Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2016; 7(2):174-183. doi:10.7150/jca.13387 This issue Cite
Review
Colorectal Carcinogenesis, Radiation Quality, and the Ubiquitin-Proteasome Pathway
Kamal Datta1 ![]() , Shubhankar Suman1, Santosh Kumar1, Albert J Fornace Jr.1, 2
, Shubhankar Suman1, Santosh Kumar1, Albert J Fornace Jr.1, 2
1. Department of Biochemistry and Molecular & Cellular Biology and Lombardi Comprehensive Cancer Center, Georgetown University, Washington, DC USA.
2. Center of Excellence in Genomic Medicine Research (CEGMR), King Abdulaziz University, Jeddah, Saudi Arabia
Received 2015-7-29; Accepted 2015-11-1; Published 2016-1-1
Abstract
Adult colorectal epithelium undergoes continuous renewal and maintains homeostatic balance through regulated cellular proliferation, differentiation, and migration. The canonical Wnt signaling pathway involving the transcriptional co-activator β-catenin is important for colorectal development and normal epithelial maintenance, and deregulated Wnt/β-catenin signaling has been implicated in colorectal carcinogenesis. Colorectal carcinogenesis has been linked to radiation exposure, and radiation has been demonstrated to alter Wnt/β-catenin signaling, as well as the proteasomal pathway involved in the degradation of the signaling components and thus regulation of β-catenin. The current review discusses recent progresses in our understanding of colorectal carcinogenesis in relation to different types of radiation and roles that radiation quality plays in deregulating β-catenin and ubiquitin-proteasome pathway (UPP) for colorectal cancer initiation and progression.
Keywords: APCMin/+, Intestinal tumor, Space radiation, High-LET radiation, Tumorigenesis, Proteasome, β-catenin, HZE-particles
Introduction
Qualitatively, ionizing radiation (IR) has been categorized depending on energy deposited in tissues per unit distance traversed into low and high linear energy transfer (low- and high-LET) radiation [1-4]. While low-LET radiation, such as γ-rays and x-rays is prevalent on earth, high-LET energetic heavy ion radiation is mostly encountered in outer space but has also recently been used in radiotherapy. Constituents of radiation environment in space encountered by astronauts during space travel are mostly high-energy particles such as protons and heavy ions including 16O, 12Si, and 56Fe. Notably, protons are the major contributor to the dose equivalent of solar particle event (SPE) originating from the sun; heavy ions are the significant contributors to the dose equivalent of galactic cosmic radiation (GCR) originating from outside the solar system. While low-LET radiation is sparsely ionizing and is less damaging relative to high-LET radiation, high-LET heavy ion radiation not only produces densely ionizing primary tracts but it also generates higher numbers of secondary ionization events known as delta rays and has a higher relative biological effectiveness (RBE) compared to low-LET radiation [1-4].
Therefore, high-LET heavy ion radiation exposure is predicted to pose greater carcinogenic risk to space travellers than low-LET radiation and is one of the major concerns for future space missions especially long duration space missions such as mission to Mars [5, 6]. Additionally, heavy ions such as 12C due to their better dose conformity and higher RBE values relative to conventional radiotherapy are making significant contribution to cancer treatment but concerns have been sounded about normal tissues exposure and thus long-term risk to GI tissues [7, 8]. It is important to note that epidemiological studies in human and laboratory studies in animal models have established low-LET radiation as a risk factor for colorectal carcinogenesis [1, 9, 10]. Considering their high-LET characteristics, heavy ions are expected to present greater risk of gastrointestinal (GI) pathologies including colorectal cancer (CRC), and our own recent observations in mouse models suggest greater risk of CRC after 56Fe radiation exposure [5, 10-12].
