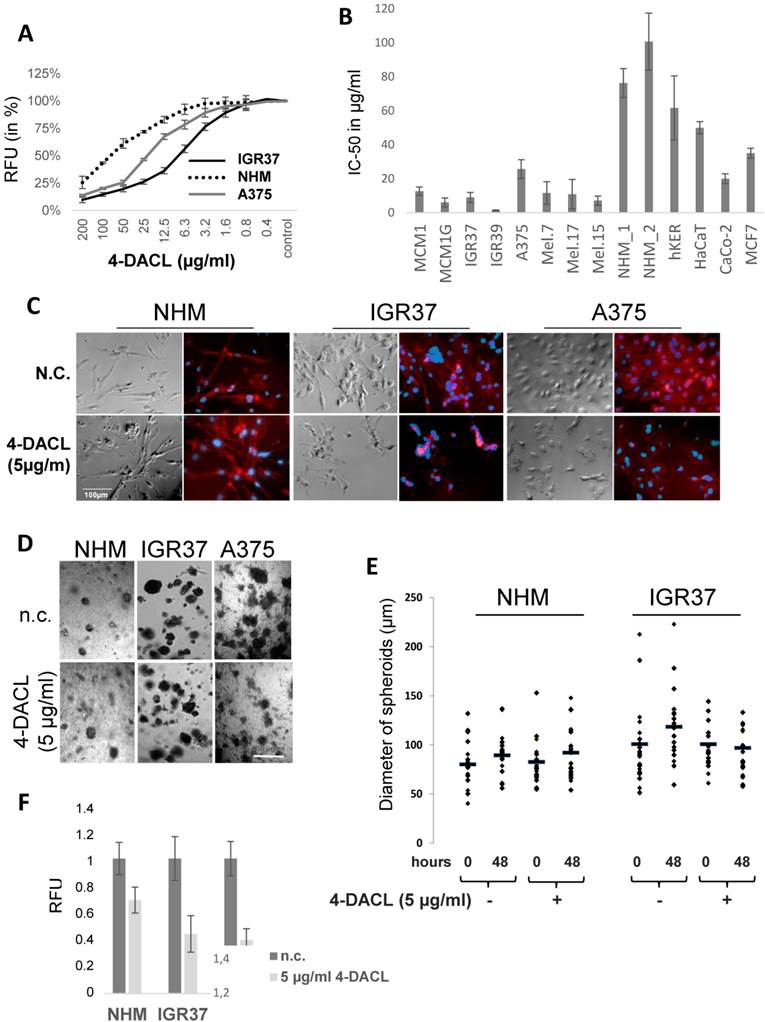
Since 4-DACL exhibits structural similarities to compounds such as mitoxantron, doxorubicin etc. which are known to intercalate into the DNA, we initially tested whether 4-DACL intercalates into or binds to the DNA.18,19 Therefore, a replacement assay was performed as described in Material and Methods and replacement of the Hoechst dye calculated. In comparison to ethidium bromide (EtBr) (<0.5µg/ml), 4-DACL showed Hoechst replacement only in very high concentrations (>25 µg/ml; Figure 3A). To verify these data, we next performed a topoisomerase I assay. The basis of this DNA unwinding assay is that compounds, which intercalate into DNA or bind in the groove, block topo I activity leading to supercoiled DNA whereas topo I activity (no intercalator or groove binder) leads to DNA relaxation. As expected, 4-DACL indicated no DNA intercalation or groove binding up to a concentration of 1mg/ml (no supercoild DNA), whereas with EtBr supercoiled DNA could be found up to a concentration of 1µg/ml (Figure 3B).
4-DACL shows no DNA intercalation but a slight mitochondrial formation of reactive oxygen species (ROS). (A) Cells were fixed (3.5% PFA), permeabilized (0.5% TX100) and stained with Hoechst and Hoechst binding measured with a multiplate reader. Thereafter cells were treated with different concentrations of 4-DACL, DMSO (neg.contr.) and Ethidiumbromide (pos. contr.), washed, measured and Hoechst replacement calculated. (B) Supercoiled DNA was incubated with different concentrations of 4-DACL or EtBr 1 h later Topoisomerase I was added and relaxation of the supercoiled DNA allowed for 30 min at 37°C before DNA was loaded on an agarose gel. Control contain DNA and topoisomerase, but no compound. (C) Supercoild DNA was incubated with different concentrations of DMSO, mitoxantron and 4-DACL and assay performed as in (B). (D) As in (C) but with EtBr. (E) IGR37 cells were treated with TBHP (pos. ctrl), DMSO (neg. ctrl) or different concentrations of 4-DACL for 1.5 h and ROS was detected using CellRox green (mitochondrial) and deep-red (cytoplasm). (*, p<0.05; **, p<0.01, as compared with n.c.).
Since chemotherapeutics such as mitoxantrone, doxorubicin, daunorubicin etc. are known to block topoisomerase II activity we next investigated whether 4-DACL showed the same mode of action. Therefore, topo II assay was performed and DNA relaxation examined. As depicted in figure 3C and D, 4-DACL showed no DNA changes compared to mitoxantrone or EtBr (Figure 3C and D).
Previous studies have implicated that anthraquinones are highly redox active molecules, which can form a redox cycle with their semiquinone.29 Therefore, we investigated the influence of 4-DACL on reactive oxygen species (ROS) production in melanoma cells. CellROX Green and Deep Red assay were performed to differentiate between mitochondrial and cytosolic ROS. As depicted in figure 3E, 4-DACL showed a concentration-dependent increase in mitochondrial but not in cytoplasmic ROS.
ROS can function as mediators of different intracellular signaling pathways that regulate numerous cellular processes, including cell cycle regulation, apoptosis or necrosis.44,45 Therefore, we have examined several signaling pathways deemed to be important for ROS-mediated inhibition of cancer cell proliferation, apoptosis and necrosis.
