Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2016; 7(4):418-426. doi:10.7150/jca.13019 This issue Cite
Research Paper
E3 Ubiquitin Ligase Siah-1 is Down-regulated and Fails to Target Natural HBx Truncates for Degradation in Hepatocellular Carcinoma
Jing Zhao1, Jing Wu2, Hao Cai3, Dan Wang3, Long Yu3 ![]() , Wen-Hong Zhang2
, Wen-Hong Zhang2 ![]()
1. Department of General Surgery, Huashan Hospital, Shanghai Medical College, Fudan University, Shanghai 200040, China;
2. Department of Infectious Diseases, Huashan Hospital, Shanghai Medical College, Fudan University, Shanghai 200040, China;
3. The State Key Laboratory of Genetics Engineering, Fudan University, Shanghai 200433, China.
Received 2015-6-23; Accepted 2015-11-14; Published 2016-1-20
Abstract
Hepatocellular carcinoma (HCC) is a common worldwide malignancy with high morbidity and mortality. Hepatitis B viral (HBV)-encoded X protein (HBx) and natural HBx variants play important roles in HBV-associated HCC development. HBx is an unstable protein that can be degraded in vivo. Our previous study found that the E3 ubiquitin ligase Siah-1 could target HBx for poly-ubiquitylation and proteasomal degradation and attenuate the transcriptional activity of HBx. However, in HCC patients, high expression levels of HBx and HBx variants are frequently observed and are associated with HCC progression. The mechanism underlying their up-regulation is largely unknown. In this study, we screened for Siah-1 mutations in 270 HCC samples and 9 HCC cell lines, and examined Siah-1 mRNA and protein expression in a subset of paired HCC specimens. Our results demonstrate that Siah-1 is highly conserved, as no somatic mutation was identified, with the exception of one synonymous transition from G to A at codon 67. Both the mRNA and protein levels of Siah-1 were significantly down-regulated in HCC tissues compared with their adjacent normal counterparts. We constructed three natural HBx truncates that were identified in our HCC cases. We found that Siah-1 failed to decrease the stability of these HBx variants and was unable to inhibit the transcriptional activity of these HBx truncates at heat shock elements (HSEs). Moreover, Siah-1 had weaker association with three HBx mutants than full length HBx. Therefore, our findings suggest that down-regulation of Siah-1, but not its mutations, and natural HBx variants resistant to Siah-1-induced degradation may be a novel mechanism for HCC development.
Keywords: hepatocellular carcinoma, HBx, Siah-1, ubiquitylation and proteasomal degradation, HBx truncates.
Introduction
Hepatocellular carcinoma (HCC) is one of the most common malignancies, with high incidence and mortality around the world [1]. Chronic hepatitis B virus (HBV) infection is an important risk factor for HCC development [1]. The HBV X gene tends to be integrated in host hepatocytes, and it encodes the HBx protein, which is 154 amino acids (aa) long [2]. HBx is a promiscuous transactivator that can regulate HBV replication, disrupt host gene expression, affect intracellular signal transduction, accelerate cell proliferation, inhibit apoptosis, and drive HCC cell migration and invasion [3]. Our previous study found that HBx could down-regulate miR-375 and miR-136 expression, which elevated the levels of oncoprotein AEG-1 and promoted HCC cell migration [4]. Accumulating evidence demonstrates that most integrated HBx genes in HCC patients have mutations or deletions in the carboxyl-terminus encoding region [5, 6]. Some HBx C-terminal truncates (HBx-Cts) have been extensively studied, and these truncates have been shown to affect HCC development through multiple mechanisms. For instance, a naturally occurring HBx-Ct with a 24 aa deletion at the C-terminus could increase C-Jun transcriptional activity and promoted the transcription of matrix metalloproteinase 10 (MMP10) to enhance invasive ability [7]. Moreover, HBx-Cts can bind to miRNA promoters to inhibit the transcription of several growth-suppressive miRNAs, such as miR-26a, miR-29c, miR-146a and miR-190, thereby enhancing cell proliferation [8].
Full length HBx is a short-lived protein in vivo, and many factors can decrease its protein level, such as the 26S proteasome complex [9], heat shock protein 40 (Hsp40) [10], HBV core proteins [11], tumor suppressor p53 [12], and transcription factor Id-1 [13]. We also identified two E3 ligases, MDM2 and Siah-1, which could destabilize HBx [14, 15]. Despite the ability of MDM2 to induce HBx digestion via the proteasome-dependent mechanism, MDM2 had no effect on HBx ubiquitination [14]. In contrast, we found Siah-1 could facilitate the poly-ubiquitylation modification of HBx, and induce HBx to undergo proteasomal degradation and inhibit its transcriptional activation at glucocorticoid response elements (GREs), heat shock response elements (HSEs) and cAMP response elements (CREs) [15]. Various factors can down-regulate HBx in cells, but high levels of HBx and HBx mutants are common events in HCC cases, which imply that HBx and its variants may be resistant to degradation. The mechanisms underlying these observations need to be elucidated.
Siah-1 is the human homolog of Drosophila seven in absentia (sina) and encodes an evolutionarily conserved RING domain E3 ubiquitin ligase [16]. Through its specific association with substrate proteins, Siah-1 promotes the ubiquitin-mediated proteasomal degradation of many proteins [17]. For example, Siah-1 can form complexes with Skp1, Eb1, SIP and pAPC to induce the ubiquitin-proteasomal degradation of β-catenin and thereby reduce malignant cell proliferation and oncogenesis [18]. As Siah-1 plays important roles in mediating protein degradation, Siah-1 has also been widely investigated in cancer development. Two inactive Siah-1 missense mutations were discovered in 95 gastric cancer specimens: a C to T transition at nucleotide 92 (Ser to Phe) and an A to C transition at nucleotide 622 (Ile to Leu), both of the Siah-1 mutants could not degrade β-catenin and trigger apoptosis [19]. However, Siah-1 mutations were only screened for in a relatively small set of HCC specimens (35 samples), and only one G to A synonymous transition at proline 50 was detected in one case [20].