Colon along with small intestinal epithelium have high cellular turnover and is replaced every 2-5 days via proliferation and differentiation of adult somatic stem cells located at the crypt base [13-16]. Notably, the canonical Wnt pathway with its transcriptional co-activator β-catenin is a major regulator of the self-renewing and rapidly cycling colonic epithelium [10, 17, 18]. Binding of Wnt ligands to the Frizzled and low-density lipoprotein receptor-related protein (LPR) allows translocation of the cytoplasmic Axin, a negative regulator of β-catenin, to the plasma membrane. Subsequently, a series of complex signaling events prevent formation of the β-catenin degradation complex comprising of Axin, adenomatosis polyposis coli (APC), protein phosphatase 2A (PP2A), glycogen synthase kinase 3β (GSK3β) and casein kinase 1α (CK1α) allowing cytoplasmic accumulation of β-catenin and later translocation to the nucleus [10, 17, 18]. In the nucleus, β-catenin along with transcription factor TCF4 binds to promoters and upregulates transcription of target genes such as cMyc and cyclin D1. On the contrary, absence of Wnt ligand binding to Frizzled/LPR allows formation of the β-catenin degradation complex leading to its phosphorylation by CK1α and GSK3β, and phosphorylated β-catenin is then ubiquitinated and targeted for proteasomal degradation - thus allowing regulation of its cellular level [10, 17-20].
The ubiquitin-proteasome pathway (UPP), which is involved in protein degradation and is active in the cytoplasm as well as in the nucleus, plays an important role in cellular proteolysis and thus in protein turnover allowing spatial and temporal regulation of protein levels in cells [21-24]. The target protein is polyubiquitinated by a multistep enzymatic process, and ubiquitin receptors are believed to subsequently guide ubiqutinated proteins to the proteasome for unfolding and degradation. Post-translational phosphorylation acts as a trigger for protein ubiquitination and proteasomal degradation. In our colorectal cancer mouse model system we have demonstrated that exposure to high-LET heavy ion radiation leads to decreased phosphorylation and thus increased accumulation of β-catenin in tumor samples with implications for higher and more aggressive intestinal tumorigenesis after such radiation exposure [10]. Considering that the β-catenin level is actively regulated via the UPP, radiation affects both β-catenin and UPP, and both the pathways are implicated in colorectal carcinogenesis [25, 26], this review will discuss recent updates on effects of radiation quality on β-catenin and UPP in relation to colorectal carcinogenesis.
Radiation exposure on the Earth and in outer space
Radiation environment on the Earth constitutes natural as well as man-made sources and both contribute (310 mrem or 3.1 mGy from each source) equally to the average annual radiation dose of 620 mrem or 6.2 mGy in the USA [27]. While radon is a major constituent of the natural radiation sources, medical procedures accounts for the major part of man-made radiation sources. Radon, whose decay is alpha particle emission and is considered high-LET, has limited tissue penetration capabilities and deposits much of its energy in a localized area. However, due to its gaseous nature, radon exposure has been implicated in increased risk of lung cancer [28]. On the contrary, medical procedures mostly involve photons such as x-rays and γ-rays with significant tissue penetration capabilities and are low-LET [27, 29]. The radiation environment in outer space is qualitatively different than that on the Earth and three major sources of radiation contribute to radiation dose accrued by astronauts travelling beyond low-Earth orbit (LEO) [1, 3, 10, 30, 31]. Crossing the Van Allen's belt exposes traveling astronauts to trapped radiation of high-energy electrons in the outer belt and of high-energy electrons and protons in the inner belt. The second radiation source in space is solar particle events (SPEs) or commonly referred to as solar storms, which are sporadic, and mostly consist of high-energy protons. Conversely, galactic cosmic radiation (GCR), which is ambient in space, originates from outside the solar system, and a major part of its dose equivalent is contributed by the high-energy heavy ions such as 56Fe, 28Si, and 16O, and 12C, which are high-LET. Commonly, space radiation exposures will occur at low doses and dose rates, but protracted exposure during long-duration space missions may lead to enough dose accumulation especially of heavy ion radiation to raise long-term health concerns [32, 33]. Indeed, calculation from the Radiation Assessment Detector (RAD) in the Mars Science Laboratory showed that a round trip to Mars would expose astronauts to a significant cumulative space radiation dose [34]. Additionally, radiation dose and dose rate of SPE is dependent on the intensity of solar activity and it has been predicted that the radiation dose to astronauts during an SPE could reach up to 2 Gy behind shielding [10, 32, 33]. Much of space radiation is high-energy and highly penetrating producing primary as well as secondary ionization tracks in tissues and has been shown to cause greater cellular damage relative to low-LET radiation on earth. Therefore, GI tissue with a highly proliferative epithelial compartment is vulnerable to long-term consequences such as cancer after space radiation exposure.