4-DACL decreases NF-κB activation in malignant melanoma cells
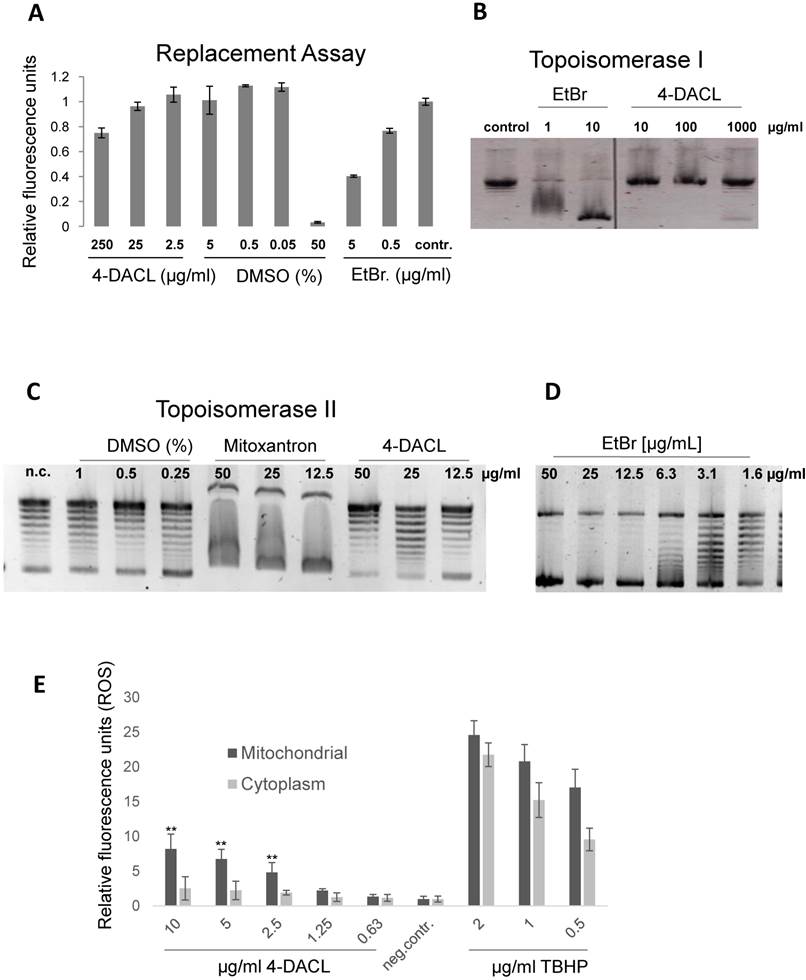
In human melanoma, NF-κB activity has been shown to be constitutively activated, leading to the deregulation of gene transcription and promotion of tumor progression.30,31 We investigated whether 4-DACL can influence NF-κB activity in human melanoma cell lines. IGR37, A375 and MCM1 were transfected with a NF-κB reporter gene (5x NF-κB-Luc), the cells treated with different concentrations of 4-DACL (10-0µg/ml) and NF-κB activation stimulated 2 h later with TNFα, LPS or 10% FCS. We found that 4-DACL decreased the basal NF-κB activity in a concentration dependent manner and also reduced NF-κB activation in a highly significant manner when stimulated with TNFα, LPS or FCS for 8 h (Figure 4A-C).
4-DACL reduces NF-κB activation in melanoma cells. A 5x NF-κB-luc reporter gene was transfected into (A) IGR37, (B) A375 and (C) MCM-1 cells and 24 h later treated with 10-0.62 µg/ml 4-DACL or left untreated. 2 h after treatment cells were non-stimulated or stimulated with TNF-α, LPS or 10% FCS. Luciferase levels were normalized for a co-transfected eGFP control. (D) IGR37 and A375 cells were analyzed by western blot with antibodies against p-IKK-α/β, p-65, p-IκB-α and tubulin. (*, p<0.05; **, p<0.01, as compared with n.c.).
To further substantiate our findings, IGR37 and A375 cells were treated with 0, 1 and 5 µg/ml 4-DACL and the regulation of NF-κB family members investigated using western blot analysis. All three proteins, phospho-IKK1/2, phospho-p65 and phospho-IκB-α were significantly reduced in IGR37 and A375 cells especially when treated with 5µg/ml 4-DACL (Figure 4D).
4-DACL increases p21, p53 and Gadd45 protein levels in a time- and concentration-dependent manner
The proteins, p21 and p53, play important roles in cellular growth control and the maintenance of genomic integrity.9 Hence, we analyzed the effect of 4-DACL on the expression of the proteins p21 and p53 using western blot analysis. The treatment of 4-DACL (5µg/ml) gradually and significantly increased the protein level of p21, p53 and growth arrest and DNA-damage-inducible protein 45 (Gadd45) over a period of at least 24 h (Figure 5A). Furthermore, treatment of IGR37 and A375 with 0, 1 and 5 µg/ml 4-DACL for 24 h significantly showed concentration-dependent activation of these cell cycle inhibitors (Figure 5B). The increased protein level of p53 in 4-DACL-treated melanoma cells correlates with increased p53-reporter activity (Figure 5 C and D).
4-DACL activates p53, p21 and GADD45G in melanoma cells in a dose dependent manner. (A) IGR37 and A375 cells were treated with 5 µg/ml 4-DACL for 0, 0.5, 1, 3, 6 and 24 hours or (B) with 0, 1 and 5 µg/ml 4-DACL for 24 hours. Total protein expression of p21Waf1/Cip1, p53, and Gadd45G were detected by western blot analysis using specific antibodies. Tubulin was used as a loading control. p53-Luc reporter assays with (C) IGR37 and (D) A375 co-transfected with eGFP and p53 plasmid (p.c.). (*, p<0.05; **, p<0.01, as compared with cells incubated media alone).
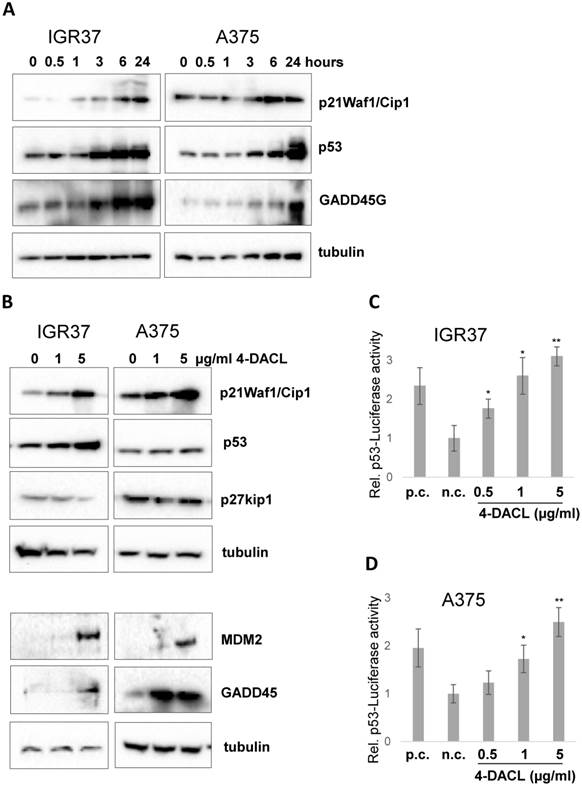
Together these data indicate that increased concentrations of 4-DACL amplify the protein levels of the cell cycle inhibitors p21, p53 and Gadd45 in malignant melanoma cells.
