Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2016; 7(3):344-352. doi:10.7150/jca.13396 This issue Cite
Research Paper
Proton Beams Inhibit Proliferation of Breast Cancer Cells by Altering DNA Methylation Status
Byungtak Kim1, Hansol Bae1, Hyunkyung Lee1, Seungyeon Lee1, Jeong Chan Park2, Kye Ryung Kim2, Sun Jung Kim1, ![]()
1. Department of Life Science, Dongguk University-Seoul, Goyang, Korea;
2. Korea Multi-purpose Accelerator Complex, Korea Atomic Energy Research Institute, Gyeongju, Korea
Received 2015-7-30; Accepted 2015-11-15; Published 2016-1-10
Citation:
Kim B, Bae H, Lee H, Lee S, Park JC, Kim KR, Kim SJ. Proton Beams Inhibit Proliferation of Breast Cancer Cells by Altering DNA Methylation Status. J Cancer 2016; 7(3):344-352. doi:10.7150/jca.13396. https://www.jcancer.org/v07p0344.htm
Other stylesAbstract
Proton beam therapy has been gaining popularity in the management of a wide spectrum of cancers. However, little is known about the effect of proton beams on epigenetic alterations. In this study, the effects of proton beams on DNA methylation were evaluated in the breast cell lines MCF-10A and MCF-7. Pyrosequencing analysis of the long interspersed element 1 (LINE1) gene indicated that a few specific CpG sites were induced to be hypermethylated by proton beam treatment from 64.5 to 76.5% and from 57.7 to 60.0% (p < 0.05) in MCF-10A and MCF-7, respectively. Genome-wide methylation analysis identified “Developmental Disorder, Hereditary Disorder, Metabolic Disease” as the top network in the MCF-7 cell line. The proliferation rate significantly decreased in proton beam-treated cells, as judged by colony formation and cell proliferation assay. Upon treatment with the proton beam, expression of selected genes (MDH2, STYXL1, CPE, FAM91A1, and GPR37) was significantly changed in accordance with the changes of methylation level. Taken together, the findings demonstrate that proton beam-induced physiological changes of cancer cells via methylation modification assists in establishing the epigenetic basis of proton beam therapy for cancer.
Keywords: Alu, breast cancer, epigenetics, LINE1, methylation, proton beam
Introduction
Proton beam radiation has been established as a highly effective modality for the local control of tumor growth [1]. The mass properties of proton particles and their unique physical characteristics allow proton therapy to spare normal tissues distal to the tumor target from incidental irradiation in various tumors [2]. For example, proton radiation doses reduced the risk of prostate cancer recurrence and the rate of metastasis [3, 4]. In another clinical study, proton beam radiation therapy used to deliver partial breast irradiation in 100 subjects produced an overall survival of 95% in a 60-month follow-up [5]. In addition, proton beam therapy has obtained successful outcomes for other cancers including lung [6], liver [7], and esophageal forms [8].
At the in vitro cell level, proton beam radiation has been used either to confirm the actual in vivo biological effects or to monitor physiological changes. Cancer stem cells from paclitaxel-resistant lung cancer cell lines showed significantly lower cell viability and invasiveness, as well as more apoptosis, by the application of protons [9]. The oncogenic properties of HT1080 human fibrosarcoma cells were decreased by proton irradiation in a dose-dependent manner in terms of cell migration and invasion [2]. Experimental studies to elucidate the molecular mechanisms underlying the cellular activity changes by proton beam are relatively sparse [10, 11]. For example, vimentin, a marker of epithelial to mesenchymal transition and the metastatic properties of melanoma, was substantially decreased in proton beam-irradiated melanoma cells [12].
CpG methylation is an epigenetic phenomenon that can regulate gene activity by altering the methylation level [13, 14]. Many tumor suppressors and oncogenes have been shown to undergo hyper- or hypo-methylation during tumor development in variable cancer types [15, 16]. Various radiation beams, such as X-rays and γ-rays, are presently being applied in medical treatments. They are known to have effects on inducing methylation status changes of cancer-related genes. [17, 18]. Despite the wealth of experimental evidence related with gene expression and cellular activity [8, 19], little study has been performed to investigate the effect of proton beams on epigenetic changes in terms of DNA methylation, a main event contributing to tumorigenesis.
In this study, global methylation changes were monitored in a pair of normal and cancer cell lines of mammary gland, MCF-10A and MCF-7, respectively, following proton beam treatment. Also, Ingenuity pathway analysis (IPA) was performed with the genes, showing CpG methylation levels significantly changed on a genome-wide methylation array. In addition, cellular activity changes of the cancer cells, including cell proliferation and colony formation capacity, were assessed upon proton beam treatment. To the extent of our knowledge, this is the first study to examine the effects of proton beams on the genome-wide methylation level of cancer cells.
Materials and methods
Cell culture and proton beam treatment
A normal breast cell line, MCF-10A, and two breast cancer cell lines, MCF-7 and MDA-MB-231, were acquired from the American Type Culture Collection (ATCC, Manassas, VA). The culture medium of the cells was as described previously [20]. The cells were grown to 80% confluence in 25 cm2 culture flasks and then treated by proton beam at 45 MeV and 8 Gy using a proton accelerator installed in the Korea Multi-Purpose Accelerator Complex (KOMAC, Gyeongju, Korea). The cells were harvested for further analysis 24 h after proton beam treatment.
