Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2016; 7(5):500-511. doi:10.7150/jca.13599 This issue Cite
Research Paper
Ganoderma lucidum Combined with the EGFR Tyrosine Kinase Inhibitor, Erlotinib Synergize to Reduce Inflammatory Breast Cancer Progression
Ivette J. Suárez-Arroyo1, Tiffany J. Rios-Fuller1, Yismeilin R. Feliz-Mosquea2, Mercedes Lacourt-Ventura1, Daniel J. Leal-Alviarez1, Gerónimo Maldonado-Martinez1, Luis A. Cubano1, Michelle M. Martínez-Montemayor1 ![]()
1. Universidad Central del Caribe-School of Medicine, Bayamón, P.R.
2. Inter American University of Puerto Rico, Bayamón, P.R.
Received 2015-8-18; Accepted 2015-12-12; Published 2016-2-5
Abstract
The high incidence of resistance to Tyrosine Kinase Inhibitors (TKIs) targeted against EGFR and downstream pathways has increased the necessity to identify agents that may be combined with these therapies to provide a sustained response for breast cancer patients. Here, we investigate the therapeutic potential of Ganoderma lucidum extract (GLE) in breast cancer, focusing on the regulation of the EGFR signaling cascade when treated with the EGFR TKI, Erlotinib. SUM-149, or intrinsic Erlotinib resistant MDA-MB-231 cells, and a successfully developed Erlotinib resistant cell line, rSUM-149 were treated with increasing concentrations of Erlotinib, GLE, or their combination (Erlotinib/GLE) for 72h. Treatment effects were tested on cell viability, cell proliferation, cell migration and invasion. To determine tumor progression, severe combined immunodeficient mice were injected with SUM-149 cells and then treated with Erlotinib/GLE or Erlotinib for 13 weeks. We assessed the protein expression of ERK1/2 and AKT in in vitro and in vivo models. Our results show that GLE synergizes with Erlotinib to sensitize SUM-149 cells to drug treatment, and overcomes intrinsic and developed Erlotinib resistance. Also, Erlotinib/GLE decreases SUM-149 cell viability, proliferation, migration and invasion. GLE increases Erlotinib sensitivity by inactivating AKT and ERK signaling pathways in our models. We conclude that a combinatorial therapeutic approach may be the best way to increase prognosis in breast cancer patients with EGFR overexpressing tumors.
Keywords: Ganoderma lucidum, Erlotinib, EGFR, synergy, drug resistance.
Introduction
Inflammatory Breast Cancer (IBC) is one of the most lethal forms of BC, where patients have 43% increased risk of death compared to women with non-IBC advanced BC [1]. IBC lethality stems from its ability to invade the vascular and lymphatic systems via generation of tumor emboli that are responsible for the inflammatory phenotype, and for metastases [2]. IBC tumors are categorized into subtypes based on estrogen and progesterone receptor (ER/PR) presence or absence, presence and extent of Human Epidermal Growth Factor Receptor 2 (HER2) amplification, and Epidermal Growth Factor Receptor (EGFR) overexpression. Importantly, IBC patients with different molecular subtypes show different outcomes [3]. HER2 amplification accounts for ~40% of IBCs while EGFR overexpression occurs in ~30% of IBC cases and in ~50% of triple negative (TN)-IBCs [4]. Patients with EGFR-positive tumors have lower survival rates, and positivity is associated with higher IBC recurrence risk, making EGFR and HER2 attractive IBC targets [5].
A pharmacological approach consisting of small tyrosine kinase inhibitors (TKIs) has been developed to target EGFR and HER2. Erlotinib targets adenosine triphosphate (ATP)-binding sites, blocking the function of EGFR-TK activity, while Lapatinib inhibits EGFR and HER2-TK activity. Both prevent the activation of downstream cellular signals that promote tumor cell survival and proliferation and have been assessed as anti-IBC therapies [6]. Although promising, the high incidence of de novo or acquired TKI resistance has greatly diminished their overall effectiveness [7, 8]. Thus, studies are underway to identify agents that combined with TKIs provide a sustained response.
Activation of downstream EGFR pathways, leads to persistent activation of PI3K/AKT/mTOR and MAPK pathways and consequent development and maintenance of an EGFR resistant phenotype [9, 10]. Intrinsic and acquired Erlotinib resistance is associated with increased AKT and ERK activity [11, 12]. The combination of Erlotinib with other TKIs have been effective to overcome intrinsic and acquired Erlotinib resistance through downregulation of AKT activation [13]. However, the combination of TKIs and nutraceuticals that may sensitize cells to therapy, as well as enhance quality of life of BC survivors remains to be explored.
The medicinal mushroom Ganoderma lucidum, displays IBC cell antiproliferative and pro-apoptotic activity at doses that have no adverse effect on non-cancerous cell viability [14]. G. lucidum extract (GLE) suppresses BC cell growth and metastatic potential [15, 16] and reduces PI3K/AKT/mTOR downstream effectors and ERK1/2 expression in IBC models [17]. Ganoderma combined with taxol and cisplatin, enhance the growth-inhibitory effects in HER2+ cells [18]. However, GLE and TKI effects in IBC have not been studied.
The present study was designed to investigate GLE's therapeutic efficacy in combination with EGFR/HER2 TKIs and to elucidate how the combination contributes to the IBC cellular response. Our findings are the first to show that GLE synergizes with Erlotinib in EGFR-overexpressing IBC cells to overcome intrinsic and acquired Erlotinib resistance by targeting AKT and ERK1/2. Our data evidence GLE's potential to synergize with conventional therapies.
Materials and methods
Cell lines and reagents
SUM-149 and SUM-102 cells were obtained from Dr. Steven Ethier, Medical University of South Carolina (Charleston, SC) [19]. MDA-MB-231 were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). KPL-4 and MDA-IBC-3 cells were kindly provided by Dr. Kurebayashi (Kawasaki Medical School, Japan) [20] and by Dr. Wendy Woodward, University of Texas MD Anderson Cancer Center (Houston, TX), respectively. To develop Erlotinib resistant (rSUM-149), parental SUM-149 cells were cultured in media supplemented with increasing Erlotinib concentrations (0.1 µM to 10 µM) for three months, then maintained in 10 µM Erlotinib. All cells were cultured at 37 °C in 5 % CO2 using culture medium recommended by the supplier. Erlotinib and Lapatinib (LC Laboratories, Woburn, MA) 5 mM working stocks were dissolved in 100% sterile DMSO (Sigma Aldrich, St. Louis, MO). Capsules (500 mg) containing GLE fruiting body and cracked spores extract, known as ReishiMax GLp™, was purchased from Pharmanex® Inc. (Provo, UT), and a 160 mg/mL working stock (10% sterile DMSO) was used for in vitro experiments.
Cell viability assays
6x104-2x105 cells/well, were seeded and cultured for 24h. Then, cells were treated in duplicates with 2-fold serial dilutions of each treatment for 24 or 72h. Cells were fixed (cold methanol), and nuclei stained [0.4% propidium iodide, (PI)] (Sigma-Aldrich), and measured using a GloMax® Microplate Reader (Promega, Madison, WI). Cell viability was calculated as percent of surviving cells after treatment relative to vehicle wells.
Wash out assays
Cells were treated with or without GLE for 72h. The treatment was removed, cells were washed with PBS and incubated for an additional 72h with fresh media. After, cell viability was determined.
Median-effect analysis
Combination Index (CI) method is based on median-effect principle developed by T.Chou to determine the nature of drug interaction as well as the dose effect relationship of each drug and its combination [21]. The effect of drug combination is describe as: CI<1 synergism, CI=1 additive effect and CI>1 antagonism (Table 1). CIs were obtained using CompuSyn® v1.0 (Biosoft, Cambridge, UK).