In this study, we screened for Siah-1 mutations in a large number of HCC samples with 270 HCC tissue samples and detected Siah-1 expression levels in some of these HCC specimens. Our results demonstrated that Siah-1 was highly conserved, with little DNA alteration, but both Siah-1 transcriptional levels and protein expression were significantly down-regulated in HCC tissues. We found that Siah-1 could not decrease the levels of three truncated HBx variants identified in our HCC samples, and failed to inhibit their transactivation activity at heat shock elements (HSEs). Furthermore, Siah-1 showed weaker association with three HBx truncates than full length HBx. These findings provide new clues on the mechanism of Siah-1 and natural HBx variants in HCC development.
Materials and methods
Patients and samples
A total of 270 HCC tissue specimens and 9 HCC cell lines were used in this study. Informed consent was obtained from each enrolled patient according to the Fudan University Institutional Review Board. The HCC tissue specimens were collected from the Qidong Liver Cancer Institute and the Liver Cancer Institute of Zhongshan Hospital in China from 1998 to 2006. The HCC specimens were diagnosed by two individual pathologists. The corresponding adjacent normal tissues were 3 cm away from the edge of the HCC tissue without obvious tumor cells. All samples were immediately frozen in liquid nitrogen after surgery and stored at -80 °C before further analysis. The 9 HCC cell lines were Hep3B, HepG2, QGY-7703, SK-Hep1, SMMC-7721, Focus, SUN-398, PLC/PRF/5, and YY-8103.
DNA and RNA extraction
Genomic DNA was extracted from the tissue samples and HCC cell lines using a QIAamp® DNA Mini Kit (QIAGEN GmbH, Hilden, Germany). Total RNA was extracted using TRIzol reagent (Invitrogen, CA, USA) according to the manufacturer's protocol. The yield and quality of the DNA and RNA were evaluated with Nano Drop ND-2000 spectrophotometer.
50 ng genomic DNA served as a template for PCR amplification. The PCR primers used to amplify Siah-1 were as follows:
F1: 5-TTAAAAGGACTTATGGCATGT-3;
R1: 5-CCTGGTGCTTCCTGTAAAT-3;
F2: 5-CAACTTGGCTATGGAGAAAG-3;
R2: 5-AGTTCTTCGCAATCGTACAG-3;
F3: 5-CATTACAACCCTACAGGGAG-3;
R3: 5-TTCACAGAAAATAAGGCACC-3.
A total of 500 ng of RNA was reverse-transcribed into cDNA using a ReverTra Ace kit (Toyobo, Japan) according to the manufacturer's protocols. qRT-PCR was performed using a standard SYBR-Green PCR kit protocol (Takara, Japan) in an ABI 7900 Real Time PCR System. All samples were normalized to the housekeeping gene glyceraldehyde 3-phosphate dehydrogenase (GAPDH). The qRT-PCR primers for Siah-1 and GAPDH were as follows:
Siah-1 RT-F: 5-TGTTTGTAGCAACTGTCGCC-3;
Siah-1RT-R: 5-AGCCACTTTCTCCATAGCCA-3;
GAPDH RT-F: 5-AGGGCTGCTTTTAACTCTGGT- 3;
GAPDH RT-R: 5-CCCCACTTGATTTTGGAGGGA-3.
Sequencing Siah-1 genomic DNA
The PCR products were sequenced in both directions with the dideoxy-mediated chain termination method on an ABI3730 DNA sequencer (Applied Biosystems, Foster City, CA). When a DNA alteration was found in a tumor sample, DNA from the matched, neighboring, pathologically non-tumorous liver tissue was used as a control.
Cell culture and transfection
Hep3B cells and Huh-7 cells were grown in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum (FBS) at 37 oC in 5% CO2 humidified atmosphere. Cells at 80% confluency were transfected with plasmids using Lipofectamine2000 ™ reagent (Invitrogen) according to manufacturer's protocol.
Plasmids and antibodies
pCMV-Myc-HBx was kindly provided by Dr. Minghua Zhu (Changhai Hospital, the Second Military Medical University of China). Three HBx truncates plasmids were generated with TaKaRa Mutagenesis Kit to introduce stop codon TAA into pCMV-Myc-HBx corresponding site. The human Siah-1 cDNA was subcloned into PCMV-Myc vector (Clontech) via amplifying liver marathon cDNA library of human. The heat shock response element (HSE)-luc constructs were purchased from Clontech.
Specific antibodies we used included: anti-GFP (Santa Cruz, USA), anti-Siah-1 (Abcam, USA), anti-HA (CST, USA), anti-Myc and anti-β-Actin (Sigma, USA).
Western blot
Whole-cell lysates were separated by 15% SDS-PAGE, and were transferred to nitrocellulose membranes (GE Healthcare, USA). The membrane was followed by blocking with 5% non-fat milk in TBS (20 mM Tris-HCl, 137mM NaCl, pH 7.6), incubated with indicated primary antibodies and corresponding horseradish peroxidase-conjugated secondary antibody. Then, the targeted protein bands were visualized with the enhanced chemilluminescence reagents (Santa Cruz Biotechnology, USA).
