Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2016; 7(5):512-515. doi:10.7150/jca.13651 This issue Cite
Short Research Communication
Loss of MKK3 and MK2 Copy Numbers in Non-Small Cell Lung Cancer
Johanna Samulin Erdem, Vidar Skaug, Aage Haugen, Shanbeh Zienolddiny ![]()
Section of Toxicology and Biological Work Environment, Department of Biological and Chemical Work Environment, National Institute of Occupational Health, Oslo, Norway.
Received 2015-8-25; Accepted 2015-12-8; Published 2016-2-5
Abstract
Identification of genetic alterations in members of the p38 mitogen-activated protein kinase (MAPK) pathway is important as these proteins have dynamic roles in tumor progression and may serve as potential therapeutic targets in cancer. We analyzed tumor and non-tumorous lung tissue of 233 non-small cell lung cancer (NSCLC) patients for the presence of copy number alterations (CNAs) in the MAPK kinase 3 (MKK3) and MAPK-activated kinase 2 (MK2) genes. We report frequent CNAs in MKK3 and MK2 genes in NSCLC. Copy number losses were detected in 31% of NSCLC tumors (odds ratio: 7.08, 95% confidence interval: 3.2-15.6, P<0.001) for the MKK3 gene and in 28% of tumors for the MK2 gene (odds ratio: 3.68, 95% confidence interval: 1.9-7.2, P<0.001). Several of the non-tumorous tissues showed an elevated MKK3 copy number, with a concurrent loss of this in 89% of the paired tumors. MKK3 gene deletions were significantly more frequent in squamous and large cell carcinoma than in adenocarcinoma. These data demonstrate a novel loss of MKK3 and MK2 genomic copy numbers in NSCLC tumors, and suggest these genes as interesting therapeutic candidates in NSCLC.
Keywords: MKK3, MK2, p38 MAPK, copy number alterations, non-small cell lung cancer.
Introduction
The p38 mitogen-activated protein kinase (MAPK) pathway is a stress response pathway, which is activated by a wide variety of stimuli. The p38 MAPK signaling cascade is important in various biological settings other than inflammation and stress responses, including cancer promotion and progression. It affects tumorigenesis by regulating cellular proliferation, differentiation, apoptosis, migration and invasion in a cell-type and cell-context specific manner [1]. The tumor-suppressing effect of the p38 pathway is mediated on multiple levels, as the pathway regulates oncogene-induced senescence, replicative senescence, contact inhibition and DNA-damage responses through various downstream effectors including TP53 [2]. As such, dysregulated p38 MAPK signaling is associated with advanced stages and poor survival in cancer patients [3]. p38 MAPKs are activated by the upstream MAPK kinases MKK3 (MAP2K3) and MKK6 (MAP2K6), and regulate phosphorylation of major transcription factors (e.g. TP53), and protein kinases, including MAPK-activated kinase 2 (MK2; also known as MAPKAPK2) [1]. It is evident that the p38 MAPK pathway is important in lung cancer [4], and recent reports demonstrating amplifications in the MK2 gene [5] and increased MKK3 mRNA expression [6] in non-small cell lung cancer (NSCLC) make these genes interesting candidates as tumor suppressors in NSCLC.
Thus, identification of genetic alterations in members of the p38 MAPK pathway is important as they have dynamic roles in tumor progression and may serve as potential therapeutic targets in cancer [3, 7]. To address this, copy number alterations (CNAs) in MKK3 and MK2 genes in NSCLC patients were investigated and their correlation to TP53 mutational status was studied.
Materials and Methods
Cases
We enrolled 233 Caucasians of Norwegian origin (71.1% male and 88.2% smokers) with early-stage lung cancer between 1988 and 1994. All patients gave written informed consent to participate in the study. The average age was 64±10 years. The smokers had on average smoked 15.3±8.0 cigarettes/day for 41.5±11.6 years, corresponding to 31.7±18.5 total pack-years. Samples of adjacent non-tumorous lung tissue were cut from the lobectomy specimens at the time of surgery. Tumor histology was confirmed by an experienced pathologist and samples containing ≥80% of tumor cells were included: 45.6% of the tumors were classified as adenocarcinoma (AD), 39.9% as squamous cell carcinoma (SQ) and 14.4% as large cell carcinoma (LC). Tissues were snap-frozen in liquid nitrogen and kept at -80°C until further processing. The study was approved by the Regional Committee for Medical and Health Research Ethics in Norway in accordance with the WMA Declaration of Helsinki.