4-DACL decreases cell proliferation in melanoma cells
Since the previous results have shown that 4-DACL decreases NFκB activity and increases the cell cycle inhibitors p21, p53 and Gadd45 in melanoma cells (Figure 5), we next sought to examine the effect of 4-DACL on cell proliferation. Therefore, IGR37 and A375 cells were treated with 0, 0.5, 1 or 5 µg/ml 4-DACL and cell proliferation was determined after 48 and 72 hours. In both IGR37 and A375 cell lines 4-DACL decreased cell proliferation in a concentration dependent manner (Figure 6A and B). We next treated the melanoma cells with 0, 1 or 5 µg/ml 4-DACL and analyzed the expression of the proliferation marker Ki67 24 h later using Western blot. Compared to the non-treated melanoma cell lines, 4-DACL decreased Ki67 expression in a concentration dependent manner (Figure 6C). These results were confirmed by staining the treated melanoma cells (5 µg/ml 4-DACL for 6 h) with antibody against the proliferation-associated nuclear antigen Ki67 and Hoechst 33342 and fluorescence microscopy (Figure 6D).
4-DACL reduces cell proliferation in a dose- and time dependent manner in melanoma cells. IGR37 (A) and A375 (B) cells were treated with 0.5, 1 or 5µg/ml µg/ml 4-DACL or were left untreated (ctrl) and stained with cell tracker after 0, 24 and 72 h and fluorescence detected with a multiplate reader. (C) Melanoma cells were treated with 4-DACL and total protein expression of Ki67 detected 24 h later by western blot analysis. (D) Melanoma cells were immunostained with anti-Ki67 antibody, Alexa488-conjugated anti-rabbit as secondary reagent, and with Hoechst 33342 dye for nuclei staining and viewed by fluorescence and phase contrast microscopy; bar represents 100 μm (E) Melanoma cells were treated with 0, 1, 2.5 or 5 µg/ml 4-DACL for 24 hours, fixed, stained with propidium iodide solution and analyzed for cell cycle distribution using flow cytometry. (F) Flow cytometric analysis showing the cell cycle distribution of IGR37, A375 and NHM.
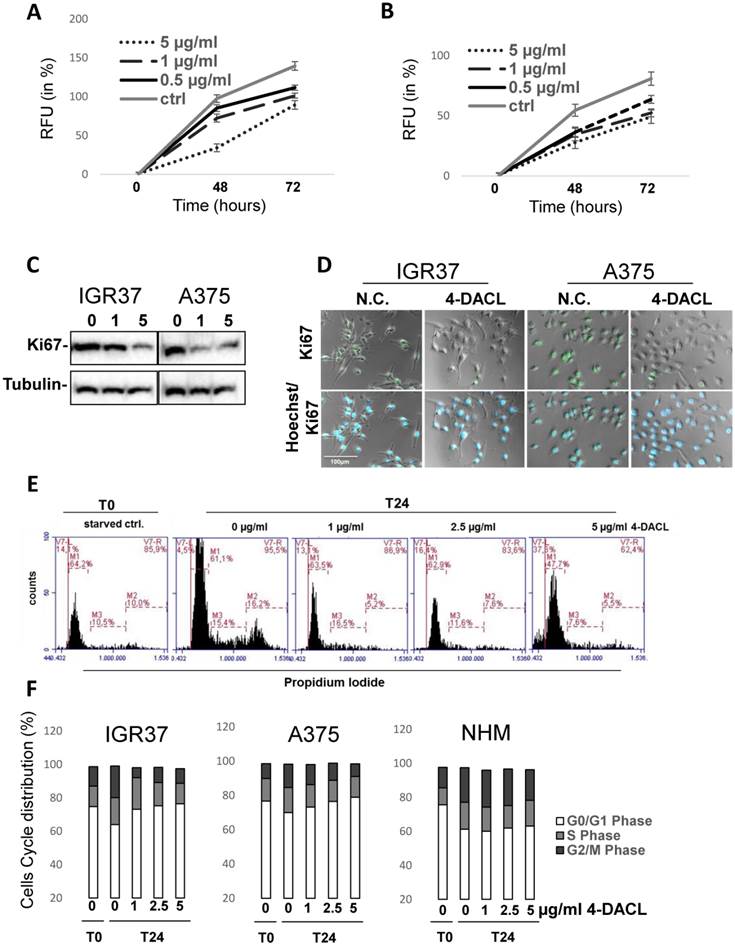
To further confirm these observations we analyzed the cell cycle distribution in IGR37, A375 and NHM cells treated with 0, 1, 2.5 or 5 µg/ml 4-DACL for 24 hours (Figure 6E-F). Compared to the untreated controls, the relative amount of cells in G0/G1 phase showed a concentration-dependent enhancement in melanoma cells (64% to 76% in IGR37 and 70% to 79% in A375) when treated with 0, 1, 2.5 and 5µg/ml 4-DACL, whereas the percentage of cells in G2 and S-phase was significantly reduced in both cell lines after 4-DACL treatment. In contrast, the relative amount of primary melanocytes in G0/G1 phase displayed no substantial enhancement (61% to 63% in NHM).
Beside the cell cycle distribution, we also found a concentration dependent increase of apoptotic cells ranging from 4.5% in non-treated cells to more than 37% when IGR37 cells were stimulated with 5 µg/ml 4-DACL (Figure 6E).
4-DACL enhances apoptosis in melanoma cells
Measuring cell metabolism, we clearly demonstrated that 4-DACL reduced the cell metabolism in melanoma cells and tumor spheroids in a concentration dependent manner (Figure 3). To further investigate the mechanism of cell death, we evaluated the AnnexinV- and 7-AAD-positive cells. As depicted in figure 7A, apoptosis (Annexin V staining) was enhanced significantly when cells were treated with 4-DACL for 16 h (1µg/ml: 8.7 %; 2.5µg/ml: 16.7%; 5µg/ml: 40.8% and 10µg/ml: 60.8 %) compared to control cells (control: 11%). 4-DACL mediates a shift from early apoptosis to late apoptosis and also from healthy cells to necrosis when melanoma cells were treated with high concentrations (Figure 7A).
4-DACL enhances apoptosis in in a dose dependent manner. (A) IGR37 cells were treated with 1, 2.5, 5 and 10 µg/ml 4-DACL for 16 h and stained with Annexin V and PI and analyzed using flow cytometry. (B) IGR37 cells were treated with 100-0.4 µg/ml cisplatin or 4-DACL for 16 h and analyzed for caspase 3/7. NHM, IGR37 and A375 cells were treated with 0-10 µg/ml 4-DACL for 16 h and caspase 3 (C), caspase 9 (D) and caspase 8 (E) activity measured. (*, p<0.05; **p<0.01, as compared with NHM cells treated with the same concentrations).