Luciferase reporter assay
Hep3B or Huh-7 cells (1×105) were seeded in 24-well plates. Each well was co-transfected with 100 ng of HSE-luc reporter plasmid and 20 ng of pRL-SV40 as an internal control. We co-transfected the cells with 200 ng of pCMV-Myc-HBx or HBx truncated plasmids as indicated in the figures. 24 h after transfection, cells were lysed with reporter lysis buffer and assayed using the Dual-Luciferase Reporter Assay kit (Promega). Each ratio represents the activity of firefly luciferase compared to that of Renilla luciferase. The mean data from three individual transfected wells are presented.
Immunoprecipitation
Cells were co-transfection with HA-Siah-1 and Myc-HBx, Myc-HBx-D3, Myc-HBx-D2, or Myc-HBx-D1, respectively. Cells were treated with proteasome inhibitor MG132 before harvest. The whole cells were washed with ice-cold PBS and lysed in 300 μl cell lysis buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% Triton, 1 mM EGTA, 1 mM Na2EDTA, 2.5 mM sodium pyrophosphate, 1mM glycerophosphate, 1 mM Na3VO4 and 1 μg/ml leupeptin) on ice. The lysate was centrifugated at 12000 rpm for 20 min at 4 oC. The supernatants were incubated with anti-HA antibody overnight on a rotating wheel at 4 oC, and thereafter with 20μl protein A/G beads incubated at 4 oC for 4 h. The beads were then washed 5 times with cold lysis buffer and resuspended in loading buffer and detected with Western blot.
Statistical Analysis
The results were presented as the mean and standard deviation (SD) from three independent experiments at least. Student's t test was used, and differences were considered to be significant when p <0.05.
Results
Mutation detection of Siah-1 gene coding sequence in HCC DNA samples
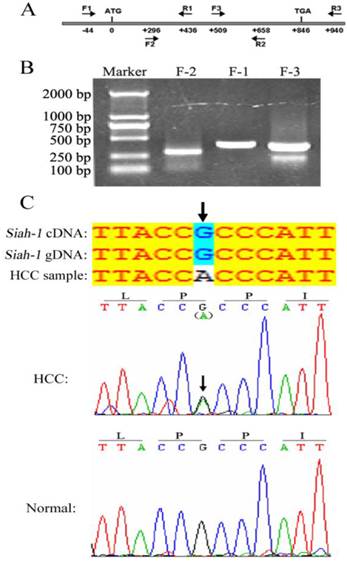
The E3 ligase Siah-1, as a p53-p21 Waf-1 inducible gene product, can promote apoptosis and tumor suppression [21]. In gastric cancer, two Siah-1 missense mutations, Ser31Phe and Ile208Leu, have been identified. These mutations failed to degrade β-catenin and drive apoptosis [19]. However, no Siah-1 mutation was discovered in a relative small set of 35 HCC samples with PCR-SSCP (single-strand conformation polymorphism) analysis [20]. As mutation screen from large cohort is more convincible and PCR-DNA sequencing assay is more exact and confirmable against PCR-SSCP method, we searched for Siah-1 mutations in a larger HCC cohort with 270 HCC tissues and 9 HCC cell lines by directly DNA sequencing. Three paired, overlapped PCR primers were designed to amplify the Siah-1 coding sequence, as shown in Fig. 1A. The PCR product bands were clear and of the expected length, as shown by DNA electrophoresis (Fig. 1B). After sequencing the PCR products, these acquired sequences were compared with the published Siah-1 cDNA and Siah-1 gDNA sequences from the NCBI website. We found only one synonymous transition, from G to A at codon 67 of Siah-1, in one HCC sample (Fig. 1C, up panel). But this mutation was not found in its adjacent normal counterpart tissue sample (Fig. 1C, bottom panel). Our results from large cohort of HCCs further confirm that Siah-1 is highly conserved, with little DNA alteration among HCC samples, and that the inactivation of Siah-1 in HCC should not be attributed to its mutation, unlike in gastric cancer.
Mutation detection of Siah-1 gene coding sequence in HCC DNA samples. (A) A schematic indicating the three paired PCR primers locations. (B) An agarose gel electrophoresis graph of PCR products amplified from HCC DNA samples using the above primers. (C) Sequencing results from the PCR products. The sequences were compared with the published Siah-1 genomic DNA (gDNA) and cDNA sequences. There was a G to A synonymous transition at codon 67 in one HCC DNA sample but not in the adjacent non-tumor tissue.