Genome-wide methylation analysis
Chromosomal DNA from cells grown in 25 cm2 culture flasks was isolated using a ZR-Duet DNA/RNA MiniPrep (Zymo Research, Irvine, CA) with a final elution in 40 μl of distilled water. A quantity of 50 ng of chromosomal DNA was treated with bisulfite and applied to the Infinium HumanMethylation450 Beadchip to monitor genome-wide methylation (> 485,000 CpG sites) (Illumina, San Diego, CA). The methylation level was presented as a methylation index (β) as described previously [21]. Briefly, β values ranging from 0 (no methylation) to 1 (100% methylation) were calculated; the higher the value, the greater the level of methylation in proton beam-treated samples relative to non-treated cells. The results of the array have been deposited on the Gene Expression Omnibus database (http://www.ncbi.nlm.nih.gov/geo/) and are accessible with the accession number GSE70024.
Pyrosequencing
One microgram of genomic DNA from proton beam-treated and non-treated cell samples was sodium bisulfite-converted using an EZ DNA methylation kit (Zymo Research). Then, the methylation level of LINE1 and Alu was determined through pyrosequencing by PyroMark ID (Qiagen, Valencia, CA) as explained previously [22]. To verify efficiency of bisulfite DNA conversion, non-CpG cytosines were adopted as internal controls. The methylation level was denoted by methylated cytosines over total cytosines as percentage. Pyrosequencing was performed on five cultured cell samples independently treated by proton beam, and a minimum of two assays per sample were performed.
Cell proliferation and colony formation assay
Cell proliferation was evaluated using a cell counter kit-8 (CCK-8, Dojindo, Japan) according to the manufacturer's instructions. Briefly, 2 × 103 cells were plated into a 96-well flat-bottomed plate with 100 μl media. Following proton beam treatment, cells were incubated for 24, 48, 96, and 120 h. After adding 10 μl of CCK-8 solution to each well of the plate, the absorbance at 450 nm was measured using a microplate reader. In colony formation assay, a 60 mm cell culture dish was used with 3 ml medium and 200 cells per dish. Two weeks after proton beam treatment, cells were fixed with a mixture of acetic acid/methanol (1:7), and then stained with crystal violet (1%).
Pathway analysis
The functional categorization and canonical pathway for the gene pool obtained from the CpGs affected by proton beam treatment were constructed with the IPA software tool. Obtaining p-values for individual networks was achieved by Fisher's exact test considering the hypergeometric distribution, as described previously [20]. The functional network that demonstrated the highest confidence was appointed as the top network.
Quantitative reverse transcription-polymerase chain reaction (qRT-PCR)
cDNA was synthesized from 2 μg of total RNA using a ReverTra Ace qPCR RT Master Mix (Toyobo, Japan) according to the instructions of the manufacturer. qRT-PCR analysis was carried out for quantifying the expression level of selected genes. Reactions were performed in triplicate at a minimum for each sample with a Kapa SYBR qPCR Kit (Kapa Biosystems, Woburn, MA) with primers complementary to specific genes (Supplementary Table S1) on an ABI 7300 instrument (Applied Biosystems). GAPDH was used to normalize RNA quantity, and 2-ΔCt method was applied for the gene expression quantification process.
Data mining and statistical analysis
The gene expression-based outcome for breast cancer online (GOBO) tool (http://co.bmc.lu.se/gobo) is designed for prognostic validation of genes in a pooled breast cancer data set comprising 1,881 cases from 11 public microarray data sets [23]. It was used in the present study to validate the methylation and expression analysis of CPE in breast cancer.
In the methylation array data, adjusted p-values <0.05 were screened for further analysis. The filtered genes were then characterized as differentially methylated when they showed a difference greater than 2-fold in methylation levels between proton beam-treated and non-treated cells in order to curtail false positive outcomes. The differences of mean level of methylation for Alu and LINE1, and the expression level between the proton beam-treated and non-treated cells, were detected by Student's t-test using SPSS for Windows, version 17.0 (SPSS Inc., Chicago, IL). P-values < 0.05 were considered statistically significant.
