Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2016; 7(3):251-261. doi:10.7150/jca.13689 This issue Cite
Research Paper
Molecular Mechanism of Cinnamomum verum Component Cuminaldehyde Inhibits Cell Growth and Induces Cell Death in Human Lung Squamous Cell Carcinoma NCI-H520 Cells In Vitro and In Vivo
Shu-mei Yang1,2, Kuen-daw Tsai1,2,3, Ho-Yiu Wong1, Yi-Heng Liu1, Ta-Wei Chen1, Jonathan Cherng4, Kwang-Ching Hsu5, Yao-Uh Ang5, Jaw-Ming Cherng 5 ![]()
1. Department of Internal Medicine, China Medical University Beigang Hospital, Yunlin, Taiwan ROC
2. School of Chinese Medicine, College of Chinese Medicine, China Medical University, Taichung, Taiwan ROC
3. Institute of Molecular Biology, National Chung Cheng University, Chiayi, Taiwan ROC
4. Faculty of Medicine, Medical University of Lublin, Lublin, Poland
5. Department of Internal Medicine, Saint Mary's Hospital Luodong, Yilan, Taiwan ROC
Received 2015-8-29; Accepted 2015-11-13; Published 2016-1-5
Citation:
Yang Sm, Tsai Kd, Wong HY, Liu YH, Chen TW, Cherng J, Hsu KC, Ang YU, Cherng JM. Molecular Mechanism of Cinnamomum verum Component Cuminaldehyde Inhibits Cell Growth and Induces Cell Death in Human Lung Squamous Cell Carcinoma NCI-H520 Cells In Vitro and In Vivo. J Cancer 2016; 7(3):251-261. doi:10.7150/jca.13689. https://www.jcancer.org/v07p0251.htm
Other stylesAbstract
Cinnamomum verum is used to make the spice cinnamon and has been used as a traditional Chinese herbal medicine. We evaluated the effects and the molecular mechanisms of cuminaldehyde (CuA), a constituent of the bark of Cinnamomum verum, on human lung squamous cell carcinoma NCI-H520 cells. Specifically, cell viability was evaluated by colorimetric assay; cytotoxicity by LDH release; apoptosis was determined by Western blotting, and morphological analysis with, acridine orange and neutral red stainings and comet assay; topoisomerase I activity was assessed using assay based upon DNA relaxation and topoisomerase II by DNA relaxation plus decatentation of kinetoplast DNA; lysosomal vacuolation and volume of acidic compartments (VAC) were evaluated with neutral red staining. The results show that CuA suppressed proliferation and induced apoptosis as indicated by an up-regulation of pro-apoptotic bax and bak genes and a down-regulation of anti-apoptotic bcl-2 and bcl-XL genes, mitochondrial membrane potential loss, cytochrome c release, activation of caspase 3 and 9, and morphological characteristics of apoptosis, including blebbing of the plasma membrane, nuclear condensation, fragmentation, apoptotic body formation, and comet with elevated tail intensity and moment. In addition, CuA also induced lysosomal vacuolation with increased VAC, cytotoxicity, as well as suppressions of both topoisomerase I and II activities in a dose-dependent manner. Further study revealed the growth-inhibitory effect of CuA was also evident in a nude mice model. Taken together, the data suggest that the growth-inhibitory effect of CuA against NCI-H520 cells is accompanied by downregulations of proliferative control involving apoptosis and both topoisomerase I and II activities, and upregulation of lysosomal with increased VAC and cytotoxicity. Similar effects were found in other cell lines, including human lung adenocarcinoma A549 cells and colorectal adenocarcinoma COLO 205 (results not shown). Our data suggest that CuA could be a potential agent for anticancer therapy.
Keywords: Cuminaldehyde, anticancer, NCI-H520 cells, topoisomerase I, topoisomerase II, lysosomal vacuolation, cytotoxicity, xenograft
Introduction
According to Toh et al., lung squamous cell carcinoma characterizes 8.8% of cases in lung cancer in never-smokers [1]. However, the malignancy is not sensitive to traditional chemotherapeutic agents and there is a need for better treatment of the disease.
The genus Cinnamomum belongs to the Lauraceae family and comprises over 250 aromatic evergreen trees distributed mostly in Asia. Cinnamomum verum is a small evergreen tree in the genus and native to Sri Lanka. The bark of this plant is used to make the spice cinnamon and has long been used as a traditional Chinese herbal medicine for various conditions, such as improvement of the complexion to make it more youthful, alleviation of fever, inflammation, cough, induction of perspiration, and treatment of circulatory disorders [2, 3]. In our ongoing study to identify anti-cancer agents from natural resources, cuminaldehyde (CuA), a constituent of the bark of the plant, was discovered to have a growth-inhibitory effect in human lung squamous cell carcinoma NCI-H520 cells.
Cancer is a hyperproliferative disorder. Numerous genetic and epigenetic changes being needed to drive normal cells toward neoplastic transformation. These alterations control various signaling pathways that cooperate to endow cancer cells with a wide range of biological capabilities necessary for growing, disseminating and finally killing their host [4]. Although anticancer drugs may act differently, apoptosis is the most common and preferred mechanism through which many anticancer agents kill and eradicate cancer cells [5].
Topoisomerases are enzymes that regulate the topological states of DNA and play important roles in maintaining genomic integrity [6]. These enzymes relax supercoiled DNA by transient protein-linked cleavages of either one (topoisomerase I) or both (topoisomerase II) of the sugar-phosphate backbones of double-stranded DNA strands [7]. In addition to apoptosis, topoisomerase is another major target of anticancer agents [8-11].
This diversity of mechanisms of carcinogenesis suggests that there are probably multiple processes that could be effective targets for the prevention of cancer. In an attempt to understand the effects and underlying mechanisms of CuA in NCI-H520 cells, we performed a series of experiments to answer the following questions: a) what are the effects of CuA on the growth in NCI-H520 cells? b) what are the effects of CuA on topoisomerase I and II activities? c) how are these activities affected? Our results indicate that CuA inhibited the growth in NCI-H520. In addition, CuA inhibited both topoisomerase I and II activities, and induced lysosomal vacuolation with increased VAC. Finally, CuA induced apoptosis, resulting in the suppression of NCI-H520 cell growth.
Materials and methods
Reagents
RPMI 1640 and fetal calf serum were purchased from GIBCO BRL (Gaithersburg, Md, USA). Cuminaldehyde (CuA) and dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich, Inc. (St. Louis, Mo, USA). Antibodies to Bax, Bak, Bcl-2, Bcl-XL, and β-actin were purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA).
Cell culture
Human lung squamous cell carcinoma NCI-H520 cells (American Type Culture Collection HTB-182, American Type Culture Collection, Manassas, VA, USA) were cultured in RPMI 1640 medium, supplemented with 10% (v/v) FBS, 10 U/mL penicillin, 10 μg/mL streptomycin, and 0.25 μg/mL amphotericin B at 37°C with 5% CO2.
XTT assay for cell viability
A number of methods have been developed for assaying cell proliferation, such as DNA synthesis and metabolic activity. Although measurement of DNA synthesized in the presence of a label is one of the most reliable and accurate assay types, the obvious downsides to this method are the hazards and hassle of using of radioactive materials. Another method measuring proliferation is the metabolic activity of cells. The assay is based on the cleavage of the tetrazolium salt (such as XTT and MTT) to formazan by mitochondrial dehydrogenases in metabolically active cells that subsequently changes the color of the media. The absorption of the media-containing dye solution can be read using a spectrophotometer.