Siah-1 was expressed at low levels in HCC samples
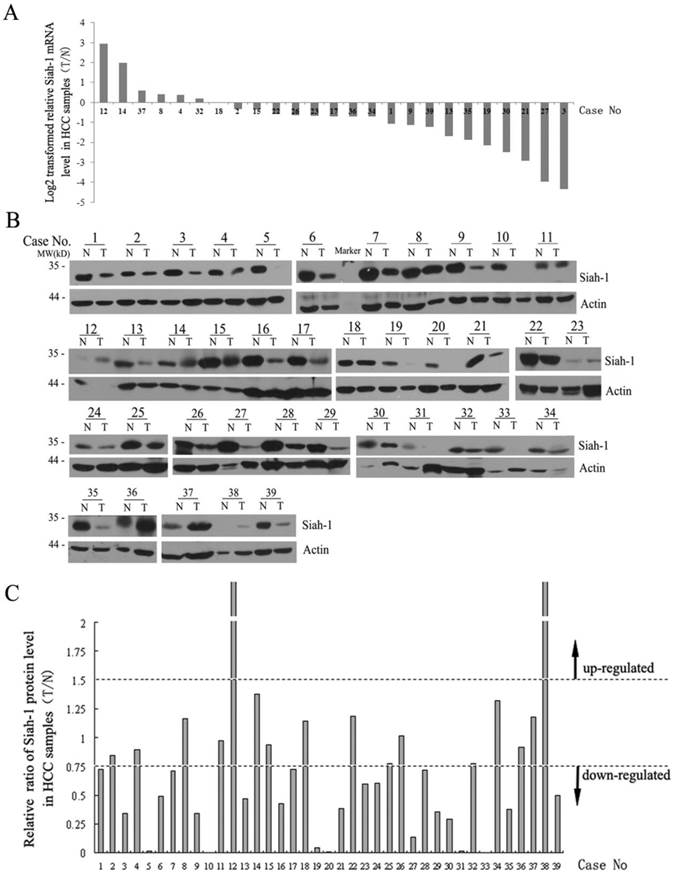
Next, we detected the mRNA and protein levels of Siah-1 in a subset of the paired HCC tissue samples via qRT-PCR and Western blot analysis. As shown in Fig. 2A, Siah-1 transcriptional levels were significantly decreased in 18 of 25 (72%) paired HCC samples (Fig. 2A). Western blot assays indicated that 23 of 39 (58.97%) paired HCC specimens had low Siah-1 protein levels compared with their normal tissue counterparts, 14 of 39 (35.90%) paired HCC had equivalent Siah-1 expression, and only 2 of 39 (5.13%) paired HCC samples exhibited increased Siah-1 protein expression (Fig. 2B and Fig. 2C). These results indicate that the down-regulation of Siah-1, instead of its mutation, may associate with HCC development, as low Siah-1 expression would attenuate its capacity to degrade oncoprotein substrates such as HBx and β-catenin.
Siah-1 was expressed at low levels in HCC samples. (A) qRT-PCR results showed the Siah-1 mRNA levels in 25 paired HCC tissue specimens. GAPDH was used as an internal control. The relative fold change of the Siah-1 mRNA level was calculated using the 2-ΔΔCt method, and the values were log2-transformed, as indicated. (B) Western Blot analysis of Siah-1 protein levels in 39 paired HCC tissues with Siah-1 and β-actin antibodies. β-actin was used as a loading control. N: non-tumor tissue, T: tumor tissue. (C) A histogram showing the relative ratio of Siah-1 protein levels in HCC tissues, which were obtained by scanning the Western blot bands and quantifying their density using Quantity One (Version 4.4.1). Siah-1 levels were standardized to the corresponding β-actin levels in N and T, and the Siah-1 level in T was compared as a ratio to its level in N. A relative ratio of a Siah-1 protein level more than 1.5 or less than 0.75 in an HCC sample was defined as up-regulation or down-regulation, respectively, as the arrow indicates.
Siah-1 failed to degrade natural HBx truncates in HCC samples
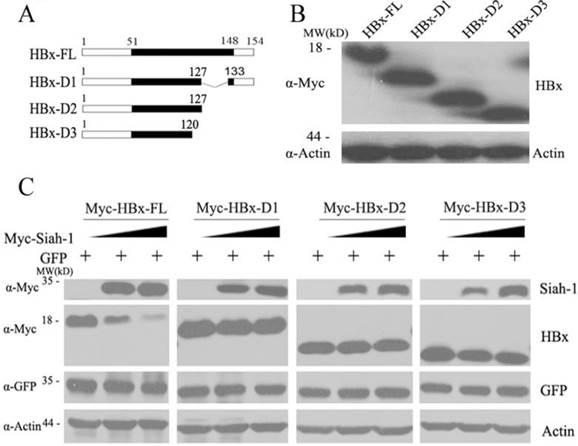
HBx plays important roles in promoting hepatocarcinogenesis[3]. Many studies, in addition to ours, have demonstrated that integrated HBx genes are frequently mutated or deleted at the C-terminus [5, 6]. C-terminally truncated HBx variants have oncogenic roles and enhance HCC progression [7, 8]. Our previous study found that Siah-1 can facilitate the poly-ubiquitylation and proteasomal degradation of HBx [15]. However, whether Siah-1 can influence the stability of natural HBx truncates needs to be clarified. Therefore, we constructed three natural HBx variants that were identified in our HCC samples: HBx-D1, HBx-D2 and HBx-D3 (Fig. 3A). These three HBx variant plasmids were expressed in Hep3B cells (Fig. 3B). When Siah-1 was co-expressed with HBx and the three HBx variants plasmids, we found that Siah-1 could degrade full length HBx proteins in a dose-dependent manner, but Siah-1 failed to destabilize HBx truncates. These findings suggest that high frequency of HBx truncates in HCC may protect HBx variants from degradation mediated by the E3 ligase Siah-1, and the accumulation of HBx variants may enhance their potential to promote HCC development.
Siah-1 failed to degrade natural HBx truncates in HCC samples. (A) A scheme representing full length HBx (HBx-FL) and the three HBx truncates. These HBx truncates were named HBx-D1, HBx-D2 and HBx-D3. (B) Western blot detected the levels of HBx-FL and the three HBx-Ct proteins using an anti-Myc antibody in Hep3B cells, with β-actin as the loading control. (C) Western blot measured the effects of Siah-1 on HBx and HBx variant protein levels. Hep3B cells were co-transfected with increasing amounts of pCMV-Myc-Siah-1, a constant amount of pCMV-Myc-HBx and the three pCMV-Myc-HBx-variant plasmids, as indicated. Western blot analysis was performed using Myc, GFP and β-actin antibodies, as indicated. β-actin was used as the loading control. The pGFP-N3 expression plasmid was used as an internal control for transfection efficiency.