Synergy and antagonism scale in drug combination studies [21].
| Synergism | Antagonism | ||
|---|---|---|---|
| CI Value | Effect Description | CI Value | Effect Description |
| <0.1 | Very strong synergism | 1.10-1.20 | Slight antagonism |
| 0.1-0.3 | Strong synergism | 1.20-1.45 | Moderate antagonism |
| 0.3-0.7 | Synergism | 1.45-3.3 | Antagonism |
| 0.7-0.85 | Moderate synergism | 3.3-10 | Strong antagonism |
| 0.85-0.9 | Slight synergism | >10 | Very strong antagonism |
CI values <1, =1, >1 indicate synergism, additive effect and antagonism.
Cell proliferation assay
For 5-bromo-2'-deoxyuridine (BrdU) incorporation detection (ELISA BrdU Cell Proliferation Assay Kit, Cell Signaling Technology, Danvers, MA), 5x104 SUM-149 cells/well were seeded and incubated overnight. After 24h, cells were treated with Erlotinib and/or GLE for 72h, then 10 µM-BrdU was added and after a 22h incubation the cells were fixed, DNA was denatured and fixed. BrdU mouse mAb was added to detect BrdU incorporation via colorimetric detection (450nm). Cell proliferation was calculated as percent of proliferating cells after treatment relative to vehicle wells.
Migration and invasion assays
Cell migration and invasion were measured using Corning® FluoroBlok™ Cell Culture Inserts and BD BioCoat Matrigel™ Invasion Assay (BD Biosciences, San José, CA) [14]. 2.5x105 quiescent cells/well were seeded in the top chambers, then treated with vehicle, 0.1 µM-Erlotinib, 0.05 mg/mL-GLE or Erl/GLE, and incubated at 37ºC to allow migration or invasion toward 10% FBS medium (chemoattractant). After 72h, cells on the upper membrane surface were removed with a cotton swab and cells attached to the bottom surface of the membrane were fixed and stained [14]. Cells were quantified with ImageJ (NIH, Bethesda, MD). Data were calculated as percent of migrated or invaded cells after treatment relative to vehicle.
Three-dimensional (3D) cell culture
105 SUM-149 or rSUM-149 cells seeded on MatTek (MatTek Corp., Ashland, MA) coverglass bottom dishes were labeled with Cell-Tracker™-Green-5-chloromethylfluorescein diacetate dye (Molecular Probes, Carlsbad, CA). Cells were overlaid with Matrigel:serum free media (1:1) and incubated at 37°C for 90min. Next day, cells were treated with vehicle, 0.1 µM-Erlotinib, 0.05 mg/mL-GLE or Erl/GLE for 72h. rSUM-149 cells were treated with vehicle, 2.0 µM-Erlotinib, 0.2 mg/mL-GLE or Erl/GLE for 72h. Spheroid formation was monitored daily via an inverted microscope. Micrographs at 200X magnification were digitally captured using an Olympus fluorescence microscope.
Immunoblotting
BC cells and tumors treated with Erlotinib and/or GLE were lysed and equal total protein was resolved via SDS-PAGE and immunoblotted with the indicated antibodies (Cell Signaling Technolog, or Abgent) [14].
In vivo study
Female SCID mice [21d, Charles River Laboratories International (Wilmington, MA)] housed under specific pathogen-free conditions, were given 2920X Teklad Global Rodent Diet (Harlan Laboratories, Indianapolis, IN) and sterile water ad libitum. Mice were housed and handled in accordance with UCC-IACUC guidelines. Cell inoculations were performed as described by us [22]. One-week post-inoculation, mice were randomly divided into vehicle [Erlotinib (n=9) and Erlotinib+GLE (n=10)] and experimental groups [Erlotinib (n=9) and Erlotinib+GLE (n=10)]. GLE was dissolved in 10% ethanol and Erlotinib in 0.5% methyl cellulose [23]. Mice were gavaged daily with each vehicle or 25 mg/kg_BW-Erlotinib or 25 mg/kg_BW-Erlotinib + 14 mg/kg_BW-GLE for 13wks. Mice treated with Erlotinib+GLE vehicle and experimental doses were gavaged with Erlotinib in the morning, then 6h later with GLE. Mouse weights and tumor volume (calipers measurements) were measured weekly. Tumor volume (mm3) was calculated: [π/6(L)(W)(H)]. Relative tumor volume was calculated as [(average tumor volume ratio on week “n”)/average tumor volume on week-1].
Statistical Analysis
Statistical analyses for in vitro studies were done using GraphPad Prism® v.6.0 (San Diego, CA) via one-way ANOVA with Dunnett's multiple comparisons estimator or two-way ANOVA with Bonferroni's correction. Quantified data are expressed as mean ± SEM. P≤0.05 was considered significant. IC50s were calculated from dose response curve fittings using the non-linear regression parameter: dose - response - inhibition (log [inhibitor] vs normalized response). CIs were obtained using CompuSyn® v.1.0 (Biosoft, Cambridge, UK). For in vivo statistical analysis, a 13wks statistical model with four comparison groups (Vehicle Erlotinib, Erlotinib, Vehicle Erlotinib+GLE, Erlotinib+GLE) was used. Analytical blocks for mouse weight were: block-1 [Vehicle Erlotinib vs Erlotinib] and block-2 [Vehicle Erlotinib+GLE vs Erlotinib+GLE]. For statistical analysis of tumor volume, analytical blocks were: block-1 [vehicle Erlotinib vs Erlotinib]; block-2 [vehicle Erlotinib+GLE vs Erlotinib+GLE] and block-3 [Erlotinib vs Erlotinib+GLE]. Normality of all variables was evaluated via the Shapiro-Wilk estimator, (tumor volume is the dependent variable). At a bivariate level, comparisons were made between each week independently in the individual blocks to assess differences in groups using the independent samples t test. To account for the variable time-horizon as a whole statistical unit to analyze tumor volume, a General Linear Model (GLM) Repeated Measures ANOVA approach per block was used. A Mauchly's test of sphericity was performed to assess if our models had or not the assumption of compound symmetry. If non-significant, we report the univariate results with an Epsilon correction; if significant; we report the multivariate results using Pillai's trace estimator. Explained factors were used to evaluate the time effect in our models. A test of between-subjects effect was applied to perceive statistical differences between the groups per block. Estimated marginal means (EMM) are reported. P≤0.05 was considered significant. In tumor volume statistical analysis, comparisons were made between each week independently in the individual blocks to assess differences in groups at a bivariate level using the independent samples t test. Others and we have reported that SUM-149 cells tend to form a ''diffuse'' tumor during the first weeks post-inoculation [22, 24]. Therefore to account for the lag time in tumor growth observed for the first 7wks of treatment, EMM were calculated taking weeks 8-13 as a unit, while weeks 1-7 were used as baseline. For tumor weight, a comparison statistical model with three blocks was used: block-1 [vehicle Erlotinib vs Erlotinib]; block-2 [vehicle Erlotinib+GLE vs Erlotinib+GLE] and block-3 [Erlotinib vs Erlotinib+GLE]. A normality diagnostics was done via Shapiro-Wilk estimator, and to detect median changes (dependent variable), a Kruskall-Wallis test was used. Post-hoc comparisons included three Mann-Whitney tests using Bonferroni correction (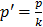 , where p'= the adjusted p value, p=0.05 and k=number of post-hoc test). IBM Statistical Package for Social Sciences (IBM-SPSS, Chicago, IL) v.22.0 for Windows was used for in vivo data.