Figure 1
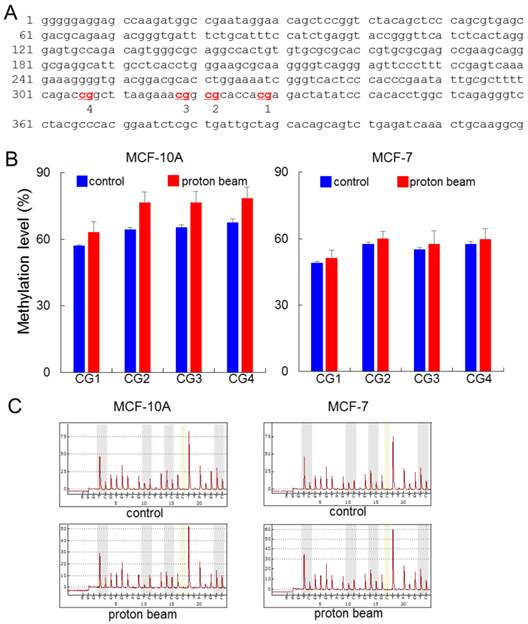
Effect of proton beam treatment on global methylation level in the MCF-10A and MCF-7 cell lines. Four CpG sites on the LINE1 in MCF-10A and MCF-7 cell lines were analyzed for their methylation level by pyrosequencing after treatment with a proton beam. (A) The sequence of the LINE1 gene adopted in this study (GenBank accession no. X58075.1). The four analyzed CpG sites are indicated in red and numbered. (B) The methylation level of the CpGs in the two cell lines are shown in bar graphs. Quintuplicated independent samples were analyzed, and average numbers are provided with the associated standard errors. (C) Representative pyrosequencing diagrams for the proton beam-treated and untreated cells.
Results
Proton beam treatment induces less global hypermethylation in MCF-7 cells than in MCF-10A cells
To examine whether the proton beam exerts its effect on the cellular activities of the cancer cell through epigenetic changes, breast cancer cell line MCF-7 was chosen and treated with the proton beam alongside normal breast cell line MCF-10A. Two highly repetitive chromosome elements, Alu and LINE1, were selected to determine the global methylation change induced by the proton beam treatment. The two elements exist on the chromosome with a high abundance (LINE1, ~105 copies/genome; Alu, ~106 copies/genome), and their hypomethylation has been frequently reported in diverse tissues, leading to tumorigenesis [24, 25].
The pyrosequencing analysis indicated that LINE1 was hypermethylated at all of the four CpG sites examined. The range of increase was from 6.1 to 11.2% by proton treatment in the MCF-10A cell line (p < 0.05) (Figure 1). Meanwhile in the MCF-7 cancer cell line, although the CpG sites underwent hypermethylation, their altered level was limited compared with the MCF-10A, as the breadth of increase only ranged from 2.0 to 2.5%. To determine whether the resistance to methylation change appears in cancer cells, another cancer cell line, MDA-MB-231, was examined; the results showed even less methylation changes than MCF-7 (Supplementary Figure S1). These results indicate that the proton beam acts in a tumor-specific manner. In contrast to LINE1, Alu did not show any significant methylation changes throughout the CpG sites in the three cell types (Supplementary Figure S2).
Developmental disorder, hereditary disorder, metabolic disease pathway is entailed in network of proton beam-induced methylation-altered genes
To evaluate the effect of proton beam treatment on the epigenomic change of the MCF-7 cell, genome-wide methylation profiles were monitored through microarray analysis. A set of 1,040 CpG sites matching our criteria of methylation profiles (i.e., methylation level change equal to or higher than 2-fold after proton beam treatment) was selected for further analysis. As shown in Figure 2, 826 and 214 CpGs were hyper- and hypo-methylated, respectively, in the proton beam-treated cells. The CpGs appeared throughout the chromosomes, with 770 being found in the coding region, 572 in the promoter region, and 204 in the intergenic region.
The 572 CpGs at the promoter sites in the MCF-7 cell were submitted to the IPA software tool to examine the functional inter-relatedness. The results indicated the “Developmental Disorder, Hereditary Disorder, Metabolic Disease” pathway as the top network and the “Inflammatory Disease, Skeletal and Muscular Disorders, Cancer” pathway as the second most prevalent network, suggesting their potential roles in tumorigenesis (Figure 3 and Table 1). Of note, a group of genes regulated by, or regulating, NF-κB (complex) featured most prominently in the top network, although NF-κB itself did not undergo methylation change. Included in the group are CASP8AP2, COMMD1, PRAS, FAF1, FAM46A, SLC37A4, SLC2A12, and NKIRAS2; all were remarkably hypermethylated by the proton beam. Among those, CASP8AP2 and PRAS are known to be upregulated in human cancer [26, 27]. The transcripts displaying the highest change of methylation level within this network were LRCH3 (hypermethylated, 3.5-fold increase) and GTF2B (hypomethylated, 2.7-fold decrease). LRCH3 is a member of the LRCH proteins that are a family of cytoskeletal regulators; their relevance to cancer is yet to be determined [28]. GTF2B is a ubiquitous transcription factor involved in the regulation of many cancer-related genes [29].