Cells were seeded in 96-well culture plates (1 x 104 cells/well). After being incubated for 24 h, the cells were treated with different concentrations of CuA for 12, 24, and 48 h. The cell viability was determined by the Cell Proliferation Kit II (XTT) (Roche Applied Science, Mannheim, Germany) following the manufacturer's protocol. The absorbance was measured using the Tecan infinite M200 spectrophotometer (Tecan, Männedorf, Switzerland) at 492 nm with a reference wavelength at 650 nm.
LDH-Cytotoxicity Assay
The first morphological evidence of apoptosis starts with retraction of the cell, loss of adherence, then convolution of the cytoplasm and plasma membrane, blebbing. Finally, the cell is fragmented into apoptotic bodies, leading to the release of the cell content into the surrounding media [12]. One method of assaying loss of membrane integrity is measuring the release of cytosolic enzyme lactate dehydrogenase (LDH) into the bathing medium [13]. This assay was originally used to quantify cell death occurring via necrosis [14], but has since been shown to accurately measure apoptosis [15-17].
Cells were seeded in 96-well culture plates (1 x 104 cells/well). After being incubated for 24 h, cells were treated with different concentrations of CuA for 48 h. LDH activity was determined using the LDH-Cytotoxicity Colorimetric Assay Kit from BioVision (Milpitas, CA, USA) following the manufacturer's protocol. The absorbance of the samples at 490 nm was measured using the Tecan infinite M200 spectrophotometer (Tecan, Männedorf, Switzerland). Results are represented as the percentage of change in activity compared with the untreated control.
Nuclear fragmentation assay
Apoptosis is the most common and preferred mechanism through which many anti-cancer agents kill tumor cells [5]. In addition, apoptosis also is the major mechanism of cancer cell death induced by selected polyphenols [18-21]. At the nuclear level, apoptosis is characterized by the activation of endogenous endonucleases with subsequent cleavage of chromatin DNA into fragments. Nuclear fragmentation assay was performed to investigate the possible mechanism of growth-inhibitory effect of CuA against NCI-H520 cells.
Acridine orange (AO) is a nucleic acid-selective metachromatic dye useful for cell cycle determination. When AO intercalates into dsDNA, the dye fluoresces green. On the other hand, it fluoresces orange when interacting with ssDNA or RNA. Apoptotic cells (with a larger fraction of DNA in the denatured form) display an orange fluorescence and a reduced green emission when compared to non-apoptotic interphase cells. In addition, when AO enter acidic compartments such as lysosomes, the dye becomes protonated and sequestered. In these low pH conditions, the dye will emit an orange light when excited by blue light [22]. Nuclear fragmentation assay is based on the characteristics of AO and observed under a fluorescence microscope. Briefly, the cells were treated with different concentrations of CuA for 48 h and stained with 5 μg/mL AO at room temperature. The cells were then observed under a fluorescence microscope [23].
Comet assay
Comet assay, a gel electrophoresis-based method that can be used to evaluate DNA damage in individual eukaryotic cells, is sensitive, versatile, and relatively simple to perform. The limit of sensitivity is approximately 50 strand breaks per diploid mammalian cell. The assay was performed following the procedure of Olive et al. [24].
Assay for volume of acidic compartment
Increase in volume of acidic compartment (VAC) is a common phenomenon of cells that undergo either apoptotic or necrotic cell death. Elevated VAC could also be a hallmark of dying cells [25]. To understand the pathogenetic effects of CuA in NCI-H520 cells, the VAC assay for lysosomes was carried out as described previously [23].
Immunoprecipitation assay
Apoptosis is regulated by two major signaling pathways: the extrinsic and the intrinsic pathways. One pathway is initiated by death receptors such as Fas, and the other is activated by molecules (such as cytochrome c) released from mitochondria (which are regulated by various proteins, such as those encoded by the Bcl-2 family genes). Both pathways eventually converge, leading to activation of the central effectors of apoptosis: a group of cysteine proteases, called the caspases, which cleave a number of different substrates [26]. Furthermore, tumors arise more frequently through the intrinsic pathway than the extrinsic pathway because of sensitivity [27]. To investigate the mechanisms involved in CuA-induced apoptosis, we initially analyzed the changes in the levels of the pro-apoptotic proteins Bax and Bak, and the anti-apoptotic proteins Bcl-2 and Bcl-XL.
Cells were treated with different concentrations of CuA for 48 h. Mitochondrial and cytoplasmic fractions were separated using the Cytochrome c Releasing Apoptosis Assay Kit (BioVision, Milpitas, CA, USA) following the manufacturer's protocol. For Western blotting, the cells were lysed on ice for 40 min in a solution containing 50 mM Tris, 0.1% SDS, 1% Triton X-100, 150 mM NaCl, 2 mM EGTA, 2 mM Na3VO4, 10 mM NaF, 12 mM β-glycerolphosphate, 16 μg/mL benzamidine hydrochloride, 10 μg/mL phenanthroline, 10 μg/mL aprotinin, 10 μg/mL leupeptin, 10 μg/mL pepstatin, and 1 mM phenylmethylsulfonyl fluoride [28]. The cell lysate was centrifuged at 12,500 ×g for 20 min. Fifty μg of total protein, as determined by BioRad Protein Assay (BioRad, Hercules, CA, USA), were resolved on 10% polyacrylamide gel electrophoresis and then transferred to Immobilon P membrane using a semidry transfer system (BioRad, Hercules, CA, USA). The membrane was blocked with 5% nonfat dry milk in TBS [20 mM Tris-HCl (pH 7.4) and 250 mM NaCl] for 1 h at room temperature and then incubated with the desired primary antibody in TBS containing 3% nonfat milk at 4°C overnight. The membrane was washed three times with TBS and incubated with appropriate peroxidase-conjugated secondary antibody, and the immunoreactive proteins were detected using the Amersham ECL Western Blotting Detection Kit (GE Healthcare, Buckinghamshire, UK) following the manufacturer's protocol.
Assay for caspase activity
Mitochondrial proteins known as SMACs (second mitochondria-derived activator of caspases) are released into cytosol following the increased permeability of the mitochondria membranes. SMAC binds to IAPs (inhibitor of apoptosis proteins), and thereby deactivates them, which in turn abrogates the IAPs from the inhibiting activity of a group of cysteine proteases called the caspases [26, 29] that carry out the degradation of the cell.
To further investigate the mechanisms involved in CuA-induced apoptosis, we analyzed the changes in activities of key caspases involved in apoptosis. The assay used here is based on the detection of the chromophore AFC after cleavage from the labeled substrate DEVD- and LEHD-AFC by caspase-3 and caspase-9, respectively. Free AFC emits a yellow-green fluorescence (λmax = 505 nm). NCI-H520 cells were treated with different concentrations of CuA for 24 h and caspase-3 and 9 activities were detected using the Fluorometric Assay Kit from BioVision (Milpitas, CA, USA) following the manufacturer's protocol. The AFC light emission was quantified using the Tecan infinite M200 spectrophotometer (Tecan, Männedorf, Switzerland). Results are represented as the percentage of change in activity compared with the untreated control.