, where p'= the adjusted p value, p=0.05 and k=number of post-hoc test). IBM Statistical Package for Social Sciences (IBM-SPSS, Chicago, IL) v.22.0 for Windows was used for in vivo data.
Results
EGFR/HER2 overexpressing cells are sensitive to GLE
Because EGFR and HER2 are important in BC progression, we tested GLE's effectiveness in EGFR (SUM-102 and SUM-149) and HER2+ cells (MDA-IBC-3 and KPL-4). The median inhibitory GLE concentration [IC50] was 0.09 and 0.18 mg/mL in SUM-102 and SUM-149 cells, respectively Fig. 1(A,B) and Fig. S1(A,B). GLE also affected SUM-149 cell proliferation (Fig. 1C). HER2+ cell IC50s were: MDA-IBC-3 (0.66 mg/mL) and KPL-4 (0.19 mg/mL). Washout experiments demonstrated that SUM-102, SUM-149 and KPL-4 cells did not recover from GLE treatment (Fig. S1C).
Effect of Erl/GLE in EGFR-overexpressing cells. A, B. SUM-102 and SUM-149 cells were treated with GLE for 72h. Cell viability was calculated as in materials and methods. IC50 was obtained from dose response curve fittings using non-linear regression. C. SUM-149 cells were treated with GLE for 72h, to measure BrdU incorporation. Significance against vehicle (*) (P≤0.05). SUM-149 cells were treated with Erlotinib, GLE or Erl/GLE for 72h. D, E. Cell viability and CIs were calculated. Significance against Erlotinib (*) or GLE (ᶲ) (P≤0.05). Columns represent means ± SEM. CIs were calculated based on the IC50 at a constant Erl:GLE ratio (1:1000) and at NCR (Erlotinib dilutions+0.05 mg/mL-GLE). CIs and fraction affected (Fa) calculated from the Erlotinib (E) and GLE (G) combinations were obtained using CompuSyn®. Fa-CI plot shows the interaction between drugs in function of Fa (original software output). CI=1 (dashed line), CIs<1 (synergism). Experiments were repeated at least three times.
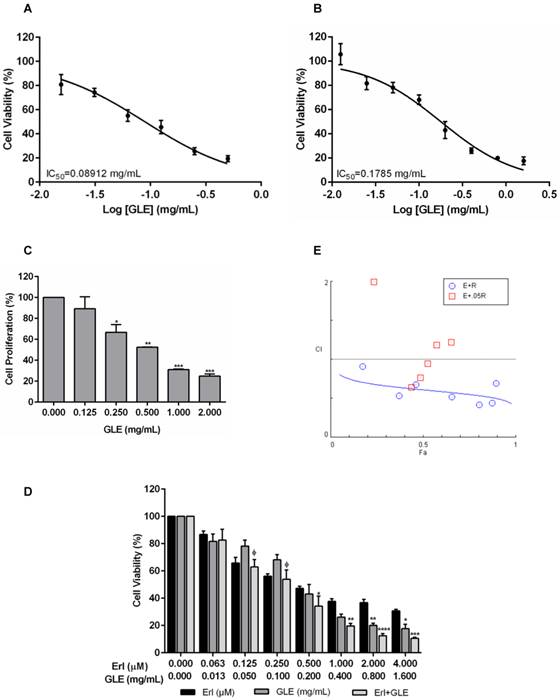
Synergic Erl/GLE effect in EGFR-overexpressing IBC cells
Chou's method has been used to determine the levels of synergism between natural products extracts (NPE) and between NPEs and anti-cancer drugs [25]. To test the efficacy of GLE combined with TKIs, EGFR and HER2+ cells were treated with Erlotinib or Lapatinib, GLE, or the combination of drugs simultaneously (Erl/GLE or Lap/GLE). Lap/GLE did not affect MDA-IBC-3 cell viability when compared to each drug, while significantly decreased KPL-4 cell viability. However, cell viability increased when cells were treated with higher Lap/GLE concentrations (Fig. S2A). In contrast, Erl/GLE significantly affected SUM-149 (Fig.1D), where at concentrations less than the IC50s of each, significantly decreased cell viability. When SUM-149 cells were treated with Erl/GLE at doses greater than the IC50s of each, the cells were significantly sensitive compared with Erlotinib. These results suggest that a synergic effect occurs between Erlotinib and GLE at low and at high doses. Since, we didn't observe a synergic Lap/GLE effect in HER2+ cells; we continued our study testing the efficacy of Erl/GLE in EGFR-overexpressing cells. To confirm the synergic Erl/GLE effect in SUM-149 cells, we determined the dose effect relationship of the combination calculating the CI. The CIs were calculated at a constant ratio (CR) of Erl:GLE (1:1000) and at NCR (Erlotinib concentrations plus 0.05 mg/mL-GLE). All combined doses at a constant Erl:GLE ratio were synergistic (Fig.1E). However, at a NCR, just three Erl/GLE showed synergy, while other concentrations show an antagonistic effect (CIs>1). Table 2 shows the affected fraction of SUM-149 cells versus CI values in constant and NCR. In SUM-149 cells, Erl/GLE CI was 0.61 at ED50 and reduced as the affected fractions increased indicating synergism, substantiating our results. Also, Erl/GLE (Dm=0.1) has greater potency than Erlotinib (Dm=0.6) or GLE (Dm=0.2).
Synergistic effect of Erlotinib and GLE in SUM-149 cells.
| Combination Index | |||||
|---|---|---|---|---|---|
| Constant Ratio | |||||
| Treatment | ED50 | ED75 | ED90 | ED95 | Dm |
| Erl+GLE | 0.60914 | 0.54950 | 0.49573 | 0.46221 | 0.10815 |
| Erl (μM) | 0.59348 | ||||
| GLE (mg/mL) | 0.17742 | ||||
| Non-constant Ratio | |||||
| Dose Erl (μM) | Dose GLE (mg/mL) | Effect | CI | Description | |
| 0.06 | 0.05 | 0.23473 | 1.99448 | A | |
| 0.13 | 0.43761 | 0.64429 | S | ||
| 0.25 | 0.48714 | 0.76154 | MS | ||
| 0.50 | 0.52588 | 0.94897 | SlS | ||
| 1.00 | 0.57428 | 1.18357 | SlA | ||
| 2.00 | 0.65307 | 1.21960 | MA | ||
| 4.00 | 0.67108 | 2.00307 | A | ||
Dm means the median effect dose or the potency of each drug such as IC50, ED50. A=Antagonism, MA=Moderate Antagonism, MS=Moderate Synergism, S=Synergism, SlA=Slight Antagonism, SlS=Slight Synergism.
Erl/GLE combination inhibits cell motility and disrupts cell aggregation
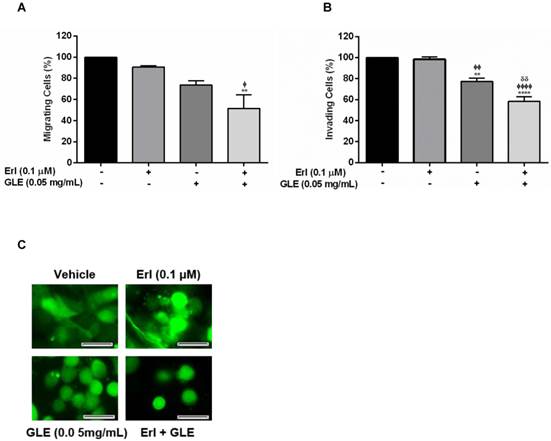
EGFR overexpression in IBC is associated with rapid tumor growth rate, invasion and metastasis. Thus, we first examined the motility capacity of SUM-149 cells using a non-lethal and synergistic combination of Erlotinib-0.1 µM and 0.05 mg/mL-GLE. As expected, Erl/GLE significantly reduced migration compared to vehicle or Erlotinib (Fig. 2A). Similarly, Erl/GLE significantly inhibited the SUM-149 invasive capacity compared to vehicle, Erlotinib and GLE (Fig. 2B). A lower GLE concentration significantly decreased invasion, in accordance with our previous published results where a higher GLE dose for shorter treatment time resulted in 80% impairment of SUM-149 cell invasion [14]. However, 0.01 µM-Erlotinib+0.05 mg/mL-GLE had no significant reduction in cell migration (Fig. S3). Since, IBC is characterized for its ability to invade via generation of tumor emboli [26] we studied the effect of Erl/GLE in a 3D-culture model. Vehicle as well as 0.1µM Erlotinib treated SUM-149 cells maintained cell aggregates as stated in published studies, while 0.05 mg/mL GLE or Erl/GLE disrupted them (Fig. 2C) [23]. These data suggest that Erl/GLE might display anti-invasive IBC effects.