HBx variants maintained their capacity to transactivate heat shock elements (HSEs), but Siah-1 could not inhibit their transcriptional activity
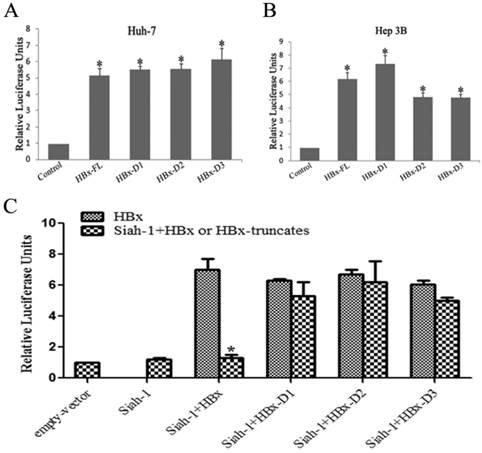
Our previous study found that HBx overexpression could markedly transactivate HSEs, and Siah-1 could inhibit this HBx-induced effect [15]. In the present study, we observed that three truncated HBx variants maintained their ability to transactivate HSEs, similar to full length HBx, in both the Huh-7 and Hep3B cell lines (Fig. 4A and 4B). When Siah-1 was co-expressed with HBx and three HBx variants, the results indicated that Siah-1 could repress the HBx-mediated transactivation of HSEs, but Siah-1 failed to effectively inhibit the transcriptional activity of three HBx variants (Fig. 4C). These results suggest that Siah-1 inability to degrade HBx variants and to repress their transcriptional activity may render HBx truncates to expedite HCC progression.
HBx variants maintained their capacity to transactivate heat shock elements (HSEs), but Siah-1 could not inhibit their transcriptional activity. (A and B) Luciferase reporter assays detected the transactivation activity of HBx and the three truncated HBx variants at HSEs in Huh-7 and Hep3B cells. (C) Luciferase reporter assays were conducted to test the effects of Siah-1 on HBx and the three truncated HBx variants and their transactivation activities at HSEs in Hep3B cells. Reporter HSE plasmids, pCMV-Myc empty vector, pCMV-Myc-HBx-FL and the three pCMV-Myc-HBx-variants were co-transfected with the pCMV-Myc-Siah-1 plasmid, as indicated. Data represent the mean values ± SD (n=3); *p < 0.05.
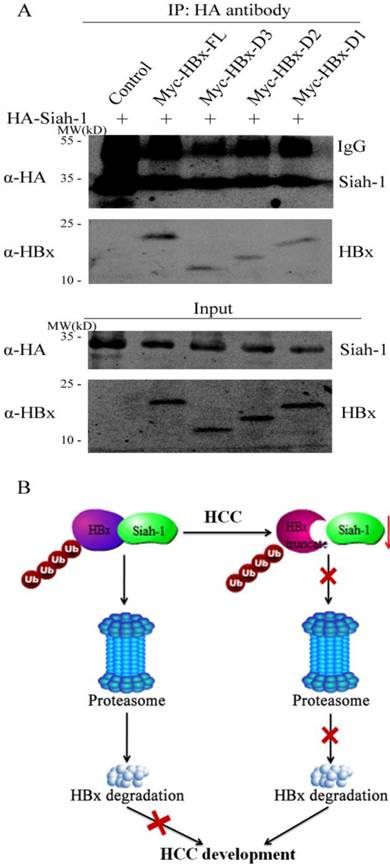
Siah-1 had weaker association with HBx truncates than full length HBx
The interaction between Siah-1 and its substrates is critical for Siah-1 induced poly-ubiquitin modification and subsequently proteasomal digestion. Our previous study revealed that Siah-1 could bind to HBx and co-localized in the cytoplasm [15]. Accordingly, we detected whether HBx truncates had effect on their association with Siah-1 via the immunoprecipitation assay. To maintain the protein level, the proteasome inhibitor MG132 was used to treat the transfected cells. As shown in Fig. 5A, when the cell lysate was immunoprecipitated with anti-HA antibody, full length HBx had stronger protein band signal than three HBx mutants. These results implicate that HBx variants may reduce their capacity to associate with E3 ligase Siah-1. Therefore, our findings suggest that both Siah-1 down-regulation and HBx mutations in HCC may attenuate their interaction, which have HBx and its variants to evade Siah-1 mediated degradation and promote HCC development (Fig. 5B).
Siah-1 had weaker association with HBx truncates than full length HBx. (A) The plasmids of HA-Siah-1 and pCMV-Myc empty vector, Myc-HBx, Myc-HBx-D3, Myc-HBx-D2, or Myc-HBx-D1, were co-transfected into Huh-7 cell line. The immunoprecipitation assay was performed with anti-HA antibody, and anti-HBx antibody was used to detect HBx and HBx truncates. IgG was the heavy chain of antibody. (B) Schematic representation of Siah-1 and HBx in HCC development. The down-regulation of Siah-1 and HBx variations may attenuate their interaction and make HBx to evade Siah-1-induced degradation.
Discussion
The E3 ubiquitin ligase Siah-1 can directly associate with its substrates and serve as an essential RING domain subunit within a larger E3 complex [22]. Siah-1 has an important binding groove structure in its C-terminus that prefers to interact with conserved PxAxVxP peptide motifs [23]. In addition to cellular proteins, Siah-1 can also target some viral proteins for proteasomal degradation, such as the herpes virus-encoded ORF45 protein[24], herpes simplex virus immediate-early protein ICP0 [25] and the hepatitis B virus-encoded HBx protein [15]. Siah-1 has been found to be involved in many diseases, including neurodegenerative disorders and cancers.