Mitochondrial membrane potential assay
Mitochondrial dysfunction is involved in the induction of apoptosis and is central to the apoptotic pathway. Indeed, the opening of the mitochondrial permeability transition pore induces depolarization of the transmembrane potential (ΔΨm) and release of apoptogenic factors [30]. To further investigate the role of mitochondria in CuA-induced apoptosis, we analyzed the changes in ΔΨm.
Mitochondrial membrane potential was determined using the mitochondrial-specific fluorescent probe JC-1 (Invitrogen, Carlsbad, CA, USA) based on the method of Reers et al. [31]. JC-1 exists as monomer when membrane potentials (ΔΨm) are lower than 120 mV and it fluoresces green (540 nm) following excitation by blue light (490 nm), and as dimer (J-aggregate) at membrane potentials greater than 180 mV and it fluoresces red (590 nm) following excitation by green light (540 nm). NCI-H520 cells were treated with CuA under the indicated conditions, harvested, and stained with 25 μM JC-1 at 37°C for 30 min, and analyzed using a fluorescence microscope and spectrophotometer. Changes in the ratio of red (590-nm emission) to green (540-nm emission) fluorescence are indicative of the mitochondrial membrane potential changes [31].
Assays for topoisomerase I and II activities
Topoisomerases are enzymes that regulate the topological states of DNA and play important roles in maintaining genomic integrity [6]. In addition to apoptosis, topoisomerase is another major target of anticancer agents [8-11]. Assays for topoisomerase I and II activities were performed using the method of Har-Vardi et al. [32].
In vivo tumor xenograft study
Male nude mice (BALB/c Nude; 6 weeks old) were purchased from the National Science Council Animal Center (Taipei, Taiwan, ROC) and maintained in pathogen-free conditions in accordance with relevant guidelines and regulations for the care and use of laboratory animals of China Medical University. NCI-H520 cells (5 x 106 cells in 200 μL) were injected subcutaneously (SC) into the flanks of the nude mice. Tumors were allowed to develop until they reached about 75 mm3, at which time treatment was started. Twelve mice were randomly separated into two groups. The mice in the CuA-treated group were injected intratumorally with CuA (10 mg/kg body weight) in a 200 μL volume daily. The control group was treated with an equal volume of vehicle. After transplantation, tumor size was monitored at weekly intervals using calipers, and tumor volume was estimated by the hemiellipsoid model formula: tumor volume = 1/2(4π/3) × (l/2) × (w/2) × h, where l = length, w = width, and h = height.
Specimens were analyzed with fluorescent TUNEL assay using the Quick Apoptotic DNA Ladder Detection Kit (Chemicon, Temecuba, CA, USA) following the manufacturer's protocol.
Statistical analysis
Data are presented as means ± standard error. The evaluation of statistical significance was determined by one-way analysis of variance (ANOVA) followed by the Bonferroni t-test for multiple comparisons. A P value less than 0.05 was considered statistically significant.
Results
Effects of CuA on cell morphological changes
When NCI-H520 cells were exposed to 20 μM of CuA for 48 h, plasma membrane blebbing was observed; cell shrinkage and cell detachment also occurred (Fig. 1B).
CuA inhibits NCI-H520 cell proliferation
We investigated the potential cell proliferation inhibitory activity of CuA in NCI-H520 cells using the XTT. As shown in Fig. 1C, CuA inhibited cell proliferation in NCI-H520 cells in a dose- and time-dependent manner. Maximal proliferation inhibition was observed at 48 h with an IC50 value of 33.29 μM.
Effect of CuA on the release of LDH in NCI-H520 cells
CuA was cytotoxic as assessed biochemically by an increase in LDH content in the supernatant in a dose-dependent manner.
Nuclear fragmentation assay
Acridine orange (AO) is a nucleic acid selective metachromatic stain useful for cell cycle determination, measuring apoptosis, detecting intracellular pH gradients and the measurement of proton-pump activity [33]. The dye differentially stains single-stranded nucleic acids orange and double-stranded nucleic acids green. Further, in living cells it serves as a pH indicator, trapped in acidic compartments, such as lysosomes, which then fluoresces a brilliant orange [34]. When NCI-H520 cells were treated with 20 μM of CuA for 48 h, the result of AO staining demonstrated that NCI-H520 cells died by apoptosis with blebbing, nuclear condensation, and fragmentation. Orange-staining lysosomal vacuoles also appeared, but no significant nuclear fragmentation in the control group was observed.
DNA strand breakage was investigated by the Single Cell Gel Electrophoresis assay (SCGE, also known as the comet assay) at 48 h following treatment with different concentrations of CuA. Fig. 2C and D demonstrate that CuA treatment resulted in elevated tail intensity and moment in NCI-H520 cells.
CuA increases volume of acidic compartments in NCI-H520 cells
Neutral Red has been used to stain lysosomes and quantify the volume of acidic compartments (VAC) in cells [23, 35, 36]. Fig. 3A and B demonstrate that the CuA treatment resulted in acidic vacuoles in NCI-H520 cells, as evidenced by positive neutral red staining. As shown in Fig. 3C, the VAC of CuA-treated NCI-H520 cells increased in a dose-dependent manner.
Given that blebbing of the plasma membrane, nuclear condensation, fragmentation, and apoptotic body formation are characteristic morphologic features of apoptosis [37], the morphological changes observed in our study suggest CuA did induce apoptosis in NCI-H520 cells (Fig. 1 and 2).
Fig 1
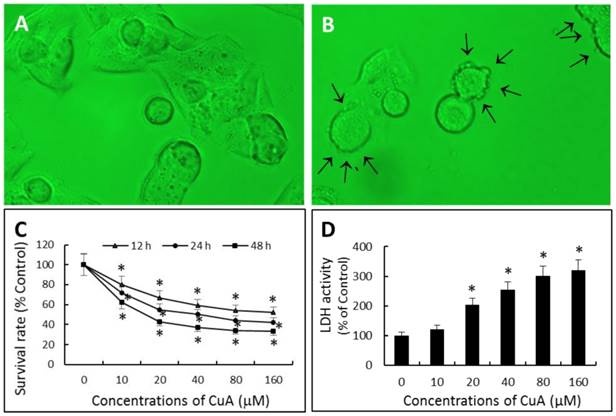
Effects of CuA on cell morphology and growth in NCI-H520 cells. (A and B) Effect of CuA on cell morphology. Cells were treated with 0 (A) or 20 μM (B) of CuA for 48 h, respectively. When NCI-H520 cells were exposed to 20 μM of CuA, plasma membrane blebbing (arrows), cell shrinkage and cell detachment were observed. (C) Effect of CuA on proliferation. NCI-H520 cells were treated with CuA at the indicated conditions. Cell proliferation inhibitory activity was evaluated by the XTT assay. (D) Effect of CuA on the release of LDH in NCI-H520 cells. The cell culture medium was collected after 48 h of treatment with the indicated concentrations of CuA. Light absorptions were analyzed at 340 nm using Tecan infinite M200 spectrophotometer. Data are expressed as means ± standard error of mean, n = 3. *Indicates a significant difference (p < 0.05) from control. CuA, cuminaldehyde.