Effect of Erl/GLE in cell motility and cell aggregation. Quiescent SUM-149 cells were seeded on top chambers and treated with vehicle, 0.1 µM-Erlotinib, 0.05 mg/mL-GLE or Erl/GLE for 72h. A, B. Cells migrated or invaded towards 10%FBS medium. Cells were fixed and stained, micrographs were captured, and cells quantified (ImageJ). C. SUM-149 cells were seeded on MatTek dishes, labeled and overlaid with Matrigel. Next day, cells were treated as above. Micrographs were obtained with a 20x objective magnification at the same location. These represent the average of 10 photos taken by treatment. Scale bars = 100µm. Columns represent means ± SEM. Experiments were repeated at least three times. Significance against: vehicle (*), Erlotinib (ᶲ) and GLE (ᵟ). (P≤0.05).
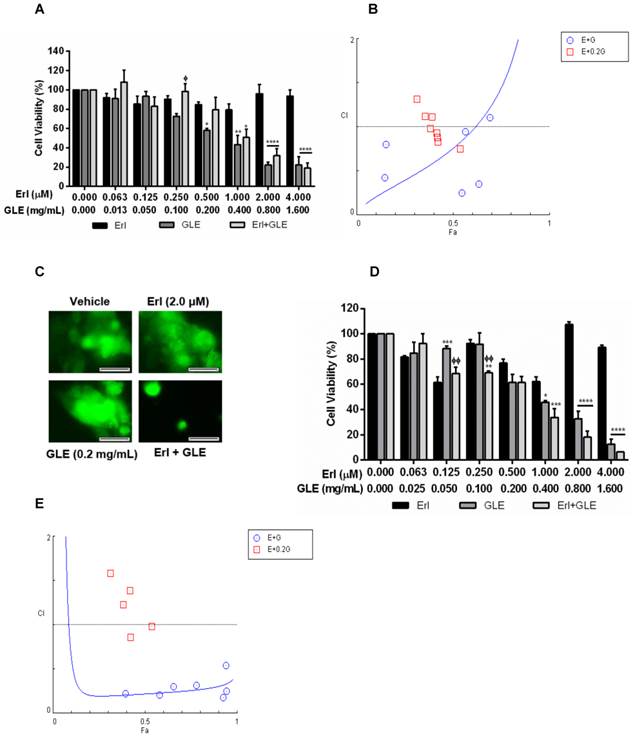
Erl/GLE synergistic effect in BC cells with acquired and intrinsic Erlotinib resistance
To study GLE's effect in IBC cells with acquired Erlotinib resistance, we successfully developed stable Erlotinib resistant cells (rSUM-149). Higher Erlotinib concentrations did not affect viability of resistant cells while GLE reduced rSUM-149 cell viability in a concentration dependent manner (Fig. 3A). However, Erl/GLE significantly decreased cell viability compared to Erlotinib but not against GLE as observed on Erlotinib sensitive SUM-149 cells. To determine the effectiveness of Erl/GLE in rSUM-149 cells, we calculated the CIs at a CR ratio (1:24;Erl:GLE), and at a NCR combining Erl+0.2 mg/mL-GLE. The Fa-CI plot (Fig. 3B) and Table 3 show that the rSUM-149 ED50 CI value was similar to that in parental cells (0.76 vs 0.61). However, the CIs increased as the effective dose increased, transforming the synergic effect into antagonistic. As expected, Dm for Erl/GLE was less (0.26) than Erlotinib (31.7) and GLE (0.33). Furthermore, only Erl/GLE disrupted rSUM-149 cell spheroid formation (Fig. 3C). Next, we studied the effect of Erl/GLE in intrinsically Erlotinib resistant BC cells by treating MDA-MB-231 cells under the same conditions as rSUM149. GLE reduced cell viability in a concentration dependent manner (Fig. 3D), while Erl/GLE significantly decreased cell viability compared to Erlotinib or GLE. In a CR, Erl/GLE, MDA-MB-231 ED50 CI value was 0.22 (synergism). The sensitivity level of these cells to GLE was 0.35 mg/mL, similar to rSUM-149 cells (0.33mg/mL). At a NCR, most of the concentrations tested with GLE were antagonists. However, two of them were synergic and one showed an additive effect (Fig. 3E and Table 3). These results suggest that an Erl/GLE synergic effect is observed in EGFR-overexpressing BC cells with acquired and intrinsic resistance.
Synergistic effect of Erlotinib and GLE in cells with Erlotinib resistance.
| Constant ratio | |||||||||||||
| Combination Indices | |||||||||||||
| Treatment | Cell line | ED50 | ED75 | ED90 | ED95 | Dm | |||||||
| Erl+GLE | rSUM-149 | 0.75667 | 1.44933 | 2.77615 | 4.31960 | 0.25833 | |||||||
| MDA-MB-231 | 0.21779 | 0.25714 | 0.30388 | 0.34043 | 0.07729 | ||||||||
| Erl (μM) | rSUM-149 | 31.7139 | |||||||||||
| MDA-MB-231 | 8.0631 | ||||||||||||
| GLE (mg/mL) | rSUM-149 | 0.32788 | |||||||||||
| MDA-MB-231 | 0.34782 | ||||||||||||
| Non-constant ratio | |||||||||||||
| rSUM-149 | MDA-MB-231 | ||||||||||||
| Dose Erl (μM) | Dose GLE (mg/mL) | Effect | CI | Description | Effect | CI | Description | ||||||
| 0.06 | 0.2 | 0.31276 | 1.31749 | MA | 0.31276 | 1.58155 | A | ||||||
| 0.13 | 0.42259 | 0.83210 | MS | 0.42259 | 0.85996 | SlS | |||||||
| 0.25 | 0.38245 | 0.98438 | AE | 0.38245 | 1.23073 | MA | |||||||
| 0.50 | 0.35481 | 1.11712 | SlA | 0.35481 | 2.08423 | A | |||||||
| 1.00 | 0.41835 | 0.88125 | SlS | 0.41835 | 1.38702 | MA | |||||||
| 2.00 | 0.41595 | 0.93012 | SlS | 0.41595 | 2.03295 | A | |||||||
| 4.00 | 0.39130 | 1.11283 | SlA | 0.39130 | 4.85018 | StA | |||||||
| 8.00 | 0.53760 | 0.75249 | MS | 0.53760 | 0.98228 | AE | |||||||
A=Antagonism, AE=Additive Effect, MA=Moderate Antagonism, MS=Moderate Synergism, SlA=Slight Antagonism, SlS=Slight Synergism, StA=Strong Antagonism.