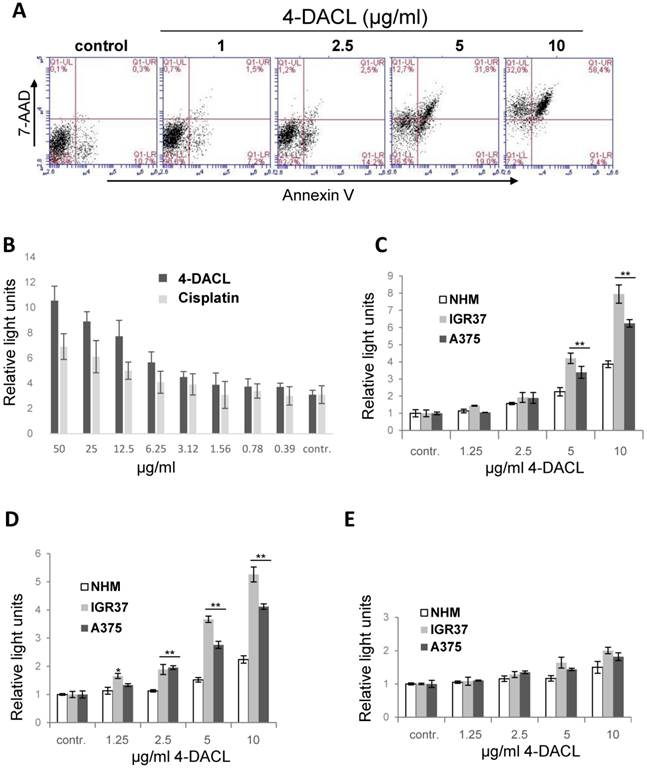
To analyse whether 4-DACL initiates the intrinsic or the extrinsic apoptosis cascades and to compare the apoptotic activity between primary melanocytes and melanoma cells, NHM, IGR37 and A375 were treated with different concentrations of 4-DACL (0-10µg/ml) and caspase-3/7, caspase-9 and -8 detected using a caspase luminescence assay. As depicted in figure 7C-E, 4-DACL shows significant and concentration-dependent induction of caspase-3/7 and caspase-9 activity after 16 hours. Moreover, caspase-3/7 and -9 activities were significantly higher in melanoma cells than in melanocytes (Figure 7B-D). Furthermore, 4-DACL increased caspase-3/7 activity more efficiently than the chemotherapeutic agent cisplatin (Figure 7B).
In summary, these results show that 4-DACL enhanced apoptosis mainly via the intrinsic pathway and is more effective in melanoma cells than in melanocytes.
Discussion
Anthraquinones are an important group of bioactive compounds with potent antitumor activity against many different cancers.32-34 Anthraquinones such as emodin, aloe-emodin or rhein have been reported to specifically block cell growth or show cytotoxicity in breast, lung, cervical, colorectal etc. whereas no cytotoxicity was observed, or only in 10-20-fold higher concentrations, in normal cells.23-25,34-37 Such specificity towards cancer cells indicates that anthraquinones have the capability to target specific signaling pathways that are constitutively active or amplified in cancer cells.
In this report, we have synthesized more than 200 pure anthraquinone derivatives and screened them for their specific anti-melanoma activity (data not shown). Of all the novel derivatives, one particular compound namely (±)-4-deoxyaustrocortilutein (4-DACL), indicated potent activity against melanoma cells and only moderate toxicity to melanocytes or primary human keratinocytes (Figure 1). In line with the potent anti-melanoma properties, it was noticed that 4-DACL also markedly impaired tumor growth and metabolism in melanoma spheroids but only slightly in melanocyte spheroids (Figure 2).
The anthraquinones are a group of small, planar aromatic molecules that are capable to penetrate intact cellular membranes and form strong intercalation complexes with DNA.38-40 A well-known member of this group is the anticancer agent mitoxantrone, which has been found to penetrate the plasma membrane, induce DNA intercalation, targets topoisomerase II and induce free radical generation.38 We propose the hydrophobic molecular properties of 4-DACL enable an easy penetration of the cell membrane allowing a covalent linkage.
We next demonstrated that 4-DACL showed DNA intercalation only at high drug concentrations (above 25 µg/ml), and no topoisomerase II suppressive effect up to 10 µg/ml (Figure 3).
Quinones are highly active molecular oxidants, involved in the biochemically induced formation of reactive oxygen species (ROS), either via single electron transfer mechanisms leading to semiquinone radical species, or via reduction of molecular oxygen by bio-reductively formed hydroquinone-species.23,29,42 The molecular structure of 4-DACL is similar to the quinones of the vitamin K series such as 2,3-dimethyl-1,4-naphthoquinone or mitochondrial ubiquinone, both of which have been reported to be able to induce ROS production.43 Thus, we hypothesized that 4-DACL may exhibit a ROS-generating potential, similar to those of the biogenic quinones. By measuring the mitochondrial and cytosolic ROS, we found that 4-DACL shows a concentration-dependent increase in mitochondrial ROS in melanoma cells (Figure 3).
ROS have various inhibitory or stimulatory roles in NF-κB signaling.46 The central role of NF-κB signaling is the inhibition of apoptosis, cell cycle regulation and metastasis. NF-κB is constitutively activated in a large number of melanoma cells suggesting NF-κB as one of the main molecular targets for molecules involved in chemoprevention.31,47-49 Additionally, NF-κB plays an important role in mediating resistance of tumor cell lines to chemotherapeutics by up-regulation of anti-apoptotic proteins. 50-52
Therefore, we analyzed the NF-κB signaling pathway in melanoma cells after 4-DACL treatment. NF-κB reporter in IGR37, A375 and MCM1 cells demonstrated that 4-DACL decreased NF-κB activity in non-treated but also in TNF-α, LPS and FCS treated cells in a dose dependent manner (Figure 4). Furthermore, the results proved that decreased NF-κB activity upon 4-DACL treatment is caused by increased de-phosphorylation of IκB-α kinases IKK-α/β, p-65 and IκB-α. Consistent with our findings Kumar et al.24 reported that the anthraquinone emodin inhibits NF-κB activation by suppressing ΙκΒ-α degradation. Li et al.53 demonstrated that emodin inhibits the expression of IKK-α/β, which are essential in phosphorylation and dissociation of IκB-α from NF-κB. Aloe-emodin, another anthraquinone derivative, has anti-tumor properties through the p53 and its downstream p21 pathway.25 We demonstrated that 4-DACL caused a time- and concentration-dependent expression of the cell cycle inhibitor proteins p21Waf1/Cip1, p53 and Gadd45 in human melanoma cells (Figure 5). Many cellular stimuli resulted in the regulation of both NF-κB and the tumor suppressor p53 and p21.54-56 The transcription factor p65, for example, inhibits p53-dependent transactivation, while p53 expression can also suppress p65 transcriptional activity.57 Furthermore, the promoter of the cyclin-dependent kinase inhibitor p21 harbours p53-responsive elements and an NF-κB binding site.55
Several observations suggest that NF-κB and p53 play a role in cell division and cell death.58 The tumor suppressor proteins, p21 and p53 counteract the oncogenic transformation by activation of cell-cycle arrest and apoptosis. Expression of p21 causes inactivation of cyclin-dependent kinase activity, which in turn blocks the cell cycle at the G1 and G2 phases.55
It has been reported that NF-κB was activated during the G0/G1 transition and could regulate cyclin D1 expression and G1- to S- phase transition.59
The decreased NF-κB signaling and the increased expression of the tumor suppressor proteins p53 and p21 due to 4-DACL treatment raised the question, whether 4-DACL treatment leads to reduced proliferation in melanoma cells. Therefore, we analyzed cell growth and Ki67 expression and found that 4-DACL altered melanoma cell proliferation in a concentration dependent manner (Figure 5). Both cell proliferation and Ki67 expression was significantly reduced in melanoma cells treated with 4-DACL (Figure 5). Furthermore, cell cycle distribution analysis revealed that the population of melanoma cells, but not melanocytes, in the G0/G1 phase increased by 4-DACL treatment in a concentration dependent manner, indicating that 4-DACL specifically inhibits cell proliferation by the induction of cell cycle arrest in G1 phase. Consistent with our findings there are several reports clearly demonstrating that anthraquinone derivative such as emodin or 3-Bromo-1-hydroxy-9,10-anthraqinone (BHAQ) induced G0/G1 arrest and apoptosis in breast cancer cells.60,61 Chihara et al.62 demonstrated that emodin and aloe-emodin causes cell cycle arrest in G0/G1 and G2/M phase, although we have not found any significant G2/M arrest when melanoma cells were treated with 4-DACL (Figure 5).