Siah-1 can mediate the ubiquitination and proteasomal degradation of synphilin-1 [26] and regulate the mono-ubiquitination of α-synuclein[27]. Synphilin-1 and α-synuclein are two major components of Lewy bodies, which have been associated with Parkinson's disease development [28]. Thus, Siah-1 may limit the availability of synphilin-1 to bind to α-synuclein and thereby attenuate the formation of Lewy bodies [29]. Siah-1 mutation screening has been performed in 209 familial and sporadic PD patients; however, no disease-causing mutation was found [30]. In cancer development, Siah-1, as a p53-p21 Waf-1 inducible gene, is activated during tumor suppression [21, 31]. Siah-1 promotes β-catenin degradation in a non-canonical manner by forming a complex with Skp1, Eb1, SIP and pAPC, which in turn reduces malignant cell proliferation [18]. Two missense mutations of Siah-1, Ser31Phe (S31F) and Ile208Leu (I208L), have been identified in 95 gastric cancer tissue samples [19]. These two mutants disabled the ability of Siah-1 to target β-catenin for degradation and failed to induce apoptosis [19]. Monia et al conducted a mutation screen of the Siah-1 coding sequence in 12 HCCs, 19 breast carcinomas, 9 prostate carcinomas, 7 colon carcinomas and 6 human cell lines that were derived from colon cancer carcinomas [32]. They found lack of Siah-1 mutation in tumors [32]. Koichi et al conducted a Siah-1 mutation analysis in 35 HCCs and only found one SNP [20]. Given that the above studies carried out merely in 35 and 12 HCC samples and their HCC specimens for detecting Siah-1 mutation were limited to those occurring loss of heterozygosity (LOH) at 16q12 [20, 32], we searched for Siah-1 mutations in a larger set of HCC samples with 270 HCC tissues and 9 HCC cell lines which were not specifically collected for LOH at 16q12. One synonymous transition from a G to A at codon 67 was found in one HCC tissue sample (Fig. 1C). Our result from large HCC cohort without specific LOH selection is more convincible to show that Siah-1 mutations are rare events in HCCs. These findings imply that Siah-1 is highly conserved and that Siah-1 gene mutations do not significantly account for disease development.
Subsequently, we detected the expression of Siah-1 in a subset of available paired HCC samples. Our results revealed that the Siah-1 transcriptional level was markedly reduced in 18 of 25 (72%) HCCs (Fig. 2A) and Western Blot indicated that Siah-1 protein levels were clearly decreased in 23 of 39 (58.97%) HCCs compared with adjacent normal tissues (Fig. 2B and 2C). Though previous studies have ever described Siah-1 levels in HCCs, Koichi et al and Monia et al only detected Siah-1 mRNA levels without examining its protein levels [20, 32]. As for the existence of complex post transcriptional regulation in vivo, the mRNA level is not always consistent with its protein expression. Brauckhoff et al have analyzed the cytoplasm and nuclear distribution of Siah-1 in HCC tissues via immunohistochemistry (IHC) assay [33], however, IHC method cannot reflect directly the alteration of total protein levels against Western blot. Therefore, our results further demonstrate that not only the transcriptional level but also total protein levels of Siah-1 are significantly down-regulated in HCC cases. There are some clues for the explanation of Siah-1 reduction in HCC. Siah-1 is located on chromosome 16q12.1, a region that frequently occurs LOH in cancers [20, 32]. The LOH at 16q12.1 was found to be associated with Siah-1 decrease in HCC [20]. In addition, Siah-1 is a p53-inducible gene, the high frequent p53 mutations in HCC may lead to the down-regulation of Siah-1. Our previous study also found that the p53 R249S mutant was unable to induce cellular Siah-1 expression [15]. In the present study, we did not examine the LOH status, p53 mutation in our HCC samples due to limited availability of the tissue specimens.
HBx plays important roles in HBV-related HCC development and progression [3]. Accumulating evidence has shown that the integrated HBx genes isolated from HCC patients are often mutated or deleted at their distal C-terminal region [5, 6]. Our group also detected the HBx status in the same HCC DNA that was used in the Siah-1 mutation screen [5]. In total, we found that 121 aa sites had mutations and that there were 48 C-terminal truncations in 287 HCC specimens. There were 133 aa site mutations and 21 HBx C-terminal truncations in 195 peri-carcinoma liver tissues [5]. In spite of lacking specific site mutations, HBx C-terminal truncations were identified in a significant number of our HCC samples [5]. Some of the HBx C-terminus truncations (HBx-Cts) have been verified to promote HCC progression. For instance, the C-truncated 34 aa mutant (HBxΔ120) can effectively transform the human immortalized liver cell line MIHA and increase the tumorigenicity of Hep3B and MIHA cells [34]. Moreover, HBxΔ120 cells exhibit differential expression of critical genes that are implicated in regulating the cell cycle and apoptosis compared with cells expressing full length HBx [34]. The HBx C-deleted 27 aa truncation HBxΔ127 was reported to enhance cell proliferation through the alteration of a positive feedback loop involving 5-lipoxygenase and fatty acid synthase [35]. The HBxΔ120 and HBxΔ127 variants were also detected in our HCC samples, with ratios of 1/287 and 2/287, respectively. Next, we constructed three truncated HBx variants, as shown in Fig. 3A. HBx-D1 was lacking the 128 to 132 aa region and was found in 2/287 of our HCC samples. The HBx-D2 and HBx-D3 variants contained the same mutations as the aforementioned HBxΔ127 and HBxΔ120 variants, respectively. When co-expressed with Siah-1, we found that the three truncated HBx variants resisted Siah-1-mediated degradation (Fig. 3C). The three truncated HBx variants maintained transactivation capacity at HSEs similar to full length HBx (Fig. 4A and 4B). However, co-expressing Siah-1 failed to attenuate their transactivation ability, whereas Siah-1 could inhibit the ability of HBx to transactivate HSEs (Fig. 4C). Moreover, these HBx mutants had weaker association with Siah-1 than full length HBx protein (Fig. 5A). Thus, we propose that the down-regulation of Siah-1 and highly frequent HBx truncations in HCC decrease the availability of Siah-1 to interact with HBx, which have HBx and it variants to evade Siah-1-induced degradation, and the consequent accumulation of HBx and its variants may enhance the carcinogenic potential to promote HCC development (Fig. 5B).