Effect of Erl/GLE in BC cells with acquired and intrinsic Erlotinib resistance. A, B. rSUM-149 cells were treated with Erl, GLE or Erl/GLE for 72h and cell viability and CIs were calculated based on the IC50 of each at a constant Erl:GLE (1:24) and at NCR (Erl+0.2 mg/mL-GLE). C. rSUM-149 cells were seeded in 3D (see Fig.2) and treated with vehicle, 2.0 µM-Erlotinib, 0.2 mg/mL-GLE or Erl/GLE for 72h. Micrographs were obtained with a 20x objective magnification at the same location. These represent the average of 10 photos taken by treatment. Scale bars = 100µm. D, E. MDA-MB-231 cells were treated with Erl, GLE or Erl/GLE for 72h and cell viability and CIs were calculated based on the IC50 of each at a constant Erl:GLE (1:47) and at NCR (Erlotinib+0.2 mg/mL-GLE). Columns represent means ± SEM. Experiments were repeated at least three times. Significance against: Erlotinib (*) or GLE (ᶲ) (P≤0.05).
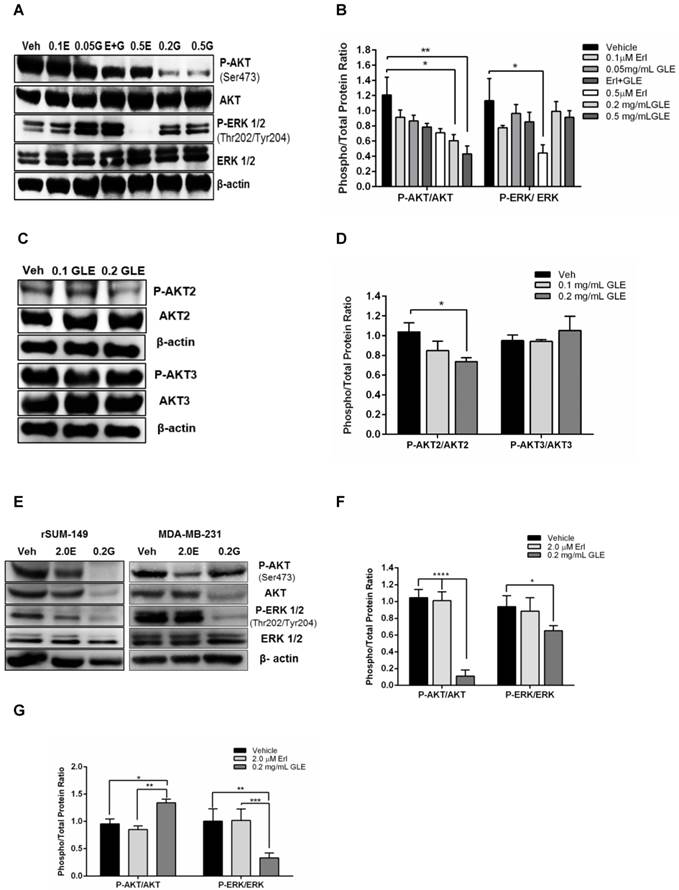
GLE downregulates AKT and ERK activity
Because resistance to Erlotinib is known to be associated with the activation of prosurvival molecules, AKT and ERK, we examined whether GLE would decrease the expression of these proteins and whether it could overcome Erlotinib resistance by inhibiting their activation. SUM-149 Erlotinib sensitive cell immunoblots show that 0.2mg/mL, 0.5mg/mL GLE (P<0.05, P<0.01, respectively) significantly reduced P-AKT compared to vehicle (Fig. 4A,B). However, there was no difference in AKT activation comparing Erl/GLE, and each alone. Our findings are consistent with those published by other groups that Erlotinib inhibition effects are ERK dependent [23]. Recent studies have focused on the role of the three AKT isoforms in BC, where AKT1 and AKT3 have been associated with IBC invasion and metastases, respectively [27]. Meanwhile, PTEN-deficient tumors depend on AKT2 for maintenance and survival [28]. Our previous results showed that GLE decreased AKT phosphorylation on Ser473. GLE reduces the activation of AKT2 at Ser474 (P<0.05) but it does not affect AKT3 (Fig. 4C,D). In order to study the effect of GLE in the expression and activation of the prosurvival proteins in Erlotinib resistant cells, we used rSUM-149 and MDA-MB-231. In cells with acquired resistance (rSUM-149), GLE decreased AKT and its phosphorylation and ERK1/2 activation (P<0.0001, P=0.03) (Fig. 4E,F). However, GLE only inhibits ERK activation (P=0.001) in cells with intrinsic resistance to Erlotinib (MDA-MB-231). Moreover, GLE increased the activation of AKT at Ser473 (Fig. 4E,G). This data suggests that GLE targets two important resistance associated proteins.
GLE downregulates AKT and ERK activity. A. SUM-149 cells were treated with vehicle, 0.1 and 0.5 µM-Erlotinib, 0.05 and 0.2 mg/mL-GLE or 0.1 µM-Erlotinib+0.05 mg/mL-GLE for 72h. B. Densitometric analysis of immunoblotting bands. C. SUM-149 cells were treated with vehicle, 0.1 or 0.2 mg/mL GLE for 72h. D. Densitometric analysis of immunoblotting bands. E. rSUM-149 and MDA-MB-231 cells were treated with vehicle, 2.0 μM-Erlotinib and 0.2 mg/mL-GLE for 72h. F, G. Densitometric analysis of immunoblotting bands. Immunoblots with the primary antibodies listed on the left side of the panel, β-actin is loading control. Densitometric analyses were done using Image J software. Data is expressed as phospho/total protein ratio. Columns represent means ± SEM. Experiments were repeated at least three times.
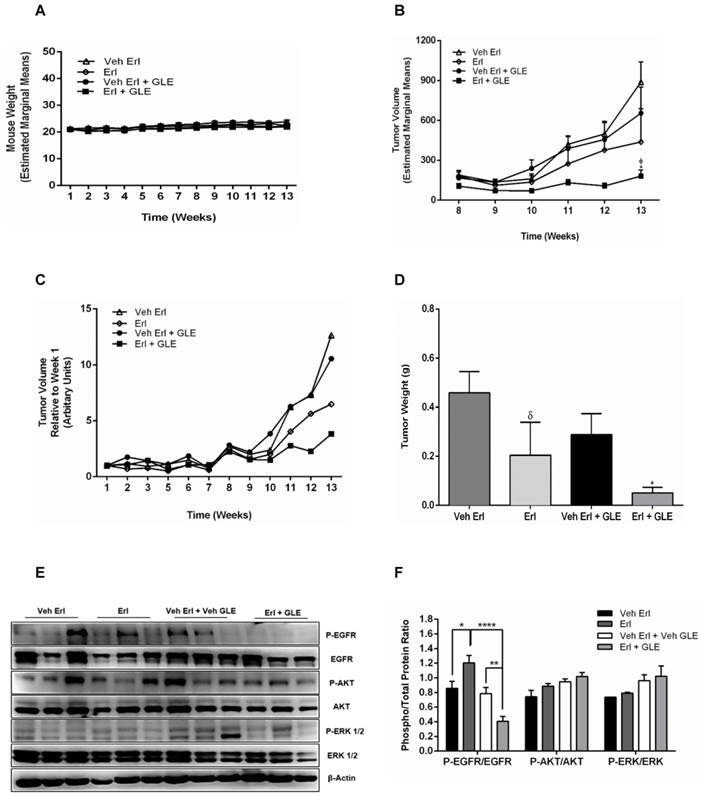
Erl/GLE combination inhibits tumor formation in vivo
We previously showed that 28 mg/kg_BW GLE reduces tumor volume and weight in a xenograft IBC model [22]. Because our new data shows that the Erl/GLE synergize to reduce cell viability, spheroid formation as well as cell motility, we sought to determine the Erl/GLE in vivo efficacy. In this study, we used the recommended GLE dose/BW (1000 mg/daily) based on an average adult woman (70 kg). There were no differences in body weights (Fig. 5A), indicating that treatments were not toxic to mice. In our in vitro studies, Erlotinib and GLE were given simultaneously which resulted in a synergic effect. In our in vivo model we administered Erlotinib in the morning, followed by GLE in the afternoon. Tumor volume was significantly reduced (P≤0.05) in Erl/GLE treated mice compared with vehicle (~28%) or Erlotinib (~41%) (Fig. 5(B,C)]. Erl/GLE treated mice showed 17% lower tumor weight (P≤0.05), and tumor weight decreased by 45% in mice treated with Erlotinib vs. vehicle (Fig. 5D). These data evidence that the order in which GLE and Erlotinib were administered does not influenced their synergic effect observed in our in vitro studies. Erl/GLE decreased the activation of EGFR vs. vehicle (P≤0.01) and vs. Erlotinib (P<0.0001) (Fig. 5E,F). These results suggest that Erl/GLE is better than Erlotinib in in vivo models reducing tumor volume and weight.