Table 1
Genes appearing at the top network in MCF-7 with differential methylation by proton beam.
| Symbol | Accession | Description | Fold change |
|---|---|---|---|
| CASP8AP2 | NM_012115 | CASP8-associated protein 2 | 2.1 |
| CBX5 | NM_012117 | Chromobox homolog 5 (HP1 alpha homolog, Drosophila) | -2.2 |
| CENPA | NM_001809 | Centromere protein A | 2.3 |
| CEP97 | NM_024548 | Centrosomal protein 97kDa | -2.8 |
| COMMD1 | NM_016144 | Copper metabolism (Murr1) domain-containing 1 | 2.3 |
| ERMAP | NM_001017922 | Erythroblast membrane-associated protein | 2.7 |
| FAF1 | NM_007051 | Fas (TNFRSF6) associated factor 1 | 2.4 |
| FAM46A | NM_017633 | Family with sequence similarity 46, member A | 3.0 |
| GMFB | NM_004124 | Glia maturation factor, beta | 2.2 |
| GTF2B | NM_001514 | General transcription factor IIB | -2.7 |
| GTF2H4 | NM_001517 | General transcription factor IIH, polypeptide 4, 52kDa | 2.1 |
| HIST1H2AD | NM_021065 | Histone cluster 1, H2ad | 2.1 |
| HIST3H2A | NM_033445 | Histone cluster 3, H2a | -2.1 |
| HIST3H2BB | NM_175055 | Histone cluster 3, H2bb | -2.1 |
| HMGCL | NM_000191 | 3-hydroxymethyl-3-methylglutaryl-Coenzyme A lyase | -2.3 |
| LRCH3 | NM_032773 | Leucine-rich repeats and calponin homology (CH) domain-containing 3 | 3.5 |
| NKIRAS2 | NM_001001349 | NFKB inhibitor interacting Ras-like 2 | 2.0 |
| RDM1 | NM_001163120 | RAD52 motif 1 | -2.2 |
| RPS10-NUDT3 | NM_001202470 | Ribosomal protein S10 and nucleoside diphosphate linked moiety X type motif 3 | 3.1 |
| RRAS | NM_006270 | Related RAS viral (r-ras) oncogene homolog | 2.7 |
| RSU1 | NM_012425 | Ras suppressor protein 1 (RSU1) | 2.2 |
| SLC2A12 | NM_145176 | Solute carrier family 2 (facilitated glucose transporter), member 12 | 2.2 |
| SLC37A4 | NM_001164280 | Solute carrier family 37 (glucose-6-phosphate transporter), member 4 | 2.0 |
| SUGT1 | NM_006704 | SGT1, suppressor of G2 allele of SKP1 (S. cerevisiae) | 2.3 |
| SUPT20H | NM_001014286 | Family with sequence similarity 48, member A | 2.1 |
| TCEB3 | NM_003198 | Transcription elongation factor B (SIII), polypeptide 3 (110kDa, elongin A) | 2.1 |
| UBE2B | NM_003337 | Ubiquitin-conjugating enzyme E2B (RAD6 homolog) | 2.7 |
| YBX3 | NM_001145426 | Cold shock domain protein A | 2.2 |
| ZNF8 | NM_021089 | Zinc finger protein 8 | 2.5 |
| ZW10 | NM_004724 | ZW10, kinetochore associated, homolog (Drosophila) | 2.0 |
Figure 2
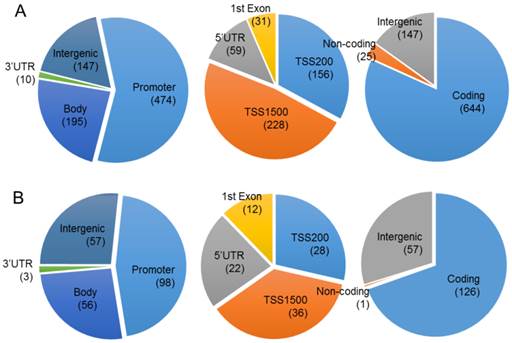
Chromosomal distribution of the CpGs in which methylation level was significantly changed by proton beam treatment. Genome-wide methylation analysis of CpGs in the MCF-7 cell line was carried out on a 450K microarray, and CpGs showing significant changes by proton beam treatment compared with untreated cells were adopted for the analysis. Venn diagrams show the allocation of the CpGs on chromosome, which were hypermethylated (A) and hypomethylated (B). The values in parentheses indicate the number of CpGs in each category. TSS, transcription start site; UTR, untranslated region.
Figure 3
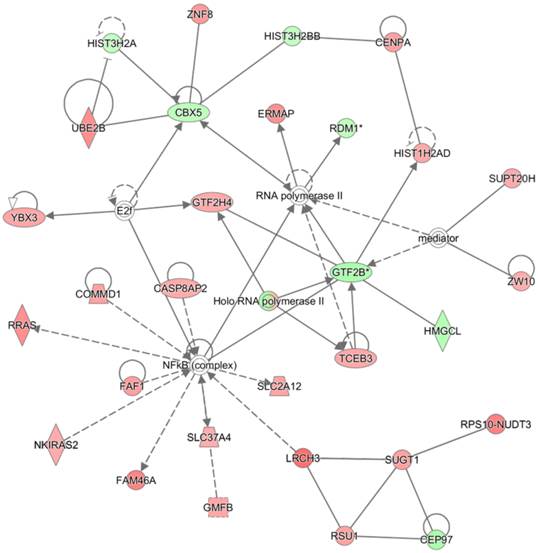
The highest confidence network constructed with differentially methylated genes by proton beam treatment on the MCF-7 cell. The CpGs in which methylation level was significantly changed by proton beam treatment were analyzed through the Ingenuity pathway analysis software tool. The analysis indicated “Developmental Disorder, Hereditary Disorder, Metabolic Disease” as the network of highest confidence. Red-colored genes are hypermethylated in the proton beam treated cells, and green-colored genes indicate hypomethylation, with intensity signifying the magnitude of methylation change. Solid and dashed lines represent direct and indirect interactions, respectively, with each interaction supported by at least one literature reference.