Beside the induction of the cell cycle arrest in G0/G1 phase, cell cycle distribution analysis additionally indicated that 4-DACL treatment provoked a concentration dependent increase of apoptotic melanoma cells, demonstrating that 4-DACL not only blocked proliferation but also induced apoptosis. To prove this, AnnexinV/7-AAD assay and caspase 3/7, -9 and -8 were performed. Apoptosis and at high concentrations also necrosis were significantly enhanced after 4-DACL treatment (Figure 6). We also demonstrated that 4-DACL primarily activated apoptosis via caspase-9 and only in very high concentrations via caspase-8, indicating that 4-DACL triggers apoptosis mainly via the intrinsic pathway (Figure 6).
In summary, we show in this study, that 4-DACL, a novel synthesized anthraquinone derivative, demonstrates potent in vitro activity against melanoma cells and melanoma spheroids and only moderate toxicity to primary cells. This specific activity against melanoma cells was attributable to its ability to block NF-κB signaling and to promote the expression of the cell cycle inhibitors p21 and p53, causing cell cycle arrest in G0/G1 phase and apoptosis via the intrinsic pathway.
These results demonstrate that 4-DACL has promising characteristics to block tumor growth, and progression suggesting 4-DACL as a novel promising molecule for the treatment of melanoma.
Materials and Methods
(±)-4-Deoxyaustrocortilutein (racemic 2,5-dihydroxy-7-methoxy-2-methyl-1,2,3,4-tetrahydroanthracene-9,10-dione)
4-DACL was synthesized as described by Burns et al.3 and purified by column chromatography (DCM/ethyl acetate 50/1 to 1/1) to give 107 mg (0.37 mmol, 70 %) as orange powder. Thin layer chromatography was performed on aluminium plates precoated with Silicagel 60 F254 (Merck). Rf= 0.25 (DCM/EA 50/1)
Analytical data:
1H (CDCl3, 400.13 MHz): 1.34 (br.s, 1H, 2-OH), 1.41 (s, 3H, 2-Me), 1.65 (m, 1H, H-3a), 1.89 (m, 1H, H-3b), 2.55 (ddd, 1H, J = 19.9 + 3.5 +2.7 Hz, H-1a), 2.75 (dm, 1H, J = 19.9 Hz, H-1b), 2.75 (m, 2H, H-4a+b), 3.89 (s, 3H, OMe), 6.61 (d, 1H, J = 2.5 Hz, H-6), 7.15 (d, 1H, J = 2.5 Hz, H-8), 12.34 (s, 1H, 5-OH)
13C (CDCl3, 100.63 MHz): 20.65 (C-4), 29.66 (2-Me), 37.17 (C-1), 33.67 (C-3), 55.94 (OMe), 67.85 (C-2), 105.90 (C-6), 107.51 (C-8), 109.42 (C-10a), 133.71 (C-8a), 142.67 (C-1a), 144.13 (C-4a), 164.14 (C-5), 165.74 (C-7), 183.95 (C-9), 188.02 (C-10).
m.p. 207.5oC (dec.)
Biochemicals and Antibodies
Purified non-labeled mouse/rabbit mono-/polyclonal antibodies were anti-p21waf/cip1, p53, GADD45G, MDM2, p27Waf1/kip1, Ki67, pIKK1/2, p-p65, p-IκB-α (Cell Signaling Technology). HRP-conjugated secondary anti-mouse and anti-rabbit antibodies were obtained from Life Technologies and Cell Signaling Technology, Inc., respectively. Anti-α-tubulin-HRP conjugated antibody was purchased from Cell Signaling Technology, Inc. Anti-rabbit eAlexa Fluor 488, Alexa Fluor 568 phalloidin and Hoechst 33342 dye were purchased by Thermo Fisher Scientific.
TNF-α were purchased from PeproTech Inc. and LPS from Sigma-Aldrich.
5x NFκB-luc-Reporter was obtained from Agilent Technologies, p53-luc Reporter, p53 control vector and eGFP plasmid were purchased from Life Technologies. Caspase3/7, 8 and 9 assay and CellTiter-Blue cell viability Assay from Promega. Cisplatin and mitoxantron were acquired from Sigma-Aldrich.
Cells and cell cultures
Human primary melanocytes were obtained from Provitro and cultivated in melanocyte growth medium (Provitro) with 10 % FCS (PAA Laboratories) and 1% Penicillin/Streptomycin (Sigma-Aldrich). Human melanoma cell lines A375 were purchased from Sigma-Aldrich, IGR39 and IGR37 from ATCC, while MCM1 and MCM1G (melanoma) were provided by Prof. Peter Petzelbauer (Department of Dermatology, Medical University of Vienna, Austria). Primary tumor metastasis-derived cell lines Mel.7, Mel.17 and Mel.15 were isolated and cultivated as described previously.63 In all experiments cell culture medium was supplemented with 10 % FCS and 1 % Pen/Strep unless indicated otherwise in the figure description.
Spheroids were generated by embedding the cells in matrigel (Corning, USA; Geltrex, LifeTechnologies) for ten days and spheroids were used for viability assay and microscopy.