Colorectal carcinogenesis and radiation
In the USA, CRC is still a common cancer and is one of the leading causes of cancer-related mortality [35]. While life-style related factors are known to play a major role in the CRC development [36], low-LET radiation exposure as a risk factor for colorectal carcinogenesis has been established from long-term follow up studies in the atom bomb survivor cohort and radiation facility workers [37-40]. Additionally, studies in prostate and childhood cancer radiotherapy patients showed significantly increased risk of second malignancy in colon supporting the notion that therapeutic radiation is a risk factor for CRC as well [41-43]. Diagnostic radiation exposures, especially procedures with multiple prolonged exposures such as CT, have also been predicted to pose colon cancer risk above the unexposed population [29, 44]. At the molecular level, radiation has been proposed to impart its carcinogenic effects via microsatellite instability (MSI) and chromosomal instability (CIN) leading to loss of function of repair genes such as MLH1 or tumor suppressor genes such as APC considered critical for colorectal carcinogenesis [45, 46]. Although, effects of heavy ion radiation on GI cells at the genome level has yet to be studied, it is expected that heavy ions due to their qualitatively greater damaging capabilities than low-LET radiation even in the deep-sited tissues will cause greater MSI and CIN. Indeed, energy deposition from x-rays decreases exponentially as it traverses a tissue and the reverse can be true for particle radiation encountered in space [47, 48]. Therefore, it is projected that there will be greater perturbation of molecular pathways critical for colorectal carcinogenesis after space radiation exposures and there are increasing reports supporting such a view [10, 49-54].
Wnt/β-catenin pathway
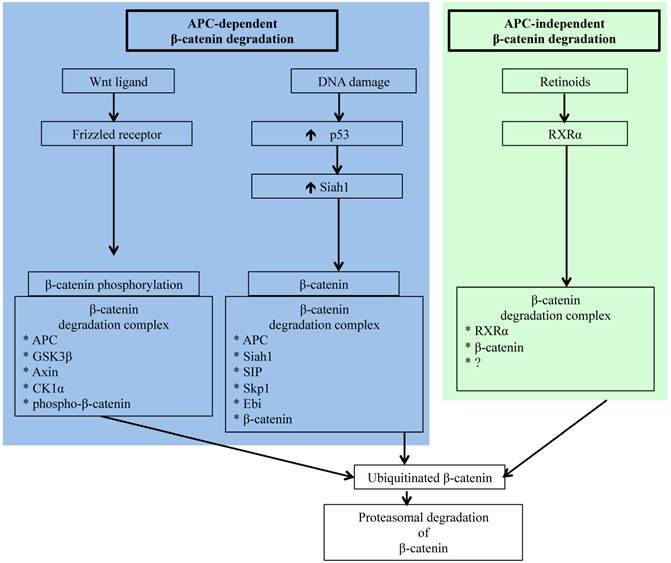
The Wnt/β-catenin pathway also known as the canonical Wnt signaling pathway is a highly conserved pathway and is involved in varied cellular processes beginning at embryogenesis and organismal development to homeostasis and tissue maintenance in adult life [55, 56]. For colorectal epithelium, the Wnt/β-catenin pathway not only plays important roles in renewal and maintenance of stem cells present at the crypt bottom, but it also ensures regulated epithelial cell proliferation, differentiation, and migration to the crypt top, and finally shading into the lumen via apoptosis. Therefore, perturbed Wnt/β-catenin signaling has catastrophic consequences on the colorectal epithelial cell turnover leading to varied human pathologies including cancer [55-57]. In the absence of Wnt ligand binding of Frizzled receptor, cytosolic β-catenin complexes with APC, GSK3β, Axin, and casein kinase 1α (CK1α) to form what is known as the β-catenin destruction complex (Figure 1). In this multi-protein complex, β-catenin is phosphorylated at the amino terminal by GSK3β and CK1α allowing recognition and ubiquitination of β-catenin by ubiquitin ligases and subsequent degradation of the β-catenin in the proteasome [55, 56]. Conversely, binding of Wnt ligand to Frizzled receptor prevents destruction complex formation leading to β-catenin accumulation. Therefore, proteasomal degradation allows regulation of cellular β-catenin level and prevents translocation of β-catenin to the nucleus for joining with TCF4 to transcribe proliferative factors such as cMyc and cyclin D1.