In vivo Erl/GLE effect. 1x106 SUM-149 cells mixed with Matrigel (1:1) were injected into the mammary fat pad of SCID mice. Mice were gavaged daily with vehicles (open triangles and open diamonds) or experimental groups (closed circles and closed squares for 13wks. A. Weekly mouse weight. B. Weekly tumor volume. C. Average tumor volume. D. Final tumor weight. E, F. Immunoblot with the primary antibodies listed on the left side of the panel, β-actin is loading control. Densitometric analyses were done using Image J software. Data is expressed as phospho/total protein ratio. Each lane represents a different animal. Estimated marginal means ± SEM. Significance against: (*) vehicle Erl+GLE, (ᵟ) vehicle Erlotinib. P≤0.05.
Discussion
Identification of agents that may be combined with TKIs represent a promising strategy for the development of therapies that could provide a sustained treatment response for BC patients. EGFR overexpression and/or HER2 amplification are poor IBC prognostic markers [5]. Thus, EGFR and HER2 pharmacologic inhibitors (i.e. TKIs) that interrupt intracellular signaling cascades, have rapidly emerged as targeted BC therapy. EGFR and/or HER2 TKIs, Erlotinib and Lapatinib, have progressed to clinical trials and are used but with limited results. Although, EGFR targeting drugs show signs of success in a limited number of IBC patients, failure in others may partly be due to acquisition or by the novo resistance [6, 7].
The dual tyrosine kinase inhibitor (TKI), Lapatinib, which targets both HER2 and EGFR, has shown significant activity in phase II studies dedicated to HER2+ IBC. However, despite its dual inhibition capacity, Lapatinib shows lack of efficacy in IBC patients with HER2-/EGFR+ tumors [29]. The anti-BC activity of EGFR TKI, Erlotinib is evidenced through multiple studies using non-IBC and IBC in vitro and xenograft models [23]. However, modest results have been observed in clinical trials. In two phase II trials, Erlotinib had minimal activity in women with metastatic BC with only two partial response cases among 87 patients [30, 31]. Although, Erlotinib is not a routine treatment for IBC patients, the promising effects of Erlotinib have been demonstrated in this type of BC [32]. Erlotinib is a therapy approved to treat non-small-cell-lung cancer for patients whose tumors harbor sensitizing mutations in EGFR. Recently, the first case of acquired resistance to HER2 therapies driven by an active EGFR sensitizing mutation was documented in an IBC patient who clinically benefited from Erlotinib therapy. In this case, after treatment with different anti-HER2 therapies, a genomic profile of IBC tissue showed a mutation in EGFR, L858R. The patient was started on metronomic Erlotinib (150mg/P.O./daily), in combination with Trastuzumab obtaining promising results for a period of 8 months [33]. This case evidences the potential use of Erlotinib in patients with acquired resistance to HER2 TKIs such as Lapatinib.
Five mechanisms have been described as determinants for EGFR and HER2 inhibitor resistance. One of the most relevant causes of resistance is the independent or constitutive activation of PI3K/AKT/mTOR and MAPK intracellular molecular effectors downstream to the target protein [34]. We previously reported that GLE reduced the expression of mTOR downstream effectors in treated IBC cells at less than 24h treatment times. In that study, we demonstrated that 28 mg/Kg_BW-GLE reduced p-ERK1/2 (Thr202/Tyr204) and Ras levels in vivo [22]. In this study, we investigated the Erl/GLE effect using the TN-EGFR overexpressing IBC cell line, SUM-149 as an Erlotinib sensitive cell line, and the TN-EGFR overexpressing non-IBC cell line, MDA-MB-231 as our intrinsic Erlotinib resistance model. To study the effect of GLE using an Erlotinib acquired resistance model, we successfully developed an Erlotinib resistance cell line (rSUM-149). We investigated Erlotinib/GLE effects in vivo for longer treatment times.
Here, we show that EGFR-overexpressing cells, SUM-102 and SUM-149, and HER2+ IBC cells, MDA-IBC-3 and KPL-4, are sensitive to GLE, being EGFR-overexpressing cells the most sensitive. GLE treated cells cannot recover from treatment, demonstrating that these cells do not develop resistance to this therapy. Studies show that synergy between OSI-027 and/or rapamycin (mTORC1/C2 inhibitors) and Lapatinib results in significantly greater cytotoxicity than the single agents in TNBC cells [35]. We demonstrate that Lap/GLE did not affect MDA-IBC-3 cell viability. Nevertheless, the combination significantly increased KPL-4 cell toxicity at lower concentrations but the response was not sustained as concentrations increased, suggesting that KPL-4 cells resist the combinatorial treatment at higher doses. Synergistic TKI effects have been studied in EGFR-overexpressing BC cells with intrinsic and acquired Erlotinib resistance [13]. In contrast to IBC HER2+ cells, our results demonstrated a synergistic Erl/GLE effect in SUM-149 cells. Because rapid tumor growth, invasion and metastases are associated with EGFR overexpression, the synergism was monitored through migration and invasion capacity of SUM-149 cells. Using non-lethal doses of Erlotinib or GLE we showed that Erl/GLE was more effective in decreasing IBC cell motility. IBC displays atypical invasion processes, which comprise lymphovascular vessel blockage with tumor emboli formation (IBC cell spheroids in vitro), an IBC hallmark [26, 36]. Erl/GLE abolishes cell contacts in in Erlotinib sensitive cells and in cells with acquired Erlotinib resistance resulting in spheroid disruption and invasiveness reduction.
Acquisition of drug resistance in BC involves various factors (i.e. genomic plasticity, overexpression of plasma membrane proteins). Our results show that 0.05mg/mL GLE, results in cell disaggregation in SUM-149, but a higher concentration of GLE does not disaggregate cells with acquired Erlotinib resistance. Is possible that rSUM-149 cells may have developed an alternative mechanism to confer resistance to Erlotinib, such as overexpression of a molecule responsible for cell-cell contacts that allowed the cells to retain cell aggregation. Further studies are necessary to understand and elucidate the mechanisms used by SUM-149 cells to acquire resistance to Erlotinib and assess the ability of GLE to target those mechanisms.