Replacement Assay
2 x 104 HaCaT cells per well were grown to a monolayer in 96 well plates, fixed with 3.5 % formaldehyde and stained with Hoechst. The monolayer was measured before treatment and after 24 h treatment with different concentrations of 4-DACL, dimethylsulfoxid (DMSO) and ethidium bromide using Infinite F200 PRO multiplate reader (TECAN) at a wavelength of 360/465 nm.
Topoisomerase I and II Assay
Topoisomerase I and II assay kits were purchased from Inspiralis and performed according to manufactuers' instruction. Samples were run on 1 % agarose gel with 70 V for 2.5 h, stained with GelRed (VWR) and detected using ChemiDocTM MP Imaging System, Universal Hood III (BioRad).
ROS Assay
2 x 104 IGR37 cells per well were grown in 96 well plates and treated with tert-Butylhydroperoxid (TBHP), DMSO (neg. ctrl) or different concentrations of 4-DACL for 1.5 h. Then CellROX Green and Deep Red Reagents (Life Technologies) were added to the treated cells at a concentration of 0.01 µM and incubated for 2 h. Thereafter, the cells were washed with PBS and measured using Infinite F200 PRO multiplate reader at a wavelength of 485/535 and 610/670.
Western blotting
For Western blotting, proteins were extracted from 105 cells from each cell line. Total protein extracts were separated by 4-20% SDS-PAGE (Peqlab) and transferred with Trans-Blot® Turbo™, Transfer System to a Trans-Blot® Turbo™ TransferPack Mini Format, 0.2 µm polyvinylidene difluoride membrane (BioRad). Membranes were blocked with 5% nonfat milk in Tris-buffered saline pH 7.4 (TBS, BioRad), and immunodetection was carried out using specific antibodies (see Biochemicals and Antibodies) via chemiluminescence with ChemiDocTM MP Imaging System, Universal Hood III (BioRad).
Luciferase Reporter gene assay
A 5x NF-κB-Luc gene was co-transfected with enhanced green fluorescence protein (eGFP) plasmid into A375, IGR37 and MCM1, starved for 18h with 1% FCS, pre-treated with 10 ng/ml TNF-α, 10 ng/ml LPS or 10% FCS and stimulated with 10, 5, 2.5, 1.25, 0.62 or 0µg/ml 4-DACL. Luciferase values were determined and normalized for co-transfected eGFP values.
A p53-Luc reporter gene was co-transfected with eGFP and/or p-53 plasmid into A375 and IGR37 and cells were non-stimulated or stimulated with 0.5, 1 or 5 µg/ml 4-DACL and luciferase levels were detected 24 h later using Infinite F200 PRO multiplate reader (TECAN) and normalized for a co-transfected eGFP control.
Flow Cytometry analysis
For cell cycle distribution analysis NHM, A375 and IGR37 cells were starved for 24 h before cells were treated with 0, 1, 2.5 and 5µg/ml 4-DACL or left untreated for 24 hours. Cells were detached, washed with PBS and fixed with 70 % ice cold ethanol. Then cells were washed again with PBS and stained with propidium iodide solution (0.1 % Triton-X-100, 2 mg DNAse free RNAse A and 500 µg/ml propidium iodide) for 15 min at 37 °C. Thereafter flow cytometry analysis was performed using Accuri Flow Cytometer and CFlow plus software (BD Biosciences).
For apoptosis-necrosis detection IGR37 and A375 cells were treated with 1, 2.5, 5 and 10µg/ml 4-DACL or left untreated for 24 h. Then cells were detached, centrifuged and washed with ice cold PBS. Thereafter, cells were stained with APC-Annexin V (BD-Pharmingten) in 1x binding buffer (BD-Pharmingen) for 30 min at 4 °C. Then propidium iodide solution (Sigma-Aldrich) was added and flow cytometry analysis was performed using Accuri Flow Cytometer and CFlow plus software (BD Biosciences).
Cell viability assays and apoptosis
Cells were treated with different concentrations of 4-DACL (200-0.4µg/ml) for 24h, cell survival normalized to untreated cells (pos. contr.) and analyzed by CellTiter-Blue cell viability assay substrate. After 2 hours of incubation the cells were analyzed with Infinite F200 PRO multiplate reader (TECAN).
Caspase-Glo® 3/7, 8 and 9 Assay from Promega was used to determine cells in apoptosis. Cells were stained with celltracker and treated as in the cell viability assay. Fluorescent signal of celltracker was detected prior to addition of luciferase substrate. Luciferase was detected with Infinite F200 PRO multiplate reader (TECAN) and normalized for fluorescence values.
Cell proliferation assay
A375 and IGR37 cells were seeded in 96 well plates at an amount of 2500 cells per well. After 24 hours cells were stained with celltracker and fluorescence was detected. Then cells were treated with 5, 1, or 0.5 µg/ml 4-DACL or were left untreated (ctrl) and after 48 and 72 hours staining and detection of cells with celltracker was repeated.
Immunostaining
Cells were seeded on 96 well plates and treated with 4-DACL or left untreated for 6 or 16 h. Cells were fixed with 3.5% formaldehyde in PBS for 30 min and permeabilized with 0.1% Triton X-100 in PBS for 10 min. Proteins were immunodetected with 5 μg/ml polyclonal anti-Ki67 antibody (for 16 h at 4°C) and Alexa Fluor-488-conjugated anti-rabbit secondary antibody and/or Allexa Fluor-568 phalloidin for 2h. Nuclei were counterstained with Hoechst 33342. Wells were washed with PBS and viewed under a fluorescence microscope (Zeiss Axio observer z1).
Statistical analysis
Statistical evaluation of the results was performed using Student's t-test. Data are expressed as mean ± SE, and significance was established. P < 0.05 (*) was considered statistically significant, and P<0.01 (**) was considered as statistically highly significant.