Schematic representation of APC-dependent and -independent ubiquitin-proteasome pathways (UPP) of β-catenin degradation. While molecular detail about the two APC-dependent β-catenin degradation complexes has been reported, we have yet to acquire a comprehensive understanding of the molecules involved in the RXRα/β-catenin degradation complex.
Ubiquitin-proteasome pathway (UPP) and colorectal carcinogenesis
Protein degradation by the proteasome due to specific tagging of proteins with ubiquitin (a small protein marker) that are destined for degradation is a highly selective and critical process for cellular functioning. Protein degradation through UPP involves ubiquitin protein, ubiquitinating and deubiquitinating enzymes, and proteasomes [58, 59]. Importantly, apart from regulating protein half-life through degradation, ubiquitination also regulates activity and localization of the target proteins [58]. Indeed, multiple ubiquitinating and deubiquitinating enzymes often clustered in multi-enzyme complex to control ubiquitin modification of proteins [58]. For ubiquitination, ubiquitin (Ub), conserved in all eukaryotes, is activated by E1 activating enzymes through adenylation at the C-terminus of the Ub. Subsequently, adenylated Ub is transferred to the E2 conjugating enzymes leading to formation of the E2-Ub thioester intermediate, which is used by a substrate specific E3 ligase to transfer the Ub to the target protein or to another Ub already on the target protein [26, 58-60]. Both mono- and poly-ubiquitination act as signal for proteolysis, however poly-ubiquinynation is required for proteasomal targeting and mono-ubiquitnation of a target protein is often associated with lysosomal degradation [59, 61]. It is important to note that the ubiquitin-mediated protein modification similar to phosphorylation and other post-translational modifications can be reversed by a group of deubiquitinating (DUB) enzymes, which are reported to be associated with E3 ligases thus regulating the level of ubiquitination [58].
The proteasome in mammalian systems is known as the 26S complex, which has a 20S subunit at the core and two 19S subunits at each end of the core component [26, 58-60]. The 20S core particle is a cylindrical multi-protein complex that provides the microenvironment for proteolysis of the target protein and at the same time protection of the non-target intracellular proteins from undesired proteolysis [23]. The 19S subunit is involved in controlling substrate recognition, recruitment, and ATP-dependent processing of the ubiquitinated target proteins [22, 23, 62-64]. In addition to protein recycling, the proteasome is also involved in modulation of various cellular processes such as cell cycle progression, regulation of gene expression, and stress responses by regulating turnover of specific proteins [24, 65]. Therefore, alterations in the UPP have the potential to alter cellular homeostasis, and studies have demonstrated that proteasomal activity is elevated in human cancers including CRC. Indeed, deregulation of one or more subunits of the 20S proteasome has been correlated with carcinogenesis, which could be due to effects of UPP on cell cycle regulatory factors such as cyclins and CDK inhibitors as well as on apoptotic factors such as p53 and caspases [26, 60, 66]. In addition, UPP helps maintain a balance between pro-apoptotic and anti-apoptotic signaling pathways and thus determines the ultimate cell fate. For example, Mdm2, a E3 ubiquitin ligase ubiquitinates p53 to regulate its stability, and its DNA-damage- and stress-induced responses [67]. DNA-damage-induced genomic instability has been linked to carcinogenesis and recently, ubiquitin-proteasome signaling has been reported to coordinate with the DNA damage response (DDR) signaling at the double strand DNA break (DSB) site to execute the necessary repair mechanism [68]. In addition, protein ubiquitination has also emerged as a crucial modulator of cellular energy metabolism and inflammation, key players in carcinogenesis, through its involvement in regulation of TNF/TNFR1 and NFκB signaling [69].