In this study, we explored the role of AKT and ERK behind the mechanism responsible for the synergistic effects between Erlotinib and GLE, and whether GLE could overcome intrinsic and acquired Erlotinib resistance by affecting these molecules. Enhanced EGFR downstream signaling of PI3K/AKT and MAPK pathways are often constitutively activated in drug-resistant BC and this activation is correlated with increased cell survival and poor prognosis [37, 38]. Studies show that the combination of EGFR-TKIs and other BC therapies induces synergistic effects via the inactivation of AKT and ERK [39, 40]. In addition, a report shows that the EGFR-TKI Gemcitabine, increases the cytotoxic effect of Erlotinib via inactivation of P-AKT in cells with intrinsic and acquired resistance [8, 13]. Contrary to published data by other groups, the synergic effect between Erlotinib and GLE is stronger at greater EDs in Erlotinib resistant EGFR-overexpressing cells. Our data demonstrate that GLE inhibited AKT activation in Erlotinib sensitive cells. We monitored the effect of GLE in all AKT isoforms using SUM-149 cells and we show that total protein levels were not affected but the activation of AKT2 decreases. GLE also reduces P-AKT in BC cells with acquired resistance. Furthermore, GLE decreased ERK1/2 activation in rSUM-149 cells and in cells with intrinsic Erlotinib resistance. It is interesting to highlight that GLE induced AKT phosphorylation in MDA-MB-231 cells. Many reasons may explain why GLE increases AKT activation but deceases total AKT. One of these reasons is the use of an antibody that recognizes all three AKT isoforms (AKT 1, 2 and 3), thus the treatment could affect one or more of the isoforms. Second, the increased phospho to total ratio means there is a change in the regulation that can occur because of an increase in kinase or decrease in phosphatase activity, or the induction of a feedback loop activation to circumvent the effect of GLE in these cells. Finally, the phosphorylation can be induced to compensate the loss of total protein. Through these findings, we show that sensitivity restoration by GLE occurred through inhibition of total AKT and downregulation of ERK activation. However, further studies are necessary to understand the molecular mechanisms behind the Erl/GLE synergic effect and of GLE in Erlotinib resistant cells.
Our previous in vivo study shows that double of the recommended GLE dose for humans, significantly reduced tumor growth and weight [22]. In this study, we treated mice with a non-affecting tumor size GLE dose (unpublished data) in combination with Erlotinib. As previously reported, we observed a contrasting effect on tumor growth starting at week eight in both vehicle and experimental groups [22, 24]. We show a significant reduction in tumor volume and tumor weight in combination-treated mice. Erlotinib/GLE treated tumors resulted in reduced activation of EGFR when compared to vehicle and to Erlotinib. However, we did not observe a dysregulation in AKT or ERK as we observed in our in vitro experiments. Based on our data of AKT isoforms further studies are necessary to address the effect of Erl/GLE in AKT isoforms using in vivo models. Since in our previous work we showed that GLE dysregulates mTOR [17] and mTOR influences other oncogenic pathways (MAPK and PI3K) and may activate feedback mechanisms, future studies will explore the effect of the combination in this molecule.
Based on our findings, we conclude that GLE chemosensitizes EGFR-overexpressing cells to Erlotinib overcoming intrinsic and acquired resistance to EGFR therapy. This action correlates with reduced expression and activity of downstream effectors such as AKT and ERK1/2 that have been implicated with resistance to anti-EGFR therapy. Our novel results open new opportunities for further studies to investigate the efficacy of GLE in combination with other EGFR inhibitors. These findings strongly suggest that GLE used in combination with other agents that target EGFR may enhance therapeutic responsiveness in BC patients with abnormal EGFR signaling.
Abbreviations
EGFR: Epidermal Growth Factor Receptor; ERK1/2: Extracellular signal-regulated kinase; AKT: Serine/threonine-specific protein kinase; TKI: Tyrosine Kinase Inhibitors; GLE: Ganoderma lucidum extract; IBC: Inflammatory Breast Cancer.
Supplementary Material
Supplementary Figures S1-S3.
Acknowledgements
We thank Dr. Suranganie F. Dharmawardhane, for training and technical advice, and Dr. Naoto Ueno for intermediating the acquisition of KPL-4 and MDA-IBC-3 IBC cell lines. We would like to acknowledge the UCC-SOM RCMI Animal Resource Center, UCC-SOM RCMI Common Instrumentation Area, and the UCC-SOM RCMI Optical Imaging Facility. This work was supported by NIH grants CA174307 (Suárez-Arroyo), GM111171 (Martínez-Montemayor), RR003035, MD007583 (Martínez-Montemayor), RR016470, GM103475 (UPR-pilot Martínez-Montemayor), GM110513 (Cubano), Title-V-PPOHA #P031M105050 and Title-V-Cooperative #P031S130068 U.S. Department of Education (Cubano). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the U.S. Department of Education.
Conflict of interest
The authors declare that they have no conflict of interest.
References
1. Dawood S, Ueno NT, Valero V, Woodward WA, Buchholz TA, Hortobagyi GN. et al. Differences in survival among women with stage III inflammatory and noninflammatory locally advanced breast cancer appear early: a large population-based study. Cancer. 2011;117:1819-26
2. Walker R. Rosen's Breast Pathology. J Clin Pathol. 1997;50:1036
3. Zhou J, Yan Y, Guo L, Ou H, Hai J, Zhang C. et al. Distinct outcomes in patients with different molecular subtypes of inflammatory breast cancer. Saudi medical journal. 2014;35:1324-30
4. Cabioglu N, Gong Y, Islam R, Broglio KR, Sneige N, Sahin A. et al. Expression of growth factor and chemokine receptors: new insights in the biology of inflammatory breast cancer. Annals of oncology: official journal of the European Society for Medical Oncology / ESMO. 2007;18:1021-9
5. Yamauchi H, Ueno NT. Targeted therapy in inflammatory breast cancer. Cancer. 2010;116:2758-9
6. Medina PJ, Goodin S. Lapatinib: a dual inhibitor of human epidermal growth factor receptor tyrosine kinases. Clin Ther. 2008;30:1426-47
7. Zhang D, Tari AM, Akar U, Arun BK, LaFortune TA, Nieves-Alicea R. et al. Silencing kinase-interacting stathmin gene enhances erlotinib sensitivity by inhibiting Ser(1)(0) p27 phosphorylation in epidermal growth factor receptor-expressing breast cancer. Molecular cancer therapeutics. 2010;9:3090-9
8. Yamasaki F, Johansen MJ, Zhang D, Krishnamurthy S, Felix E, Bartholomeusz C. et al. Acquired resistance to erlotinib in A-431 epidermoid cancer cells requires down-regulation of MMAC1/PTEN and up-regulation of phosphorylated Akt. Cancer research. 2007;67:5779-88
9. Maiello MR, D'Alessio A, De Luca A, Carotenuto A, Rachiglio AM, Napolitano M. et al. AZD3409 inhibits the growth of breast cancer cells with intrinsic resistance to the EGFR tyrosine kinase inhibitor gefitinib. Breast cancer research and treatment. 2007;102:275-82
10. Karachaliou N, Rosell R, Molina MA, Viteri S. Predicting resistance by selection of signaling pathways. Translational lung cancer research. 2014;3:107-15