In summary, we found that Siah-1 was significantly down-regulated in HCC, though no somatic mutations were detected. Moreover, Siah-1 presented weak association with natural HBx variants and failed to induce their degradation, and could not repress transcriptional activity of the HBx truncates. Our results illustrate a novel mechanism for Siah-1 and natural HBx truncates in HCC pathogenesis.
Acknowledgements
This study was supported by the Natural Science Foundation of China (81201521), the China Postdoctoral Science Foundation Funded Project (2013M541467), and the Foundation from Science and Technology Commission of Shanghai (13XD1401200).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Kew MC. Epidemiology of chronic hepatitis B virus infection, hepatocellular carcinoma, and hepatitis B virus-induced hepatocellular carcinoma. Pathol Biol (Paris). 2010;58:273-7
2. Peng Z, Zhang Y, Gu W, Wang Z, Li D, Zhang F. et al. Integration of the hepatitis B virus X fragment in hepatocellular carcinoma and its effects on the expression of multiple molecules: a key to the cell cycle and apoptosis. Int J Oncol. 2005;26:467-73
3. Zhang X, Zhang H, Ye L. Effects of hepatitis B virus X protein on the development of liver cancer. J Lab Clin Med. 2006;147:58-66
4. Zhao J, Wang W, Huang Y, Wu J, Chen M, Cui P. et al. HBx Elevates Oncoprotein AEG-1 Expression to Promote Cell Migration by Downregulating miR-375 and miR-136 in Malignant Hepatocytes. DNA Cell Biol. 2014
5. Wang D, Cai H, Yu WB, Yu L. Identification of hepatitis B virus X gene variants between hepatocellular carcinoma tissues and pericarcinoma liver tissues in Eastern China. Int J Clin Exp Pathol. 2014;7:5988-96
6. Poussin K, Dienes H, Sirma H, Urban S, Beaugrand M, Franco D. et al. Expression of mutated hepatitis B virus X genes in human hepatocellular carcinomas. Int J Cancer. 1999;80:497-505
7. Sze KM, Chu GK, Lee JM, Ng IO. C-terminal truncated hepatitis B virus x protein is associated with metastasis and enhances invasiveness by C-Jun/matrix metalloproteinase protein 10 activation in hepatocellular carcinoma. Hepatology. 2013;57:131-9
8. Yip WK, Cheng AS, Zhu R, Lung RW, Tsang DP, Lau SS. et al. Carboxyl-terminal truncated HBx regulates a distinct microRNA transcription program in hepatocellular carcinoma development. PLoS One. 2011;6:e22888
9. Hu Z, Zhang Z, Doo E, Coux O, Goldberg AL, Liang TJ. Hepatitis B virus X protein is both a substrate and a potential inhibitor of the proteasome complex. J Virol. 1999;73:7231-40
10. Sohn SY, Kim SB, Kim J, Ahn BY. Negative regulation of hepatitis B virus replication by cellular Hsp40/DnaJ proteins through destabilization of viral core and X proteins. J Gen Virol. 2006;87:1883-91
11. Kim JH, Kang S, Kim J, Ahn BY. Hepatitis B virus core protein stimulates the proteasome-mediated degradation of viral X protein. J Virol. 2003;77:7166-73
12. Park SG, Min JY, Chung C, Hsieh A, Jung G. Tumor suppressor protein p53 induces degradation of the oncogenic protein HBx. Cancer Lett. 2009;282:229-37
13. Ling MT, Chiu YT, Lee TK, Leung SC, Fung MK, Wang X. et al. Id-1 induces proteasome-dependent degradation of the HBX protein. J Mol Biol. 2008;382:34-43
14. Xian L, Zhao J, Wang J, Fang Z, Peng B, Wang W. et al. p53 Promotes proteasome-dependent degradation of oncogenic protein HBx by transcription of MDM2. Mol Biol Rep. 2010;37:2935-40
15. Zhao J, Wang C, Wang J, Yang X, Diao N, Li Q. et al. E3 ubiquitin ligase Siah-1 facilitates poly-ubiquitylation and proteasomal degradation of the hepatitis B viral X protein. FEBS Lett. 2011;585:2943-50
16. Hu G, Chung YL, Glover T, Valentine V, Look AT, Fearon ER. Characterization of human homologs of the Drosophila seven in absentia (sina) gene. Genomics. 1997;46:103-11
17. Hu G, Fearon ER. Siah-1 N-terminal RING domain is required for proteolysis function, and C-terminal sequences regulate oligomerization and binding to target proteins. Mol Cell Biol. 1999;19:724-32
18. Matsuzawa SI, Reed JC. Siah-1, SIP, and Ebi collaborate in a novel pathway for beta-catenin degradation linked to p53 responses. Mol Cell. 2001;7:915-26