Proton beam treatment inhibits proliferation of breast cancer cells
Proton beam application altered the methylation level of a collection of genes that were related to “Developmental Disorder, Hereditary Disorder, Metabolic Disease” or “Inflammatory Disease, Skeletal and Muscular Disorders, Cancer” pathways. This observation prompted us to examine the effect of proton beam treatment on cellular proliferation. To do this, the proliferation of the MCF-10A, MCF-7, and MDA-MB-231 cells were examined by CCK-8 cell proliferation assay as well as colony formation assay, after being treated or remaining untreated with proton beam. Findings indicated that proton beam treatment retarded the proliferation in all three cell types (Figure 4A). Of note, the anti-proliferation effect of proton beam treatment appeared later in MCF-7 and MDA-MB-231 cancer cells than in the normal MCF-10A cell line; a remarkable proliferation rate difference was observed between the proton beam-treated and non-treated samples. This difference appeared 3 days after proton beam treatment in the cancer cells, while it appeared after 2 days in the normal cell.
To confirm the anti-proliferation effect of proton beams addressed by the proliferation assay, colony formation assay was carried out with the MCF-7 cell line. As shown in Figure 4B, colony sizes were remarkably smaller in the case of the proton beam-treated cells.
Expression analysis of the differentially methylated genes
To check for the association between promoter methylation and gene expression, genes that showed hypermethylation or hypomethylation on the methylation array after proton beam treatment of the MCF-7 cells were randomly selected. Expression levels then analyzed by real-time RT-PCR. Four hypomethylated genes (MDH2, STYXL1, CPE, and GPR37) and two hypermethylated genes (FAM91A1 and POU2AF1) were selected, in which CpG sites were located at the promoter region. As shown in Figure 5, all of the genes were up- or down-regulated in line with the methylation profile, except for POU2AF1, for which expression was not significantly changed, while the CpGs were hypermethylated.
For one of the dysregulated genes, CPE, interrogating its expression using the GOBO database revealed that patients with higher expression were more likely to have a higher rate of distant metastasis-free survival (DMSF) (p < 0.05) (Figure 6). Taken together, the results indicate that proton beam-induced expression change of cancer-related genes via alteration of CpG methylation level in the cultured cancer cells may support the merits of proton beam treatment for cancer therapy with respect to epigenetics.
Figure 4
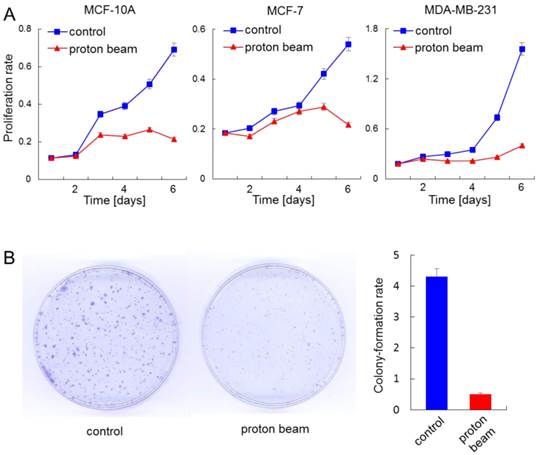
Anti-proliferation effect of proton beam treatment on mammary gland cells. The indicated cells were treated with a proton beam at a strength of 8 Gy, and their proliferation was monitored alongside the non-treated cells. (A) Cell proliferation assay using cell counting kit-8 was carried out for a normal cell line, MCF-10A, and cancerous cell lines MCF-7 and MDA-MB-231. (B) Colony formation assay was carried out for the MCF-7 cell. The bar graph denotes the ratio of colonies shown by the colony formation assay.
Figure 5
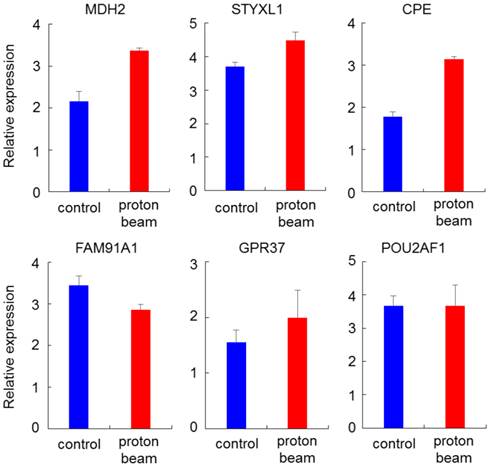
Expression quantification of methylation-altered genes by proton beam treatment through qRT-PCR in breast cancer cells. Real-time RT-PCR analysis of six genes with altered methylation levels due to proton beam treatment in MCF-7 cell. All genes, excepting POU2AF1, showed expression changes in accordance with the change of methylation. Triplicated samples were analyzed in each experiment, and the average of relative expression values are shown with standard errors.