Fig 2
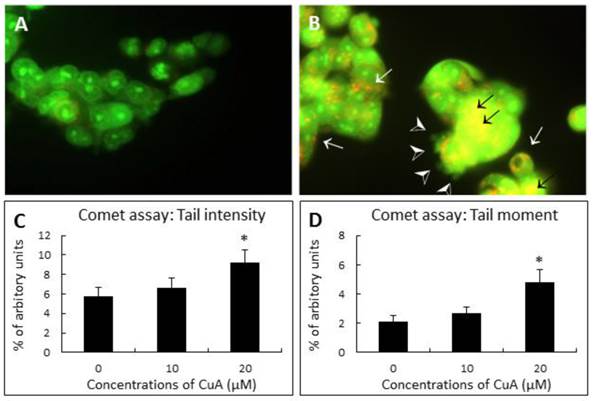
CuA-induced nuclear fragmentation in NCI-H520 cells. (A and B) Acridine orange staining. Cells were treated with CuA 0 and 20 μM, respectively, for 48 h, then stained with acridine orange. Orange vacuoles in cells showed that they were acidic. (A) Representative control group with intact green nucleus suggesting a good cell viability; (B) Representative test group treated with 20 μM CuA in which nuclear fragmentation (back arrows), lysosomal vacuolation (white arrows), and blebbing (arrow heads) were observed. (C and D) Comet assay. Effect of CuA on tail intensity (C) and moment (D) in comet assay. NCI-H520 cells were treated with CuA at the indicated conditions for 48 h. Cells were embedded in agarose and DNA was then unwound in an alkaline solution and subjected to electrophoresis. Cells were then stained with DAPI and analyzed under fluorescence microscopy using automated analytical software (Comet Assay 2.0; Perceptive Instrument, UK). Data are expressed as means ± standard error of mean, n = 350. *Indicates a significant difference (p < 0.05) from control. CuA, cuminaldehyde.
Fig 3
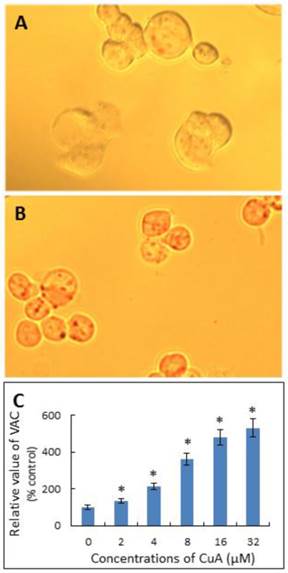
CuA increased VAC in NCI-H520 cells. NCI-H520 cells were treated with 0, or 20 μM of CuA for 48 h, then stained with neutral red. (A) Control group. There were no obvious vacuoles in cells. (B) NCI-H520 cells treated with 20 μM of CuA for 48 h. The acidic red vacuoles in cells were observed. (C) CuA increased VAC in a dose-dependent manner. NCI-H520 cells were incubated with the indicated concentration of CuA for 48 h and data were analyzed with a spectrophotometer. Data are expressed as means ± standard error of mean, n = 3. *Indicates a significant difference (p < 0.05) from control. CuA, cuminaldehyde.
CuA suppresses growth of NCI-H520 cells by execution of apoptosis through activation of the mitochondrial pathway
To investigate the mechanisms involved in CuA-induced apoptosis, we initially analyzed the changes in the levels of pro-apoptotic proteins Bax and Bak, and anti-apoptotic proteins Bcl-2 and Bcl-XL. Immunoblotting analysis revealed significant change in the expressions of both pro-apoptotic and anti-apoptotic proteins. CuA-treated NCI-H520 cells expressed increased Bax and Bak protein levels, but decreased Bcl-2 and Bcl-XL protein expressions (Fig. 4A).
Then, we further investigated the role of mitochondria in the CuA-induced apoptosis in NCI-H520 cells. Since early apoptotic cell death often involves release of cytochrome c from mitochondria into cytosol, the cytosolic fraction of cytochrome c protein was determined with Western blotting. The results show that the cytosolic fraction from the untreated NCI-H520 cells contained no observable cytochrome c, whereas it did become detectable after 48 h of treatment with CuA in a dose-dependent manner starting from a concentration of 10 μM in NCI-H520 cells. Furthermore, expression of mitochondrial cytochrome c changed in the opposite direction (Fig. 4B).
We then investigated the mitochondrial dysfunction by measuring mitochondrial membrane potential ΔΨm in CuA-treated NCI-H520 cells using the mitochondria-specific dye JC-1, both microscopically and spectrophotometrically. As shown in Fig. 4C, CuA induced the loss of mitochondrial membrane potential, as indicated by decreased ΔΨm, in a dose-dependent manner.
Caspases, or cysteine-dependent aspartate-directed proteases, are a family of cysteine proteases that play important roles in apoptosis. As shown in Fig. 4D, the activities of both caspase-3 and -9 increased in a dose-dependent manner in CuA-treated NCI-H520 cells. This is consistent with the mitochondrial depolarization and release of cytochrome c from mitochondria into the cytosol.
CuA inhibits topoisomerase I activity in NCI-H520 cells
Inhibition of topoisomerase I activity in NCI-H520 cells by CuA was achieved in the presence of increasing concentration of CuA (Fig. 5A, lanes 3-5) or camptothecin (CPT, lane 6), a known specific inhibitor of topoisomerase I [38-40]. Fig. 5A shows that the conversion of the supercoiled plasmid pUC 19 to the relaxed form, decreased in a dose-dependent manner in the presence of CuA or CPT (compare lanes 3-6 with lane 2). These results show that the DNA relaxation activity of NCI-H520 cell nuclear proteins was inhibited by CuA.
Fig 4
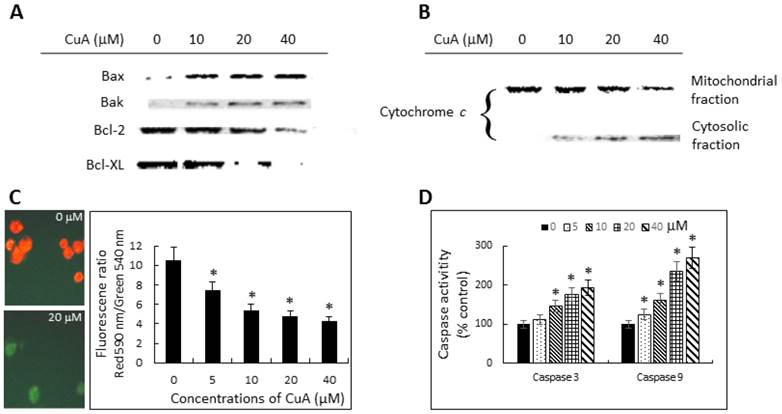
Cuminaldehyde induced apoptosis through the mitochondrial pathway in NCI-H520 cells. (A and B) Cells were treated with the indicated concentrations of cuminaldehyde for 48 h. Levels of Bax, Bak, Bcl-2, Bcl-XL, and cytochrome c were evaluated by Western blotting using specific antibodies; the cytoplasm and mitochondria fractions were extracted from cell pellets. (A) Expressions of Bcl-2 family proteins. (B) Release of cytochrome c from mitochondria into cytosol. (C) Cuminaldehyde induced mitochondrial depolarization. (C, left) Cells were treated with the indicated concentrations of cuminaldehyde for 24 h and ΔΨm was analyzed by JC-1 using fluorescence microscopy: (Left, upper part) Control cells with intact mitochondria fluorescencing red; (Left, lower part) Most cuminaldehyde-treated cells fluorescencing green, suggesting the loss of ΔΨm; (Right part) Cells were treated with the indicated concentrations of cuminaldehyde for 24 h and ΔΨm was analyzed by JC-1 using spectrophotometer. (D) Activations of caspase-3 and -9. Cells were treated with the indicated concentrations of cuminaldehyde for 24 h and activities of caspases-3 and -9 were determined fluorometrically using fluorescence-labeled synthetic substrates. Data are expressed as means ± standard error of mean, n = 3. *Indicates a significant difference (p < 0.05) from control. CuA, cuminaldehyde.