Assessment of TP53 mutations and CNA by quantitative (q)PCR
DNA was extracted using standard proteinase K digestion followed by phenol-chloroform extraction and ethanol precipitation. Exons 4-9 in the TP53 gene were sequenced for mutations in tumor tissues [8, 9]. MKK3 and MK2 copy numbers were analyzed using qPCR and SYBR Green I technology according to D'Haene et al. [10], with some modifications. The multicopy gene FTH1 was used as a reference gene. Primer sequences were: MKK3 forward primer, 5'-GGAGGCTGATGACTTGGTGA-3'; reverse primer, 5'-ACTGAGAGGGATAGGCAGGG-3'; MK2 forward primer, 5'-CGGCCTGCAGATCAAGAAGA-3'; reverse primer, 5'-CCTGGCTGGTGACCTTGTAG-3'; FTH1 forward primer, 5'-GATGATGTGGCTTTGAAGAACTTTGCCA-3'; reverse primer, 5'-CACCTCGTTGGTTCTGCAGCTTCATCA-3'. Primer specificity was determined by melting point analysis. The PCR was run in duplicate using a relative standard curve approach. The standard curve was generated by performing serial dilutions of plasmid DNA containing one copy of the area of interest for each of the assayed genes. pUC57 plasmid DNA (GenScript, Piscataway, NJ, USA) was added to each standard to maintain a constant amount of total DNA per reaction tube. Samples were normalized to the mean of control samples run on each plate. H460 and H774 cell lines (American Type Culture Collection) were used as internal controls for the MKK3 assay because H774 cells have a double deletion of the MKK3 gene [11]. A 1/1 mixture of H460 and H774 cells had half genomic MKK3 level (0.94±0.36). The genomic MKK3 level in H460 cells was determined as 2.12±0.33. Based on this, the cut-off for copy number amplification was set to 2.45 and samples with <1.55 copies were considered to have a copy number loss. The same cut-off values were used for both assays.
Statistical analysis
Statistical analyses were performed using IBM SPSS version 22.0 software. Associations between CNAs and lung tumor development were estimated by odds ratios (ORs) and their 95% confidence intervals (CIs) from conditional logistic regression adjusted for age, gender, total pack-years and histology. Effects of clinicopathological data and TNM staging on gene status were assessed by χ2 or Fisher's exact test for categorical variables and by nonparametric tests for ordinal variables. P<0.05 was considered significant.
Results and discussion
The p38 MAPK pathway has well-known functions in inflammation and stress response but also acts as a negative regulator of cell proliferation and as a tumor suppressor by its impact on several cell-cycle regulators, including TP53. Components of the p38 MAPK pathway are potential targets for novel cancer therapies [2] and CNAs in important genes of the p38 MAPK pathway have been reported in many cancer types [12]. MKK3 gene deletion is a known risk factor for breast cancer and MKK3 expression is reduced in several different tumor types, including lung cancer [6]. In our data set, analysis of tumor and non-tumorous tissues from 233 NSCLC patients demonstrated that loss of MKK3 copy number was prominent in 31.1% of NSCLC tumors compared with only 7.0% of the adjacent non-tumorous tissues (OR: 7.08, 95% CI: 3.2-15.6, P<0.001, Fig. 1A). Interestingly, 18% of the non-tumorous tissues showed an increase in copy number (≥3 copies) of the MKK3 gene (P=0.002, Fig. 1A). However, in 71.7% of these patients the extra MKK3 gene copy was not present in tumors, and 17.4% of these showed a loss of MKK3 copy number in the paired tumor tissue. The loss of MKK3 copy numbers was significantly more frequent in SQ and LC than in AD (P=0.035, Fig. 1B). MKK3 gene status was not affected by other clinicopathological parameters such as gender, age and smoking (data not shown).
MKK3 and MK2 copy number status in NSCLC. Percentage of copy number atlterations (CNAs) in (A) MKK3 and (C) MK2 genes in lung tumors and non-tumorous tissues. Distribution of (B) MKK3 and (D) MK2 gene CNAs in NSCLC of different histology. Copy number loss was defined as copy number ≤1.55; whereas, gain was defined as copy number ≥2.45.
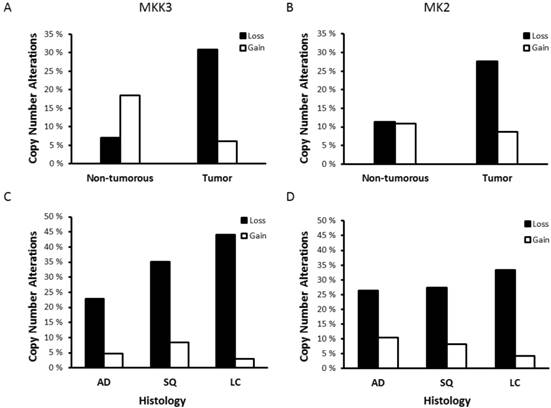
The p38 downstream target MK2 is also implicated in tumorigenesis [13]. In fact, patients with highly elevated MK2 copy numbers (>4) have increased risk of NSCLC [5]. In contrast, we demonstrated a loss of MK2 copy number in 27.7% of NSCLC tumors (OR: 3.68, 95% CI: 1.9-7.2, P<0.001, Fig. 1C). This was not affected by tumor histology (Fig. 1D), gender, age or smoking (data not shown). In contrast to the previous study showing increased MK2 copy numbers, our data are consistent with a suggested role of the p38/MK2 pathway in tumor suppression [2].