Figure 6
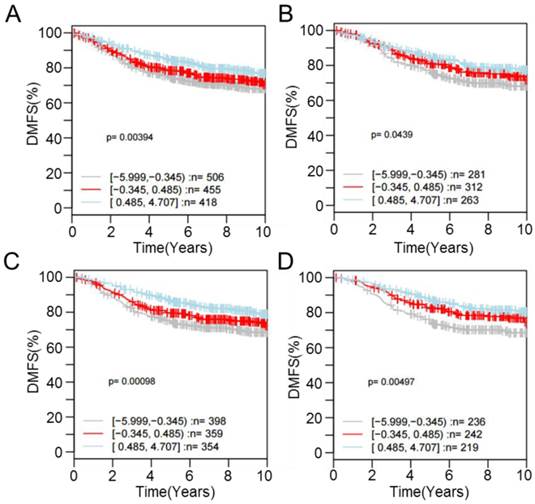
Kaplan-Meier survival analysis of CPE expression in the breast cancer. Samples (n = 1,379) were stratified into tertiles based on CPE expression level. The log-rank test was performed in all tumor samples, as well as in different tumor subtypes, using distant metastasis-free survival (DMFS) as the endpoint. High CPE expression is significantly associated with higher DMFS over time among (A) all cases regardless of tumor ER- and LN-positivity (p = 3.94E-3), (B) cases with ER-positive tumors (p = 4.39E-2), (C) cases with LN-negative tumors (p = 9.80E-4), and (D) cases with ER-positive and LN-negative tumors (p = 4.97E-3).
Discussion
Although radiation therapies have become popular in cancer treatment, their molecular mechanisms, especially with respect to epigenetics yielding cellular activity change, have been minimally elucidated. Considering the fact that proton beams affect key regulatory pathways, including apoptosis, cell proliferation, and cell signaling, and that epigenetic alterations are responsible for these phenomena, it is reasonable to consider the existence of an epigenetic route while the proton beam acts on the cell. To examine the potential of proton beams to induce methylation changes in DNA, the global marker genes Alu and LINE1, which are scattered on the chromosome in high copies, were selected and monitored for methylation changes by the proton beam. Hypomethylation of these markers has been implicated in various cancer types; this hypomethylation may cause genomic instability, eventually leading cells to cancer [30, 31]. Therefore, the hypermethylation of LINE1 observed in this study by proton beam treatment may imply that the beam has an anti-tumorigenic effect on the cells. In detail, the normal breast cell line MCF-10A showed more hypermethylation than the cancerous cell line, MCF-7, implying that the normal cell is more resistant to the proton beam with respect to genomic instability. Furthermore, methylation change of LINE1 suggests its function as a molecular marker to assure proper exposure of target cells to the proton beam. A few molecular markers and cellular activities, such as DNA strand break [32] and induction of apoptosis [33], are currently proposed.
More CpGs were hypermethylated than those hypomethylated by proton beam treatment (826 vs. 214), possibly suggesting that more genes are deactivated than are activated. Other forms of physical radiation such as X-rays [34], γ-rays [18], and ion beams [35] have also been shown to alter CpG methylation. Genome-wide screening is available for ionizing radiation. These data included 1,235 hypermethylated and 1,281 hypomethylated CpGs, and the dysregulated pathway is different from that of the proton beam, wherein the IL-10 signaling pathway and acute phase signaling involved in immune signaling were significantly overpresented [36].
Many tumor suppressor genes, including ARF6 (2.6-fold increase), NF2 (2.2-fold increase), and CDKN2C (2.7-fold increase), were hypermethylated by the proton beam, suggesting potential cell-proliferative activity of the proton beam through suppressing expression of the tumor suppressors. However, in practice, the proton beam-treated MCF-7 cells experienced growth retardation, implying an alternative molecular mechanism for this phenomenon. One possible explanation for this is the high score of decreased repair of DNA from the IPA analysis. In fact, genes involved in DNA repair such as CDK2AP1, NFE2L1, and SMUG1 were hypermethylated. It is therefore speculated that dysregulated genes responsible for DNA repair acted more effectively than genes responsible for tumorigenesis, causing the cell to undergo less proliferation as a result of the proton beam. This explanation was supported by the colony forming assay as well as the cell proliferation assay (Figure 4).
The methylation of CpGs on the promoter appears to be strongly associated with its gene expression, as judged by RT-PCR results. In particular, expression of CPE was increased with hypomethylation by proton beam. The higher survival rate of breast cancer patients with higher expression of the tumor suppression gene may support the merits of proton beam treatment for cancer therapy with respect to epigenetics.
A limitation of this study is the absence of an assessment of methylation levels with respect to varied strengths of the proton beam. Previous studies indicate different responses of gene expression and cellular activity subject to different strengths of physical radiation. For example, a study investigated the radioactive response in AG01522 human fibroblasts first exposed to 5 Gy and then followed by 2 Gy of X-ray's, in comparison to those cells that had only received a single 2 Gy dose; a low-dose X-ray triggered cellular responses that defended against subsequent high dose-induced damages [37]. However, information regarding the applied exposure conditions of proton beams is sparse, as this therapy is still under development. In addition to the 8-Gy dose adopted in this study, a wide range of proton beam strengths need to be applied to the studied cells, which will help to pinpoint the precise strength that provokes a specific epigenetic change.