Fig 5
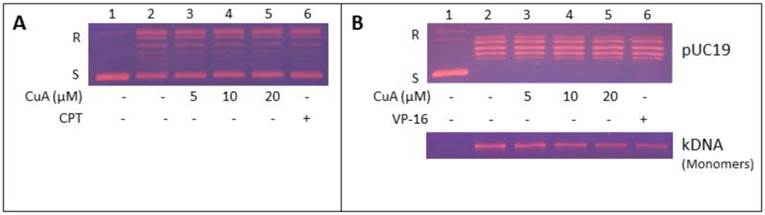
Inhibition of NCI-H520 topoisomerase I and II activities by CuA. (A) CuA inhibited topoisomerase I activity in NCI-H520 cells. Nuclear proteins from NCI-H520 cells were added to a specific topoisomerase I reaction mixture in the presence of the indicated concentrations of CuA (lanes 3-5), or 60 μM CPT (lane 6), or the vehicle (1% DMSO, lane 2). Lane 1: pUC19 DNA only. (B) CuA inhibits topoisomerase II activity in NCI-H520 cells. DNA relaxation assay (upper part) and decatenation assay (lower part): Nuclear proteins from NCI-H520 cells were added to a specific topoisomerase II reaction mixture in the presence of the indicated concentrations of CuA (lanes 3-5) or 60 μM VP-16, a specific topoisomerase II inhibitor (lane 6), or the vehicle (1% DMSO, lane 2). (Lane 1) Supercoiled pUC19 DNA (upper part) or kDNA (lower part) only. kDNA is a large network of plasmids, and when it is analyzed by gel electrophoresis, it penetrates only slightly into agarose gel (result not shown). Upon decatenation by topoisomerase II, mini circles monomers of kDNA were formed (lower part, lanes 2-6). CPT, camptothecin; CuA, cuminaldehyde; S and R, supercoiled and the relaxed forms of the pUC 19 plasmid DNA, respectively; VP-16, etoposide.
CuA inhibits topoisomerase II activity in NCI-H520 cells
Inhibition of topoisomerase II activity in NCI-H520 cells by CuA was examined in the presence of increasing concentration of CuA (Fig. 5B, lanes 3-5) or 60 μM etoposide (VP-16, lane 6), a known inhibitor of topoisomerase II [39]. Fig. 5B, upper part, shows that the conversion of the supercoiled plasmid pUC 19 to the relaxed form, decreased in a dose-dependent manner in the presence of CuA or VP-16 (compare lanes 3-6 with lane 2). These findings show that the DNA relaxation activity of NCI-H520 cell nuclear proteins was inhibited by CuA. Moreover, the effect of CuA on topoisomerase II in NCI-H520 cells was further evaluated with the decatenation assay. Decatenation activity involves the releasing of monomers (minicircle DNA) from the kinetoplast DNA (kDNA, a large network of plasmid). The nuclear proteins extracted from the NCI-H520 cells contained topoisomerase II, which converted kinetoplast DNA to monomer DNA molecules (Fig. 5B, lower part, compare lane 2 with lane 1). The conversion of kinetoplast DNA to monomers decreased in a dose-dependent manner in the presence of CuA (compare lanes 3-5 with lane 2) or VP-16 (compare lane 6 with lane 2). These results indicate that the decatenation activity of NCI-H520 cell nuclear proteins was inhibited by CuA.
CuA inhibits growth of NCI-H520 xenograft in nude mice
To determine whether CuA suppresses growth of the NCI-H520 xenograft, equal numbers of NCI-H520 cells were injected subcutaneously into both flanks of the nude mice. Tumor growth suppression was obvious in mice treated with 10 mg/kg/day of CuA, where ~45% reduction in tumor size was found, in contrast to mice treated with the vehicle (Fig. 6B). In addition, no significant effect was found when the mice were treated with 4 mg/kg/day of CuA (results not shown). None of the CuA treatments caused any significant decrease in diet consumption or body weight change (data not shown) compared with control mice. To gain insight into the mechanism of the antitumor effect of CuA in vivo, we harvested the NCI-H520 xenograft from vehicle- and CuA-treated mice and assessed cell death using the TUNEL analysis. As shown in Fig. 6A in the right parts, compared with tumors of vehicle-treated mice (upper part), elevated TUNEL-positive cells that suggest apoptosis, were found in the tumors of the CuA-treated mice (lower part).
Discussion
Epidemiological and experimental studies have consistently shown that there is a correlation between regular consumption of fruits and vegetables and prevention of developing lifestyle disorders, such as cardiovascular disorders and cancer [41, 42]. Phytochemicals, such as polyphenols and flavonoids which are abundant in fruits and vegetables, seem to possess many of the desirable qualities for preventing cancer and could have great potential as chemopreventive and antiproliferative agents [43-48]. Cinnamomum verum has been traditionally used for treating dyspepsia, blood circulation and inflammatory disorders, including gastritis [49, 50]. CuA, a constituent of the bark of the plant, could be such a natural agent. However, till now very few studies on CuA have been published, and to the best of our knowledge, there has been no report focusing on its effects on topoisomerase I and II activities. The current study thus aimed at investigating the antiproliferative activity of CuA and elucidating its underlying mechanisms.
Fig 6
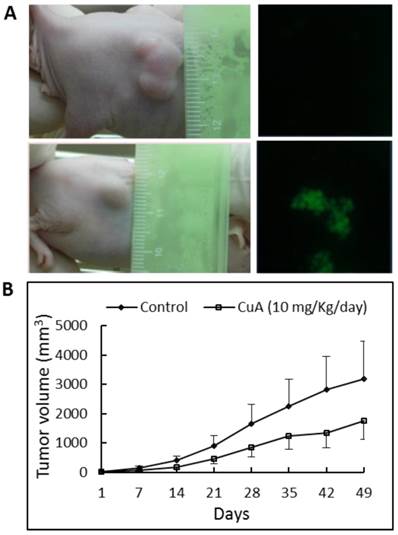
CuA suppressed growth and induced apoptosis in NCI-H520 xenograft. Animals bearing pre-established tumors (n = 6 per group) were injected intratumorally with CuA (10 mg/kg/day) or vehicle. During the 50-day CuA treatment, tumor volumes were monitored using calipers and apoptosis was assessed by TUNEL assay. (A) Left: Representative tumor-possessing nude mice from the control (upper part) and CuA-treated (lower part) groups. (A) Right: CuA induced apoptosis in NCI-H520 xenograft by TUNEL assay. Representative TUNEL assays of tumors from the control (upper part) and CuA-treated (lower part) groups. (B) Means of tumor volume measured at the indicated number of days after implantation. CuA, cuminaldehyde.