11. Yi YW, Hong W, Kang HJ, Kim HJ, Zhao W, Wang A. et al. Inhibition of the PI3K/AKT pathway potentiates cytotoxicity of EGFR kinase inhibitors in triple-negative breast cancer cells. Journal of cellular and molecular medicine. 2013;17:648-56
12. Xu ZH, Hang JB, Hu JA, Gao BL. RAF1-MEK1-ERK/AKT axis may confer NSCLC cell lines resistance to erlotinib. International journal of clinical and experimental pathology. 2013;6:1493-504
13. Bartholomeusz C, Yamasaki F, Saso H, Kurisu K, Hortobagyi GN, Ueno NT. Gemcitabine Overcomes Erlotinib Resistance in EGFR-Overexpressing Cancer Cells through Downregulation of Akt. Journal of Cancer. 2011;2:435-42
14. Martinez-Montemayor MM, Acevedo RR, Otero-Franqui E, Cubano LA, Dharmawardhane SF. Ganoderma lucidum (Reishi) inhibits cancer cell growth and expression of key molecules in inflammatory breast cancer. Nutrition and cancer. 2011;63:1085-94
15. Jiang J, Slivova V, Harvey K, Valachovicova T, Sliva D. Ganoderma lucidum suppresses growth of breast cancer cells through the inhibition of Akt/NF-kappaB signaling. Nutrition and cancer. 2004;49:209-16
16. Jiang J, Slivova V, Sliva D. Ganoderma lucidum inhibits proliferation of human breast cancer cells by down-regulation of estrogen receptor and NF-kappaB signaling. Int J Oncol. 2006;29:695-703
17. Suarez-Arroyo IJ, Rosario-Acevedo R, Aguilar-Perez A, Clemente PL, Cubano LA, Serrano J. et al. Anti-tumor effects of Ganoderma lucidum (reishi) in inflammatory breast cancer in in vivo and in vitro models. PloS one. 2013;8:e57431
18. Kuo HP, Hsu SC, Ou CC, Li JW, Tseng HH, Chuang TC. et al. Ganoderma tsugae Extract Inhibits Growth of HER2-Overexpressing Cancer Cells via Modulation of HER2/PI3K/Akt Signaling Pathway. Evidence-based complementary and alternative medicine: eCAM. 2013;2013:219472
19. Ethier SP, Kokeny KE, Ridings JW, Dilts CA. erbB Family Receptor Expression and Growth Regulation in a Newly Isolated Human Breast Cancer Cell Line. Cancer research. 1996;56:899-907
20. Kurebayashi J, Otsuki T, Tang CK, Kurosumi M, Yamamoto S, Tanaka K. et al. Isolation and characterization of a new human breast cancer cell line, KPL-4, expressing the Erb B family receptors and interleukin-6. British journal of cancer. 1999;79:707-17
21. Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006;58:621-81
22. Suarez-Arroyo IJ, Rosario-Acevedo R, Aguilar-Perez A, Clemente PL, Cubano LA, Serrano J. et al. Anti-Tumor Effects of Ganoderma lucidum (Reishi) in Inflammatory Breast Cancer in In Vivo and In Vitro Models. PloS one. 2013;8:e57431
23. Zhang D, LaFortune TA, Krishnamurthy S, Esteva FJ, Cristofanilli M, Liu P. et al. Epidermal growth factor receptor tyrosine kinase inhibitor reverses mesenchymal to epithelial phenotype and inhibits metastasis in inflammatory breast cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2009;15:6639-48
24. Carlson AL, Hoffmeyer MR, Wall KM, Baugher PJ, Richards-Kortum R, Dharmawardhane SF. In situ analysis of breast cancer progression in murine models using a macroscopic fluorescence imaging system. Lasers in surgery and medicine. 2006;38:928-38
25. Qin C, Tan KL, Zhang CL, Tan CY, Chen YZ, Jiang YY. What does it take to synergistically combine sub-potent natural products into drug-level potent combinations? PloS one. 2012;7:e49969
26. Kleer CG vGK, Merajver SD. Molecular biology of breast cancer metastasis. Inflammatory breast cancer: clinical syndrome and molecular determinants. Breast Cancer Res. 2000;2:423-9
27. Lehman HL, Van Laere SJ, van Golen CM, Vermeulen PB, Dirix LY, van Golen KL. Regulation of inflammatory breast cancer cell invasion through Akt1/PKBalpha phosphorylation of RhoC GTPase. Molecular cancer research: MCR. 2012;10:1306-18
28. Chin YR, Yuan X, Balk SP, Toker A. PTEN-deficient tumors depend on AKT2 for maintenance and survival. Cancer discovery. 2014;4:942-55
29. Johnston S, Trudeau M, Kaufman B, Boussen H, Blackwell K, LoRusso P. et al. Phase II study of predictive biomarker profiles for response targeting human epidermal growth factor receptor 2 (HER-2) in advanced inflammatory breast cancer with lapatinib monotherapy. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2008;26:1066-72
30. Dickler M, Cobleigh M, Miller K, Klein P, Winer E. Efficacy and safety of erlotinib in patients with locally advanced or metastatic breast cancer. Breast cancer research and treatment. 2009;115:115-21
31. Tan AR, Yang X, Hewitt SM, Berman A, Lepper ER, Sparreboom A. et al. Evaluation of biologic end points and pharmacokinetics in patients with metastatic breast cancer after treatment with erlotinib, an epidermal growth factor receptor tyrosine kinase inhibitor. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2004;22:3080-90
32. Ali SM, Alpaugh RK, Buell JK, Stephens PJ, Yu JQ, Wu H. et al. Antitumor response of an ERBB2 amplified inflammatory breast carcinoma with EGFR mutation to the EGFR-TKI erlotinib. Clinical breast cancer. 2014;14:e14-6
33. Ali SM, Alpaugh RK, Buell JK, Stephens PJ, Yu JQ, Wu H. et al. Antitumor Response of an ERBB2 Amplified Inflammatory Breast Carcinoma With EGFR Mutation to the EGFR-TKI Erlotinib. Clinical Breast Cancer. 2014;14:e14-e6
34. Tortora G, Bianco R, Daniele G, Ciardiello F, McCubrey JA, Ricciardi MR. et al. Overcoming resistance to molecularly targeted anticancer therapies: Rational drug combinations based on EGFR and MAPK inhibition for solid tumours and haematologic malignancies. Drug resistance updates: reviews and commentaries in antimicrobial and anticancer chemotherapy. 2007;10:81-100
35. Liu T, Yacoub R, Taliaferro-Smith LD, Sun SY, Graham TR, Dolan R. et al. Combinatorial effects of lapatinib and rapamycin in triple-negative breast cancer cells. Molecular cancer therapeutics. 2011;10:1460-9
36. Hoffmeyer MR, Wall KM, Dharmawardhane SF. In vitro analysis of the invasive phenotype of SUM 149, an inflammatory breast cancer cell line. Cancer cell international. 2005;5:11
37. Zhao M, Ramaswamy B. Mechanisms and therapeutic advances in the management of endocrine-resistant breast cancer. World journal of clinical oncology. 2014;5:248-62
38. Yang XL, Lin FJ, Guo YJ, Shao ZM, Ou ZL. Gemcitabine resistance in breast cancer cells regulated by PI3K/AKT-mediated cellular proliferation exerts negative feedback via the MEK/MAPK and mTOR pathways. Onco Targets Ther. 2014;7:1033-42
39. Atalay G, Cardoso F, Awada A, Piccart MJ. Novel therapeutic strategies targeting the epidermal growth factor receptor (EGFR) family and its downstream effectors in breast cancer. Annals of oncology: official journal of the European Society for Medical Oncology / ESMO. 2003;14:1346-63
40. Liu L, Shi H, Liu Y, Anderson A, Peterson J, Greger J. et al. Synergistic effects of foretinib with HER-targeted agents in MET and HER1- or HER2-coactivated tumor cells. Molecular cancer therapeutics. 2011;10:518-30
Author contact
![]() Corresponding author: Michelle M. Martínez-Montemayor, Ph.D. Phone: 787-798-3001; Fax: 787-740-4390; e-mail: michelle.martinezedu.
Corresponding author: Michelle M. Martínez-Montemayor, Ph.D. Phone: 787-798-3001; Fax: 787-740-4390; e-mail: michelle.martinezedu.