In summary, we have presented global, as well as genome-wide, methylation change profiles of proton beam-treated breast cancer cells. These were compiled using the most comprehensive methylation measurement and analysis techniques available. Our findings support the hypothesis that proton beam treatment results in cellular activity change through a mechanism of epigenetic alteration. Genes related to cell-to-cell signaling or DNA repair are key among those affected. Further investigation into the mechanisms leading to altered methylation, as well as the association between methylation change and cell death, could replenish our understanding of the epigenetic role of proton beam treatment, and thereby contribute to treatment options for cancer that employ this technology.
Supplementary Material
Supplementary Table S1 and Figures S1-S2.
Acknowledgements
This work was supported by National Research Foundation of Korea (NRF) grants 2014M2B2A4030844 and NRF-2012R1A1A2040830.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Lukens JN, Lin A, Hahn SM. Proton therapy for head and neck cancer. Curr Opin Oncol. 2015;27:165-71
2. Ogata T, Teshima T, Kagawa K. et al. Particle irradiation suppresses metastatic potential of cancer cells. Cancer Res. 2005;65:113-20
3. Zietman AL, Bae K, Slater JD. et al. Randomized trial comparing conventional-dose with high-dose conformal radiation therapy in early-stage adenocarcinoma of the prostate: long-term results from proton radiation oncology group/american college of radiology 95-09. J Clin Oncol. 2010;28:1106-11
4. Kuban DA, Levy LB, Cheung MR. et al. Long-term failure patterns and survival in a randomized dose-escalation trial for prostate cancer. Who dies of disease? Int J Radiat Oncol Biol Phys. 2011;79:1310-7
5. Bush DA, Do S, Lum S. et al. Partial breast radiation therapy with proton beam: 5-year results with cosmetic outcomes. Int J Radiat Oncol Biol Phys. 2014;90:501-5
6. Rana S, Simpson H, Larson G. et al. Dosimetric impact of number of treatment fields in uniform scanning proton therapy planning of lung cancer. J Med Phys. 2014;39:212-8
7. Muroi H, Nakajima M, Satomura H. et al. Effectiveness of proton beam therapy on liver metastases of esophageal cancer: report of a case. Int Surg. 2015;100:180-4
8. Ishikawa H, Hashimoto T, Moriwaki T. et al. Proton beam therapy combined with concurrent chemotherapy for esophageal cancer. Anticancer Res. 2015;35:1757-62
9. Zhang X, Lin SH, Fang B. et al. Therapy-resistant cancer stem cells have differing sensitivity to photon versus proton beam radiation. J Thorac Oncol. 2013;8:1484-91
10. Romanowska-Dixon B, Elas M, Swakon J. et al. Metastasis inhibition after proton beam, beta- and gamma-irradiation of melanoma growing in the hamster eye. Acta Biochim Pol. 2013;60:307-11
11. Calugaru V, Nauraye C, Cordelieres FP. et al. Involvement of the Artemis protein in the relative biological efficiency observed with the 76-MeV proton beam used at the Institut Curie Proton Therapy Center in Orsay. Int J Radiat Oncol Biol Phys. 2014;90:36-43
12. Kedracka-Krok S, Jankowska U, Elas M. et al. Proteomic analysis of proton beam irradiated human melanoma cells. PLoS One. 2014;9:e84621
13. Schubeler D. Function and information content of DNA methylation. Nature. 2015;517:321-6
14. Rhee I, Jair KW, Yen RW. et al. CpG methylation is maintained in human cancer cells lacking DNMT1. Nature. 2000;404:1003-7
15. Tahara T, Shibata T, Nakamura M. et al. Association between IL-17A, -17F and MIF polymorphisms predispose to CpG island hyper-methylation in gastric cancer. Int J Mol Med. 2010;25:471-7
16. Soes S, Daugaard IL, Sorensen BS. et al. Hypomethylation and increased expression of the putative oncogene ELMO3 are associated with lung cancer development and metastases formation. Oncoscience. 2014;1:367-74
17. Bae JH, Kim JG, Heo K. et al. Identification of radiation-induced aberrant hypomethylation in colon cancer. BMC Genomics. 2015;16:56
18. Kumar A, Rai PS, Upadhya R. et al. gamma-radiation induces cellular sensitivity and aberrant methylation in human tumor cell lines. Int J Radiat Biol. 2011;87:1086-96
19. Ha BG, Park JE, Cho HJ. et al. Inhibitory effects of proton beam irradiation on integrin expression and signaling pathway in human colon carcinoma HT29 cells. Int J Oncol. 2015;46:2621-8