In this study, we first examined the effect of CuA on the growth of human lung squamous cell carcinoma NCI-H520 cells. We found that CuA suppressed the proliferation of NCI-H520 cells in a dose- and time-dependent manner. Although cells can die by non-apoptotic mechanisms, apoptosis is the most common and preferred mechanism through which many chemotherapeutic agents kill and eradicate tumor cells [5]. In addition, apoptosis has been reported as the major mechanism of cancer cell death induced by selected polyphenols [18-21]. Our results suggest that CuA effectively induced apoptosis as indicated by a change in Bax/Bcl-2 ratios, loss of ΔΨm, release of cytochrome c from the mitochondria into cytosol, activations of caspase-3 and -9 (Fig. 4), and morphological characteristics of apoptosis, including blebbing of the plasma membrane, nuclear condensation, fragmentation, and apoptotic body formation as shown in various stainings and comet assay (Fig. 1 and 2).
Mitochondria are crucial to multicellular life and apoptosis often involves the release of cytochrome c from mitochondria into cytosol. Apoptosis-inducing agents that target mitochondria may affect them in various ways. They may induce the formation of membrane pores leading to mitochondrial swelling, or increase the permeability of mitochondrial membrane, resulting in the release of apoptotic effectors from mitochondrial into cytosol. The released cytochrome c binds to apoptotic protease activating factor-1 (Apaf-1) and dATP, which then bind to pro-caspase-9 to create a protein complex called apoptosome. The apoptosome cleaves the pro-caspase-9 to its active form of caspase-9. The activated caspase-9 in turn activates effector caspase-3, thereby initiating a cascade of proteolytic events [51]. Furthermore, the activities of different genes determine the cell's decision to activate its self-destruction program. Apoptosis regulator Bcl-2 is a family of evolutionarily related proteins that is involved in the regulation of mitochondrial outer membrane permeabilization pore (MOMP), and can be either pro-apoptotic (such as Bax and Bak) or anti-apoptotic (such as Bcl-2 proper and Bcl-xL). Proper execution of the decision of apoptosis requires the coordinated expressions of the involved genes. Regarding the key events in the induction of apoptosis, the present study demonstrates that CuA induced the release of cytochrome c from mitochondria into cytosol in a dose-dependent manner. Moreover, the up-regulated expressions of Bax and Bak taken together with the down-regulated expressions of Bcl-2 and Bcl-XL in response to CuA treatment, the collapse of ΔΨm, the up-regulated activities of most upstream protease of intrinsic apoptotic pathway, caspase-9, and the effector caspase-3 suggest the involvement of these proteins in CuA-induced apoptotic cell death.
Our results also indicate that CuA induced vacuolation with elevated VAC. Increase of VAC has been reported to be a common phenomenon of cells that undergo either apoptotic or necrotic cell death and could be a hallmark of dying cells [25]. Since apoptosis is an ordered process, the increase of VAC could be responsible for the self-digestion during the course of cell death [25].
Type I topoisomerases act by creating a transient single-stranded break in the DNA double helix molecule, followed by either a single-stranded DNA passage or controlled rotation about the break. Type I topoisomerases are involved in all DNA processes that involve tracking systems and play important roles in maintaining genomic integrity [6]. Furthermore, elevated levels of topoisomerase I mRNA, protein, and catalytic activity are seen across human tumors [52].
Type II topoisomerases act by generating a transient double-stranded DNA break, followed by a double-stranded DNA passage event. Type II topoisomerases function in numerous DNA processes and are required for recombination, the separation of daughter chromosomes, and proper chromosome structure, condensation, and decondensation [6]. The enzyme is increased dramatically during cell proliferation and peaks in G2/M. The resulting transient double-stranded break could lead to fragmentation of the genome with chromosomal translocations and other DNA aberrations [7, 53].
In addition to cell cycle regulation, topoisomerase is another major target of anticancer agents [8-11]. The chemotherapeutic agent etoposide kills cells by stabilizing the transient intermediate cleavage complex. The accumulation of cleavage complexes leads to the generation of permanent DNA strand breaks that fragments the genome and results in the activation of death pathways [54]. Apoptosis has been demonstrated to be the most efficient death-pathway in tumor cells after topoisomerase II is inhibited [55]. Our findings documented that CuA inhibited topoisomerase I and II activities in a concentration-dependent manner (Fig. 5), which in part, could be a mechanism driving the cells to apoptosis. While the majority of topoisomerase inhibitors are selectivity target either topoisomerase I or II [56], our study clearly shows that CuA inhibited both topoisomerase I and II activities in NCI-H520 cells. It is generally accepted that carcinogenesis is a multi-step process. Studying the effects of CuA on VAC and topoisomerase I and II in NCI-H520 cells in this process may provide new information on the pathological process of cancer. However, further work is needed to elucidate the specific underlying mechanism of the inhibition, possible mutagenic effects, and others for clinical usage as a chemopreventive or anticancer agent against lung squamous cell carcinoma and/or other malignances.
Therapy-induced cytotoxicity and other associated side effects of anticancer drugs are major concerns of chemotherapy. Therefore, the ideal drug should selectively kill cancer cells and not damage the healthy ones. None of the CuA treatments caused any significant decrease in diet consumption or body weight change compared with control mice. These results provide convincing proof of the protective effect of CuA treatment against NCI-H520 xenograft growth in nude mice without any observable toxicity; this suggests that CuA has an antiproliferative action in NCI-H520 cells and can potentially serve as an antiproliferative agent for cancer.
Conclusion
Collectively, our data clearly indicate that CuA induced apoptosis and also suppressed tumor cells growth and the associated biomarkers. The molecular events associated with the tumor suppression effect of CuA include not only the downregulation of cell proliferative controls involving apoptosis and both topoisomerase I and II, but also the upregulation of lysosomal vacuolation with increased VAC and cytotoxicity.
In conclusion, the present study provides fundamental information on the growth suppression effect of CuA in NCI-H520 cells, which suggests a short-term model for the evaluation of potential anti-cancer pharmacological modulators against lung squamous cell carcinoma. In fact, similar effects were also found in other cell lines, including human lung adenocarcinoma A549 cells and colorectal adenocarcinoma COLO 205 (results not shown). Our findings provide a justification for the development of CuA as a safe and effective anti-cancer agent against carcinogenesis.