Acknowledgements
This work was supported by the Austrian Forschungs Förderungs Gesellschaft (FFG; Project Basic).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Gill M, Smrdel AF, Strauch RJ Pigments of Fungi. P. Structure and Absolute Stereochemistry of Antibiotic Tetrahydroanthraquinones from the Fungus Dermocybe splendida Horak. X-Ray Structure Determination of Austrocortirubin Phenylboronate and Austrocortilutein Acetonide. J Chem Soc, Perkin Trans 1. 1990:1583-1592
2. Gill M, Gimenez A, Jhingran AG, Qureshi A. Austrocortilutein Stereoisomers from Australian Fungi of the Genus Dermocube. Phytochemistry. 1992;31:947-951
3. Burns CJ, Gill M. Pigments of Fungi. XXII. Synthesis of (±)-Austrocortirubin and Related Tetrahydroanthraquinones. Aust J Chem. 1991;44:1447-1458
4. Hurst EA, Harbour JW, Cornelius LA. Ocular melanoma: a review and the relationship to cutaneous melanoma. Arch Dermatology. 2003;139:1067-1073
5. Chin L, Garraway LA, Fisher DE. Malignant melanoma: genetics and therapeutics in the genomic era. Genes Dev. 2006;20:2149-2182
6. Chang AE, Karnell LH, Menck HR. The National Cancer Data Base Report on Cutaneous and Noncutaneous Melanoma. Cancer. 1998;83:1664-1678
7. Lomas J, Martin-Duque P, Pons M, Quintanilla M. The genetics of malignant melanoma. Front Biosci. 2008;13:5071-93
8. Sharma HW, Narayanan R. The NF-kappaB transcription factor in oncogenesis. Anticancer Res. 1996;16(2):589-96
9. Baeuerle PA, Baltimore D. NF-kappa B: ten years after. Cell. 1996;87(1):13-20
10. Xiong Y, Hannon GJ, Zhang H, Casso D, Kobayashi R, Beach D. p21 is a universal inhibitor of cyclin kinases. Nature. 1993;366(6456):701-4
11. Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell. 1993;75(4):805-16
12. el-Deiry WS. p21/p53, cellular growth control and genomic integrity. Curr Top Microbiol Immunol. 1998;227:121-37
13. Jiang H, Lin J, Su ZZ, Herlyn M, Kerbel RS, Weissman BE, Welch DR, Fisher PB. The melanoma differentiation-associated gene mda-6, which encodes the cyclin-dependent kinase inhibitor p21, is differentially expressed during growth, differentiation and progression in human melanoma cells. Oncogene. 1995;10(9):1855-64
14. Vidal MJ, Loganzo F Jr, de Oliveira AR, Hayward NK, Albino AP. Mutations and defective expression of the WAF1 p21 tumour-suppressor gene in malignant melanomas. Melanoma Res. 1995;5(4):243-50
15. Bulavin DV, Tararova ND, Aksenov ND, Pospelov VA, Pospelova TV. Deregulation of p53/p21Cip1/Waf1 pathway contributes to polyploidy and apoptosis of E1A+cHa-ras transformed cells after gamma-irradiation. Oncogene. 1999;18(41):5611-9
16. Yoshida K, Miki Y. The cell death machinery governed by the p53 tumor suppressor in response to DNA damage. Cancer Sci. 2010;101(4):831-5
17. Montano X, Shamsher M, Whitehead P, Dawson K, Newton J. Analysis of p53 in human cutaneous melanoma cell lines. Oncogene. 1994;9(5):1455-9
18. Lyu YL, Kerrigan JE, Lin CP, Azarova AM, Tsai YC, Ban Y, Liu LF. Topoisomerase IIbeta mediated DNA double-strand breaks: implications in doxorubicin cardiotoxicity and prevention by dexrazoxane. Cancer Res. 2007;67(18):8839-46
19. Weiss RB. The anthracyclines: will we ever find a better doxorubicin? Semin Oncol. 1992;19(6):670-86
20. Huang L, Zhang T, Li S, Duan J, Ye F, Li H, She Z, Gao G, Yang X. Anthraquinone G503 induces apoptosis in gastric cancer cells through the mitochondrial pathway. PLoS One. 2014;9(9):e108286
21. Xie G, Zhu X, Li Q, Gu M, He Z, Wu J, Li J, Lin Y, Li M, She Z. et al. J. SZ-685C, a marine anthraquinone, is a potent inducer of apoptosis with anticancer activity by suppression of the Akt/FOXO pathway. Br J Pharmacol. 2010;159(3):689-97
22. Zhu X, He Z, Wu J, Yuan J, Wen W, Hu Y, Jiang Y, Lin C, Zhang Q, Lin M. et al. A marine anthraquinone SZ-685C overrides adriamycin-resistance in breast cancer cells through suppressing Akt signaling. Oncol Rep. 2013;29(6):2341-7
23. Huang Q, Lu G, Shen HM, Chung MC, Ong CN. Anti-cancer properties of anthraquinones from rhubarb. Med Res Rev. 2007;27(5):609-30
24. Kumar A, Dhawan S, Aggarwal BB. Emodin (3-methyl-1,6,8-trihydroxyanthraquinone) inhibits TNF-induced NF-kappaB activation, IkappaB degradation, and expression of cell surface adhesion proteins in human vascular endothelial cells. Oncogene. 1998;17(7):913-8
25. Kuo PL, Lin TC, Lin CC. The antiproliferative activity of aloe-emodin is through p53-dependent and p21-dependent apoptotic pathway in human hepatoma cell lines. Life Sci. 2002;71(16):1879-92
26. Balch CM, Cascinelli N. The new melanoma staging system. Tumori. 2001;87:64-68
27. Sun W, Schuchter LM. Metastatic Melanoma. Curr Treat Options Oncol. 2001;2:139-202
28. Bhatia S, Tykodi SS, Thompson JA. Treatment of Metastatic Melanoma. Oncology. 2009;23(6):488-496
29. Su YT, Chang HL, Shyue SK, Hsu SL. Emodin induces apoptosis in human lung adenocarcinoma cells through a reactive oxygen species-dependent mitochondrial signaling pathway. Biochem Pharmacol. 2005;70(2):229-41
30. Dhawan P, Richmond A. A novel NF-kappa B-inducing kinase-MAPK signaling pathway up-regulates NF-kappa B activity in melanoma cells. J Biol Chem. 2002;277(10):7920-8
31. Kreiseder B, Orel L, Bujnow C, Buschek S, Pflueger M, Schuett W, Hundsberger H, de Martin R, Wiesner C. α-Catulin downregulates E-cadherin and promotes melanoma progression and invasion. Int J Cancer. 2013;132(3):521-30
32. Huang Q, Shen HM, Shui G, Wenk MR, Ong CN. Emodin inhibits tumor cell adhesion through disruption of the membrane lipid Raft-associated integrin signaling pathway. Cancer Res. 2006;66(11):5807-15
33. Zhang L, Chang JH, Zhang BQ, Liu XG, Liu P, Xue HF, Liu LY, Fu Q, Zhu M, Liu CZ. The pharmacokinetic study on the mechanism of toxicity attenuation of rhubarb total free anthraquinone oral colon-specific drug delivery system. Fitoterapia. 2015;104:86-96