Analysis of effects of MK2 and MKK3 CNAs on tumor size and on involvement of lymph nodes as classified by TNM staging were investigated by Pearson's chi-squared test. MK2 and MKK3 copy numbers were not correlated with tumor size and involvement of lymph nodes (data not shown). It should be noted that the cohort used in this study included only early-stage NSCLC without metastatic spread. Thus, the possible correlation of MK2 and MKK3 copy numbers on NSCLC metastasis cannot be excluded.
TP53 is an important tumor suppressor downstream of MKK3/p38/MK2 signaling in NSCLC. We observed no correlation between MKK3 or MK2 copy numbers and TP53 mutational status (P=0.407 and P=0.430, respectively). Others have shown that the p38-pathway regulates TP53 on a transcriptional level directly by p38 or by its downstream kinases MK2 or MK5 following DNA damage [2, 14].
In conclusion, this is the first report of loss of MKK3 and MK2 genomic copy numbers in NSCLC tumors. Our study was limited to NSCLC, and these observations may not be universal for all lung cancer phenotypes. These data highlight the importance of understanding genomic changes in novel tumor suppressors and suggest that MKK3 and MK2 may be interesting candidates with potential therapeutic value in NSCLC.
Abbreviations
AD: adenocarcinoma; CNA: copy number alteration; LC: large cell carcinoma; NSCLC: non-small cell lung cancer; OR: Odds Ratio; SQ: squamous cell carcinoma.
Acknowledgements
We thank Professor Lodve B. Stangeland (University of Bergen, Norway) for his kind contribution in collecting tissue samples, and Mayes Kasem, Tove Andreassen and Elin Thorner Einarsdottir for their excellent technical assistance. This study was supported by the Norwegian Cancer Society.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Wagner EF, Nebreda AR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer. 2009;9:537-49
2. Han J, Sun P. The pathways to tumor suppression via route p38. Trends in Biochemical Sciences. 2007;32:364-71
3. Koul HK, Pal M, Koul S. Role of p38 MAP Kinase Signal Transduction in Solid Tumors. Genes & Cancer. 2013;4:342-59
4. Greenberg AK, Basu S, Hu J, Yie T-a, Tchou-Wong KM, Rom WN. et al. Selective p38 Activation in Human Non-Small Cell Lung Cancer. Am J Respir Cell Mol Biol. 2002;26:558-64
5. Liu B, Yang L, Huang B, Cheng M, Wang H, Li Y. et al. A functional copy-number variation in MAPKAPK2 predicts risk and prognosis of lung cancer. Am J Hum Genet. 2012;91:384-90
6. MacNeil AJ, Jiao S-C, McEachern LA, Yang YJ, Dennis A, Yu H. et al. MAPK Kinase 3 Is a Tumor Suppressor with Reduced Copy Number in Breast Cancer. Cancer Res. 2014;74:162-72
7. Yong H-Y, Koh M-S, Moon A. The p38 MAPK inhibitors for the treatment of inflammatory diseases and cancer. Expert Opinion on Investigational Drugs. 2009;18:1893-905
8. Lind H, Ekstrom PO, Ryberg D, Skaug V, Andreassen T, Stangeland L. et al. Frequency of TP53 mutations in relation to Arg72Pro genotypes in non small cell lung cancer. Cancer Epidemiol Biomarkers Prev. 2007;16:2077-81
9. Kristensen AT, Bjorheim J, Ekstrom PO. Detection of mutations in exon 8 of TP53 by temperature gradient 96-capillary array electrophoresis. Biotechniques. 2002;33:650-3
10. D'Haene B, Vandesompele J, Hellemans J. Accurate and objective copy number profiling using real-time quantitative PCR. Methods. 2010;50:262-70
11. Teng DHF, Chen Y, Lian L, Ha PC, Tavtigian SV, Wong AKC. Mutation Analyses of 268 Candidate Genes in Human Tumor Cell Lines. Genomics. 2001;74:352-64
12. Dehan E, Ben-Dor A, Liao W, Lipson D, Frimer H, Rienstein S. et al. Chromosomal aberrations and gene expression profiles in non-small cell lung cancer. Lung Cancer. 2007;56:175-84
13. Johansen C, Vestergaard C, Kragballe K, Kollias G, Gaestel M, Iversen L. MK2 regulates the early stages of skin tumor promotion. Carcinogenesis. 2009;30:2100-8
14. Sun P, Yoshizuka N, New L, Moser BA, Li Y, Liao R. et al. PRAK Is Essential for ras-Induced Senescence and Tumor Suppression. Cell. 2007;128:295-308
Author contact
![]() Corresponding author: Dr. Shanbeh Zienolddiny, National Institute of Occupational Health, PO Box 8149 Dep, N-0033 Oslo, Norway. Tel +47-23195284, Fax: +47-23195203, E-mail: shan.zienolddinyno.
Corresponding author: Dr. Shanbeh Zienolddiny, National Institute of Occupational Health, PO Box 8149 Dep, N-0033 Oslo, Norway. Tel +47-23195284, Fax: +47-23195203, E-mail: shan.zienolddinyno.