Acknowledgements
This work was supported by grants from China Medical University CMUBH R103-002 and CMUBH R103-006.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Toh CK, Gao F, Lim WT, Leong SS, Fong KW, Yap SP. et al. Never-smokers with lung cancer: epidemiologic evidence of a distinct disease entity. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2006;24:2245-51 doi:10.1200/JCO.2005.04.8033
2. Hwa JS, Jin YC, Lee YS, Ko YS, Kim YM, Shi LY. et al. 2-methoxycinnamaldehyde from Cinnamomum cassia reduces rat myocardial ischemia and reperfusion injury in vivo due to HO-1 induction. Journal of ethnopharmacology. 2012;139:605-15 doi:10.1016/j.jep.2011.12.001
3. Duke JA, Duke P-AK, duCellier JL. Duke's Handbook of Medicinal Plants of The Bible. Boca Raton, London, New York: CRC Press. 2008
4. Artandi SE, DePinho RA. Telomeres and telomerase in cancer. Carcinogenesis. 2010;31:9-18 doi:10.1093/carcin/bgp268
5. Aleo E, Henderson CJ, Fontanini A, Solazzo B, Brancolini C. Identification of new compounds that trigger apoptosome-independent caspase activation and apoptosis. Cancer research. 2006;66:9235-44 doi:10.1158/0008-5472.CAN-06-0702
6. McClendon AK, Osheroff N. DNA topoisomerase II, genotoxicity, and cancer. Mutation research. 2007;623:83-97 doi:10.1016/j.mrfmmm.2007.06.009
7. Heck MM, Earnshaw WC. Topoisomerase II: A specific marker for cell proliferation. J Cell Biol. 1986;103:2569-81
8. Naowaratwattana W, De-Eknamkul W, De Mejia EG. Phenolic-containing organic extracts of mulberry (Morus alba L.) leaves inhibit HepG2 hepatoma cells through G2/M phase arrest, induction of apoptosis, and inhibition of topoisomerase IIalpha activity. J Med Food. 2010;13:1045-56 doi:10.1089/jmf.2010.1021
9. Baikar S, Malpathak N. Secondary metabolites as DNA topoisomerase inhibitors: A new era towards designing of anticancer drugs. Pharmacogn Rev. 2010;4:12-26 doi:10.4103/0973-7847.65320
10. Bandele OJ, Clawson SJ, Osheroff N. Dietary polyphenols as topoisomerase II poisons: B ring and C ring substituents determine the mechanism of enzyme-mediated DNA cleavage enhancement. Chem Res Toxicol. 2008;21:1253-60 doi:10.1021/tx8000785
11. Sudan S, Rupasinghe HP. Flavonoid-enriched apple fraction AF4 induces cell cycle arrest, DNA topoisomerase II inhibition, and apoptosis in human liver cancer HepG2 cells. Nutrition and cancer. 2014;66:1237-46 doi:10.1080/01635581.2014.951733
12. Andrade R, Crisol L, Prado R, Boyano MD, Arluzea J, Arechaga J. Plasma membrane and nuclear envelope integrity during the blebbing stage of apoptosis: a time-lapse study. Biol Cell. 2010;102:25-35 doi:10.1042/BC20090077
13. Lobner D. Comparison of the LDH and MTT assays for quantifying cell death: validity for neuronal apoptosis? J Neurosci Methods. 2000;96:147-52
14. Koh JY, Choi DW. Quantitative determination of glutamate mediated cortical neuronal injury in cell culture by lactate dehydrogenase efflux assay. J Neurosci Methods. 1987;20:83-90
15. Gwag BJ, Lobner D, Koh JY, Wie MB, Choi DW. Blockade of glutamate receptors unmasks neuronal apoptosis after oxygen-glucose deprivation in vitro. Neuroscience. 1995;68:615-9
16. Koh JY, Gwag BJ, Lobner D, Choi DW. Potentiated necrosis of cultured cortical neurons by neurotrophins. Science. 1995;268:573-5
17. Li J, Zhang J. Inhibition of apoptosis by ginsenoside Rg1 in cultured cortical neurons. Chin Med J (Engl). 1997;110:535-9
18. Miura T, Chiba M, Kasai K, Nozaka H, Nakamura T, Shoji T. et al. Apple procyanidins induce tumor cell apoptosis through mitochondrial pathway activation of caspase-3. Carcinogenesis. 2008;29:585-93 doi:10.1093/carcin/bgm198
19. Liu JR, Dong HW, Chen BQ, Zhao P, Liu RH. Fresh apples suppress mammary carcinogenesis and proliferative activity and induce apoptosis in mammary tumors of the Sprague-Dawley rat. J Agric Food Chem. 2009;57:297-304 doi:10.1021/jf801826w
20. Yoon H, Liu RH. Effect of selected phytochemicals and apple extracts on NF-kappaB activation in human breast cancer MCF-7 cells. J Agric Food Chem. 2007;55:3167-73 doi:10.1021/jf0632379
21. Zheng CQ, Qiao B, Wang M, Tao Q. Mechanisms of apple polyphenols-induced proliferation inhibiting and apoptosis in a metastatic oral adenoid cystic carcinoma cell line. Kaohsiung J Med Sci. 2013;29:239-45 doi:10.1016/j.kjms.2012.09.001
22. Darzynkiewicz Z. Differential staining of DNA and RNA in intact cells and isolated cell nuclei with acridine orange. Methods Cell Biol. 1990;33:285-98
23. Fan C, Wang W, Zhao B, Zhang S, Miao J. Chloroquine inhibits cell growth and induces cell death in A549 lung cancer cells. Bioorganic & medicinal chemistry. 2006;14:3218-22 doi:10.1016/j.bmc.2005.12.035
24. Olive PL, Banath JP. The comet assay: a method to measure DNA damage in individual cells. Nat Protoc. 2006;1:23-9 doi:10.1038/nprot.2006.5
25. Ono K, Wang X, Han J. Resistance to tumor necrosis factor-induced cell death mediated by PMCA4 deficiency. Mol Cell Biol. 2001;21:8276-88 doi:10.1128/MCB.21.24.8276-8288.2001
26. Fesik SW, Shi Y. Structural biology. Controlling the caspases. Science. 2001;294:1477-8 doi:10.1126/science.1062236
27. Mohan S, Abdul AB, Abdelwahab SI, Al-Zubairi AS, Sukari MA, Abdullah R. et al. Typhonium flagelliforme induces apoptosis in CEMss cells via activation of caspase-9, PARP cleavage and cytochrome c release: its activation coupled with G0/G1 phase cell cycle arrest. Journal of ethnopharmacology. 2010;131:592-600 doi:10.1016/j.jep.2010.07.043
28. Hsu YL, Cho CY, Kuo PL, Huang YT, Lin CC. Plumbagin (5-hydroxy-2-methyl-1,4-naphthoquinone) induces apoptosis and cell cycle arrest in A549 cells through p53 accumulation via c-Jun NH2-terminal kinase-mediated phosphorylation at serine 15 in vitro and in vivo. J Pharmacol Exp Ther. 2006;318:484-94 doi:10.1124/jpet.105.098863
29. Deveraux QL, Reed JC. IAP family proteins-suppressors of apoptosis. Genes & development. 1999;13:239-52
30. Ly JD, Grubb DR, Lawen A. The mitochondrial membrane potential (deltapsi(m)) in apoptosis; an update. Apoptosis. 2003;8:115-28