34. Lee HZ. Effects and mechanisms of emodin on cell death in human lung squamous cell carcinoma. Br J Pharmacol. 2001;134(1):11-20
35. ZhangL Chang CJ, Bacuss SS Hung MC. Suppressed transformation and induced differentiation of HER-2/neu-overexpressing breast cancer cells by emodin. Cancer Res. 1995;55:3890
36. Chan TC, Chang CJ, Koonchanok NM, Geahlen RL. Selective inhibition of the growth of ras-transformed human bronchial epithelial cells by emodin, a protein-tyrosine kinase inhibitor. Biochem Biophys Res Commun. 1993;193:1152
37. Yeh FT, Wu CH, Lee HZ. Signaling pathway for aloe-emodin-induced apoptosis in human H460 lung nonsmall carcinoma cell. Int J Cancer. 2003;106:26-33
38. Mazerski J, Martelli S, Borowski E. The geometry of intercalation complex of antitumor mitoxantrone and ametantrone with DNA: molecular dynamics simulations. Acta Biochim Pol. 1998;45(1):1-11
39. Lown JW, Morgan AR, Yen SF, Wang YH, Wilson WD. Characteristics of the binding of the anticancer agents mitoxantrone and ametantrone and related structures to deoxyribonucleic acids. Biochemistry. 1985;24(15):4028-35
40. Cheng CC, Zbinden G, Zee-Cheng RK. Comparison of antineoplastic activity of aminoethylaminoanthraquinones and anthracycline antibiotics. J Pharm Sci. 1979;68(3):393-6
41. Fox ME, Smith PJ. Subcellular localisation of the antitumour drug mitoxantrone and the induction of DNA damage in resistant and sensitive human colon carcinoma cells. Cancer Chemother Pharmacol. 1995;35(5):403-10
42. Allimuthu T. Dharmaraja and Harinath Chakrapani. A Small Molecule for Controlled Generation of Reactive Oxygen Species. Org Lett. 2014;16(2):398-401
43. Ross D, Thor H, Threadgill MD, Sandy MS, Smith MT, Moldéus P, Orrenius S. The role of oxidative processes in the cytotoxicity of substituted 1,4-naphthoquinones in isolated hepatocytes. Arch Biochem Biophys. 1986;248(2):460-6
44. Hildeman DA, Mitchell T, Kappler J, Marrack P. T cell apoptosis and reactive oxygen species. J Clin Invest. 2003;111(5):575-81
45. Ray PD, Huang BW, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 2012;24(5):981-90
46. Morgan MJ, Liu ZG. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011;21(1):103-15
47. Shattuck-Brandt RL, Richmond A. Enhanced degradation of I-kappaB alpha contributes to endogenous activation of NF-kappaB in Hs294T melanoma cells. Cancer Res. 1997;57(14):3032-9
48. Pepper C, Hewamana S, Brennan P, Fegan C. NF-kappaB as a prognostic marker and therapeutic target in chronic lymphocytic leukemia. Future Oncol. 2009;5(7):1027-37
49. Madonna G, Ullman CD, Gentilcore G, Palmieri G, Ascierto PA. NF-κB as potential target in the treatment of melanoma. J Transl Med. 2012;10:53
50. Wiesner C, Winsauer G, Resch U, Hoeth M, Schmid JA, van Hengel J, van Roy F, Binder BR, de Martin R. Alpha-catulin, a Rho signalling component, can regulate NF-kappaB through binding to IKK-beta, and confers resistance to apoptosis. Oncogene. 2008;27(15):2159-69
51. Kreiseder B, Holper-Schichl YM, Muellauer B, Jacobi N, Pretsch A, Schmid JA, Martin Rd, Hundsberger H, Eger A, Wiesner C. Alpha-catulin contributes to drug-resistance of melanoma by activating NF-κB and AP-1. PLoS One. 2015;10(3):e0119402
52. Wang CY, Cusack JC Jr, Liu R, Baldwin AS Jr. Control of inducible chemoresistance: enhanced anti-tumor therapy through increased apoptosis by inhibition of NF-kappaB. Nat Med. 1999;5(4):412-7
53. Li HL, Chen HL, Li H, Zhang KL, Chen XY, Wang XW, Kong QY, Liu J. Regulatory effects of emodin on NF-kappaB activation and inflammatory cytokine expression in RAW 264.7 macrophages. Int J Mol Med. 2005;16:41-47
54. Wang CY, Mayo MW, Baldwin AS Jr. TNF- and cancer therapy-induced apoptosis: potentiation by inhibition of NF-kappaB. Science. 1996;274(5288):784-7
55. Ma S, Tang J, Feng J, Xu Y, Yu X, Deng Q, Lu Y. Induction of p21 by p65 in p53 null cells treated with Doxorubicin. Biochim Biophys Acta. 2008;1783(5):935-40
56. Siebenlist U, Franzoso G, Brown K. Structure, regulation and function of NF-kappa B. Annu Rev Cell Biol. 1994;10:405-55
57. Webster GA, Perkins ND. Transcriptional cross talk between NF-kappaB and p53. Mol Cell Biol. 1999;19(5):3485-95
58. Ak P, Levine AJ. p53 and NF-κB: different strategies for responding to stress lead to a functional antagonism. FASEB J. 2010;24(10):3643-52
59. Guttridge DC, Albanese C, Reuther JY, Pestell RG, Baldwin AS Jr. NF-kappaB controls cell growth and differentiation through transcriptional regulation of cyclin D1. Mol Cell Biol. 1999;19(8):5785-99
60. Yan YY, Fu LW, Zhang W, Ma HS, Ma CG, Liang YJ, Liu BY, Yu JZ, Wu QZ, Dong YM. Emodin azide methyl anthraquinone derivative induced G0/ G1 arrest in HER2/neu-overexpressing MDA-MB-453 breast cancer cells. J BUON. 2014;19(3):650-5
61. Abu N, Akhtar MN, Ho WY, Yeap SK, Alitheen NB. 3-Bromo-1-hydroxy-9,10-11MCF-7 and MDA-MB231. Molecules. 2013;18(9):10367-77
62. Chihara T, Shimpo K, Beppu H, Yamamoto N, Kaneko T, Wakamatsu K, Sonoda S. Effects of Aloe-emodin and Emodin on Proliferation of the MKN45 Human Gastric Cancer Cell Line. Asian Pac J Cancer Prev. 2015;16(9):3887-91
63. Amatschek S. Lucas R, Eger A, Pflueger M, Hundsberger H, Knoll C, Grosse-Kracht S, Schuett W, Koszik F, Maurer D, Wiesner C. CXCL9 induces chemotaxis, chemorepulsion and endothelial barrier disruption through CXCR3-mediated activation of melanoma cells. Br J Cancer. 2011;104(3):469-79
Author contact
![]() Corresponding author: Christoph Wiesner, PhD, SeaLife Pharma GmbH, 3430 Tulln, Austria. Phone: +43-680-3250982; E-mail: wiesnercom.
Corresponding author: Christoph Wiesner, PhD, SeaLife Pharma GmbH, 3430 Tulln, Austria. Phone: +43-680-3250982; E-mail: wiesnercom.