19. Kim CJ, Cho YG, Park CH, Jeong SW, Nam SW, Kim SY. et al. Inactivating mutations of the Siah-1 gene in gastric cancer. Oncogene. 2004;23:8591-6
20. Matsuo K, Satoh S, Okabe H, Nomura A, Maeda T, Yamaoka Y. et al. SIAH1 inactivation correlates with tumor progression in hepatocellular carcinomas. Genes Chromosomes Cancer. 2003;36:283-91
21. Roperch JP, Lethrone F, Prieur S, Piouffre L, Israeli D, Tuynder M. et al. SIAH-1 promotes apoptosis and tumor suppression through a network involving the regulation of protein folding, unfolding, and trafficking: identification of common effectors with p53 and p21(Waf1). Proc Natl Acad Sci U S A. 1999;96:8070-3
22. Qi J, Kim H, Scortegagna M, Ronai ZA. Regulators and effectors of Siah ubiquitin ligases. Cell Biochem Biophys. 2013;67:15-24
23. Santelli E, Leone M, Li C, Fukushima T, Preece NE, Olson AJ. et al. Structural analysis of Siah1-Siah-interacting protein interactions and insights into the assembly of an E3 ligase multiprotein complex. J Biol Chem. 2005;280:34278-87
24. Abada R, Dreyfuss-Grossman T, Herman-Bachinsky Y, Geva H, Masa SR, Sarid R. SIAH-1 interacts with the Kaposi's sarcoma-associated herpesvirus-encoded ORF45 protein and promotes its ubiquitylation and proteasomal degradation. J Virol. 2008;82:2230-40
25. Nagel CH, Albrecht N, Milovic-Holm K, Mariyanna L, Keyser B, Abel B. et al. Herpes simplex virus immediate-early protein ICP0 is targeted by SIAH-1 for proteasomal degradation. J Virol. 2011;85:7644-57
26. Nagano Y, Yamashita H, Takahashi T, Kishida S, Nakamura T, Iseki E. et al. Siah-1 facilitates ubiquitination and degradation of synphilin-1. J Biol Chem. 2003;278:51504-14
27. Rott R, Szargel R, Haskin J, Shani V, Shainskaya A, Manov I. et al. Monoubiquitylation of alpha-synuclein by seven in absentia homolog (SIAH) promotes its aggregation in dopaminergic cells. J Biol Chem. 2008;283:3316-28
28. Engelender S, Kaminsky Z, Guo X, Sharp AH, Amaravi RK, Kleiderlein JJ. et al. Synphilin-1 associates with alpha-synuclein and promotes the formation of cytosolic inclusions. Nat Genet. 1999;22:110-4
29. Liani E, Eyal A, Avraham E, Shemer R, Szargel R, Berg D. et al. Ubiquitylation of synphilin-1 and alpha-synuclein by SIAH and its presence in cellular inclusions and Lewy bodies imply a role in Parkinson's disease. Proc Natl Acad Sci U S A. 2004;101:5500-5
30. Franck T, Krueger R, Woitalla D, Muller T, Engelender S, Riess O. Mutation analysis of the seven in absentia homolog 1 (SIAH1) gene in Parkinson's disease. J Neural Transm. 2006;113:1903-8
31. Amson RB, Nemani M, Roperch JP, Israeli D, Bougueleret L, Le Gall I. et al. Isolation of 10 differentially expressed cDNAs in p53-induced apoptosis: activation of the vertebrate homologue of the drosophila seven in absentia gene. Proc Natl Acad Sci U S A. 1996;93:3953-7
32. Medhioub M, Vaury C, Hamelin R, Thomas G. Lack of somatic mutation in the coding sequence of SIAH1 in tumors hemizygous for this candidate tumor suppressor gene. Int J Cancer. 2000;87:794-7
33. Brauckhoff A, Malz M, Tschaharganeh D, Malek N, Weber A, Riener MO. et al. Nuclear expression of the ubiquitin ligase seven in absentia homolog (SIAH)-1 induces proliferation and migration of liver cancer cells. J Hepatol. 2011;55:1049-57
34. Ma NF, Lau SH, Hu L, Xie D, Wu J, Yang J. et al. COOH-terminal truncated HBV X protein plays key role in hepatocarcinogenesis. Clin Cancer Res. 2008;14:5061-8
35. Wang Q, Zhang W, Liu Q, Zhang X, Lv N, Ye L. A mutant of hepatitis B virus X protein (HBxDelta127) promotes cell growth through a positive feedback loop involving 5-lipoxygenase and fatty acid synthase. Neoplasia. 2010;12:103-15
Author contact
![]() Corresponding authors: Long Yu and Wen-Hong Zhang. Prof. Long Yu, Tel: (+86) 21 65643954; Fax: (+86) 21 65643404; E-mail: longyuedu.cn; Prof. Wen-Hong Zhang, Tel: (+86) 21 52888023; Fax: (+86) 21 62489015; E-mail: zhangwenhongedu.cn.
Corresponding authors: Long Yu and Wen-Hong Zhang. Prof. Long Yu, Tel: (+86) 21 65643954; Fax: (+86) 21 65643404; E-mail: longyuedu.cn; Prof. Wen-Hong Zhang, Tel: (+86) 21 52888023; Fax: (+86) 21 62489015; E-mail: zhangwenhongedu.cn.