31. Reers M, Smiley ST, Mottola-Hartshorn C, Chen A, Lin M, Chen LB. Mitochondrial membrane potential monitored by JC-1 dye. Methods Enzymol. 1995;260:406-17
32. Har-Vardi I, Mali R, Breietman M, Sonin Y, Albotiano S, Levitas E. et al. DNA topoisomerases I and II in human mature sperm cells: characterization and unique properties. Hum Reprod. 2007;22:2183-9 doi:10.1093/humrep/dem170
33. White K, Grether ME, Abrams JM, Young L, Farrell K, Steller H. Genetic control of programmed cell death in Drosophila. Science. 1994;264:677-83
34. Mpoke SS, Wolfe J. Differential staining of apoptotic nuclei in living cells: application to macronuclear elimination in Tetrahymena. J Histochem Cytochem. 1997;45:675-83
35. Cover TL, Puryear W, Perez-Perez GI, Blaser MJ. Effect of urease on HeLa cell vacuolation induced by Helicobacter pylori cytotoxin. Infection and immunity. 1991;59:1264-70
36. Patel HK, Willhite DC, Patel RM, Ye D, Williams CL, Torres EM. et al. Plasma membrane cholesterol modulates cellular vacuolation induced by the Helicobacter pylori vacuolating cytotoxin. Infection and immunity. 2002;70:4112-23
37. Wyllie AH, Kerr JF, Currie AR. Cell death: the significance of apoptosis. Int Rev Cytol. 1980;68:251-306
38. Pommier Y. Diversity of DNA topoisomerases I and inhibitors. Biochimie. 1998;80:255-70
39. Li TK, Liu LF. Tumor cell death induced by topoisomerase-targeting drugs. Annu Rev Pharmacol Toxicol. 2001;41:53-77 doi:10.1146/annurev.pharmtox.41.1.53
40. Pommier Y, Pourquier P, Urasaki Y, Wu J, Laco GS. Topoisomerase I inhibitors: selectivity and cellular resistance. Drug Resist Updat. 1999;2:307-18 doi:10.1054/drup.1999.0102
41. Willett WC. Balancing life-style and genomics research for disease prevention. Science. 2002;296:695-8 doi:10.1126/science.1071055
42. Block G, Patterson B, Subar A. Fruit, vegetables, and cancer prevention: a review of the epidemiological evidence. Nutrition and cancer. 1992;18:1-29 doi:10.1080/01635589209514201
43. Shukla S, Meeran SM, Katiyar SK. Epigenetic regulation by selected dietary phytochemicals in cancer chemoprevention. Cancer letters. 2014;355:9-17 doi:10.1016/j.canlet.2014.09.017
44. Priyadarsini RV, Nagini S. Cancer chemoprevention by dietary phytochemicals: promises and pitfalls. Curr Pharm Biotechnol. 2012;13:125-36
45. Surh YJ. Cancer chemoprevention with dietary phytochemicals. Nature reviews Cancer. 2003;3:768-80 doi:10.1038/nrc1189
46. Yang CS, Landau JM, Huang MT, Newmark HL. Inhibition of carcinogenesis by dietary polyphenolic compounds. Annu Rev Nutr. 2001;21:381-406 doi:10.1146/annurev.nutr.21.1.381
47. Watson WH, Cai J, Jones DP. Diet and apoptosis. Annu Rev Nutr. 2000;20:485-505 doi:10.1146/annurev.nutr.20.1.485
48. Middleton E Jr, Kandaswami C, Theoharides TC. The effects of plant flavonoids on mammalian cells: implications for inflammation, heart disease, and cancer. Pharmacological reviews. 2000;52:673-751
49. Tanaka S, Yoon YH, Fukui H, Tabata M, Akira T, Okano K. et al. Antiulcerogenic compounds isolated from Chinese cinnamon. Planta medica. 1989;55:245-8 doi:10.1055/s-2006-961994
50. Reddy AM, Seo JH, Ryu SY, Kim YS, Kim YS, Min KR. et al. Cinnamaldehyde and 2-methoxycinnamaldehyde as NF-kappaB inhibitors from Cinnamomum cassia. Planta medica. 2004;70:823-7 doi:10.1055/s-2004-827230
51. Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES. et al. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479-89
52. Husain I, Mohler JL, Seigler HF, Besterman JM. Elevation of topoisomerase I messenger RNA, protein, and catalytic activity in human tumors: demonstration of tumor-type specificity and implications for cancer chemotherapy. Cancer research. 1994;54:539-46
53. Hsiang YH, Wu HY, Liu LF. Proliferation-dependent regulation of DNA topoisomerase II in cultured human cells. Cancer research. 1988;48:3230-5
54. Baldwin EL, Osheroff N. Etoposide, topoisomerase II and cancer. Curr Med Chem Anticancer Agents. 2005;5:363-72
55. El-Awady RA, Ali MM, Saleh EM, Ghaleb FM. Apoptosis is the most efficient death-pathway in tumor cells after topoisomerase II inhibition. Saudi Med J. 2008;29:558-64
56. Denny WA, Baguley BC. Dual topoisomerase I/II inhibitors in cancer therapy. Curr Top Med Chem. 2003;3:339-53
Author contact
![]() Corresponding author: Jaw-Ming Cherng, Department of Internal Medicine, Saint Mary's Hospital Luodong, Yilan, Taiwan ROC. Tel: 886-3-9388-652 Fax: 886-3-9575-653 E-mail: happy.professorcom.
Corresponding author: Jaw-Ming Cherng, Department of Internal Medicine, Saint Mary's Hospital Luodong, Yilan, Taiwan ROC. Tel: 886-3-9388-652 Fax: 886-3-9575-653 E-mail: happy.professorcom.
Citation styles
APA
Yang, S.m., Tsai, K.d., Wong, H.Y., Liu, Y.H., Chen, T.W., Cherng, J., Hsu, K.C., Ang, Y.U., Cherng, J.M. (2016). Molecular Mechanism of Cinnamomum verum Component Cuminaldehyde Inhibits Cell Growth and Induces Cell Death in Human Lung Squamous Cell Carcinoma NCI-H520 Cells In Vitro and In Vivo. Journal of Cancer, 7(3), 251-261. https://doi.org/10.7150/jca.13689.
ACS
Yang, S.m.; Tsai, K.d.; Wong, H.Y.; Liu, Y.H.; Chen, T.W.; Cherng, J.; Hsu, K.C.; Ang, Y.U.; Cherng, J.M. Molecular Mechanism of Cinnamomum verum Component Cuminaldehyde Inhibits Cell Growth and Induces Cell Death in Human Lung Squamous Cell Carcinoma NCI-H520 Cells In Vitro and In Vivo. J. Cancer 2016, 7 (3), 251-261. DOI: 10.7150/jca.13689.
NLM
Yang Sm, Tsai Kd, Wong HY, Liu YH, Chen TW, Cherng J, Hsu KC, Ang YU, Cherng JM. Molecular Mechanism of Cinnamomum verum Component Cuminaldehyde Inhibits Cell Growth and Induces Cell Death in Human Lung Squamous Cell Carcinoma NCI-H520 Cells In Vitro and In Vivo. J Cancer 2016; 7(3):251-261. doi:10.7150/jca.13689. https://www.jcancer.org/v07p0251.htm
CSE
Yang Sm, Tsai Kd, Wong HY, Liu YH, Chen TW, Cherng J, Hsu KC, Ang YU, Cherng JM. 2016. Molecular Mechanism of Cinnamomum verum Component Cuminaldehyde Inhibits Cell Growth and Induces Cell Death in Human Lung Squamous Cell Carcinoma NCI-H520 Cells In Vitro and In Vivo. J Cancer. 7(3):251-261.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.