20. Park SB, Kim B, Bae H. et al. Differential Epigenetic Effects of Atmospheric Cold Plasma on MCF-7 and MDA-MB-231 Breast Cancer Cells. PLoS One. 2015;10:e0129931
21. Kim B, Kang S, Jeong G. et al. Identification and comparison of aberrant key regulatory networks in breast, colon, liver, lung, and stomach cancers through methylome database analysis. PLoS One. 2014;9:e97818
22. Woloszynska-Read A, Mhawech-Fauceglia P, Yu J. et al. Intertumor and intratumor NY-ESO-1 expression heterogeneity is associated with promoter-specific and global DNA methylation status in ovarian cancer. Clin Cancer Res. 2008;14:3283-90
23. Ringner M, Fredlund E, Hakkinen J. et al. GOBO: gene expression-based outcome for breast cancer online. PLoS One. 2011;6:e17911
24. Chalitchagorn K, Shuangshoti S, Hourpai N. et al. Distinctive pattern of LINE-1 methylation level in normal tissues and the association with carcinogenesis. Oncogene. 2004;23:8841-6
25. Kochanek S, Renz D, Doerfler W. DNA methylation in the Alu sequences of diploid and haploid primary human cells. EMBO J. 1993;12:1141-51
26. Jeong EG, Lee SH, Lee HW. et al. Immunohistochemical and mutational analysis of FLASH in gastric carcinomas. APMIS. 2007;115:900-5
27. Sawada J, Urakami T, Li F. et al. Small GTPase R-Ras regulates integrity and functionality of tumor blood vessels. Cancer Cell. 2012;22:235-49
28. Foussard H, Ferrer P, Valenti P. et al. LRCH proteins: a novel family of cytoskeletal regulators. PLoS One. 2010;5:e12257
29. Li L, Zhang A, Cao X. et al. General transcription factor IIb overexpression and a potential link to proliferation in human hepatocellular carcinoma. Pathol Oncol Res. 2013;19:195-203
30. Xiang S, Liu Z, Zhang B. et al. Methylation status of individual CpG sites within Alu elements in the human genome and Alu hypomethylation in gastric carcinomas. BMC Cancer. 2010;10:44
31. Barchitta M, Quattrocchi A, Maugeri A. et al. LINE-1 hypomethylation in blood and tissue samples as an epigenetic marker for cancer risk: a systematic review and meta-analysis. PLoS One. 2014;9:e109478
32. Esposito G, Belli M, Campa A. et al. DNA fragments induction in human fibroblasts by radiations of different qualities. Radiat Prot Dosimetry. 2006;122:166-8
33. Baran I, Ganea C, Privitera S. et al. Detailed analysis of apoptosis and delayed luminescence of human leukemia Jurkat T cells after proton irradiation and treatments with oxidant agents and flavonoids. Oxid Med Cell Longev. 2012;2012:498914
34. Wang J, Mao X, Zhang Y. et al. [Genome-wide screening of CpG island methylation changes induced by low-dose X-ray radiation in mice]. Nan Fang Yi Ke Da Xue Xue Bao. 2014;34:228-31
35. Lima F, Ding D, Goetz W. et al. High LET (56)Fe ion irradiation induces tissue-specific changes in DNA methylation in the mouse. Environ Mol Mutagen. 2014;55:266-77
36. Halvorsen AR, Helland A, Fleischer T. et al. Differential DNA methylation analysis of breast cancer reveals the impact of immune signaling in radiation therapy. Int J Cancer. 2014;135:2085-95
37. Hou J, Wang F, Kong P. et al. Gene Profiling Characteristics of Radioadaptive Response in AG01522 Normal Human Fibroblasts. PLoS One. 2015;10:e0123316
Author contact
![]() Corresponding author: Sun Jung Kim, Ph.D. Department of Life Science, Dongguk University, Goyang 10326, Korea. Phone: 82-31-961-5129, E-mail: sunjungkedu
Corresponding author: Sun Jung Kim, Ph.D. Department of Life Science, Dongguk University, Goyang 10326, Korea. Phone: 82-31-961-5129, E-mail: sunjungkedu
Citation styles
APA
Kim, B., Bae, H., Lee, H., Lee, S., Park, J.C., Kim, K.R., Kim, S.J. (2016). Proton Beams Inhibit Proliferation of Breast Cancer Cells by Altering DNA Methylation Status. Journal of Cancer, 7(3), 344-352. https://doi.org/10.7150/jca.13396.
ACS
Kim, B.; Bae, H.; Lee, H.; Lee, S.; Park, J.C.; Kim, K.R.; Kim, S.J. Proton Beams Inhibit Proliferation of Breast Cancer Cells by Altering DNA Methylation Status. J. Cancer 2016, 7 (3), 344-352. DOI: 10.7150/jca.13396.
NLM
Kim B, Bae H, Lee H, Lee S, Park JC, Kim KR, Kim SJ. Proton Beams Inhibit Proliferation of Breast Cancer Cells by Altering DNA Methylation Status. J Cancer 2016; 7(3):344-352. doi:10.7150/jca.13396. https://www.jcancer.org/v07p0344.htm
CSE
Kim B, Bae H, Lee H, Lee S, Park JC, Kim KR, Kim SJ. 2016. Proton Beams Inhibit Proliferation of Breast Cancer Cells by Altering DNA Methylation Status. J Cancer. 7(3):344-352.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.