Carcinogenesis in colorectal epithelium like in other organs occurs as a result of complex multistep signaling pathway alterations [70], and sporadic CRC is commonly initiated through mutations in APC allowing oncogenic β-catenin accumulation leading to transition of normal epithelium to microscopic aberrant crypt foci (ACF) considered a pathologic precursor for CRC [71, 72]. Acquisition of additional gain of function mutations in oncogenes, such as K-ras, and loss of function mutations in tumor suppressor genes, such as p53 and Smad4, supports disease progression from ACF to visible adenomas and subsequently to adenocarcinoma. Importantly, however, several molecules, such as β-catenin, c-jun, c-fos, Akt, Smad4, and p53 involved in CRC initiating and promoting pathways, are regulated by the UPP [26, 60, 73-76]. However, accumulation of oncogenic β-catenin due to alterations in its regulatory pathways remains the key early event in colorectal carcinogenesis [77, 78], and apart from the already discussed APC-dependent APC-AXIN-GSK3β pathway, β-catenin is also targeted to ubiquitin-proteasome degradation either via APC-dependent p53/Siah1 pathway or via the APC-independent retinoid X receptor α (RXRα) pathway [79-82] (Figure 1). Siah1 (seven in absentia homolog 1), a mammalian homolog of the Drosophila seven in absentia (Sina) protein, is p53-inducible and similar to p53 acts as a tumor suppressor through regulation of cell cycle, proliferation, and apoptosis [79, 83, 84]. Additionally, Siah1 with its E3 ubiquitin ligase properties has been demonstrated to bind to E2 ubiquitin conjugating enzymes and ubiquitinate β-catenin for degradation in a complex requiring the presence of the APC protein [79, 84]. Therefore, Siah1 acts as a link between p53 and Wnt/β-catenin pathways and plays an important role in the cellular β-catenin turnover independent of the GSK3β. Consequently, p53 mutation, recognized as a late event in colorectal carcinogenesis and linked to disease progression, robs cells of an alternate p53/Siah1-mediated β-catenin degradation pathway leading to additional loss of control of growth and proliferation.
Since β-catenin's escape from regulatory surveillance has grave cellular consequences and two pathways involved in preventing pathogenic accumulation of β-catenin are dependent on APC, commonly mutated in CRC, it is essential that there is an APC- and p53-independent β-catenin degradation pathway. Indeed, Xiao et al. have reported on the existence of a RXRα-mediated β-catenin degradation pathway [82]. Nuclear receptor RXRα, a member of the RXR family of DNA binding receptors with anti-proliferative and anti-neoplastic function, is involved in a plethora of genomic and non-genomic actions through heterodimerization with other nuclear receptors as well as through homodimerization with itself [85, 86]. While RXRα in the presence of agonist is known to get co-degraded with its dimerization partners via UPP, molecular details of how RXRα interacts with β-catenin for proteasomal degradation is not fully understood. However, it is believed that RXRα interacts with β-catenin in a degradation complex similar to that of APC/β-catenin complex and targets β-catenin to proteasomal degradation [82]. Notably, RXRα is capable of regulating β-catenin turnover even in cells with APC and p53 mutations, and is demonstrated to hinder CRC progression [82]. Studies in animal models of human CRC have established that administration of RXRα agonist could block adenoma-carcinoma transition [85]. Considering that protein turnover is critical for cellular homeostasis, availability of multiple independent pathways for protein degradation ensure that the potentially pro-carcinogenic β-catenin level remains within physiologic limits to avert cancer initiation and progression.
Radiation stress and ubiquitin-proteasome pathway (UPP)
Both low- and high-LET radiation are known to alter cellular protein turnover through alterations at the genomic [82, 86], epigenomic [87], transcriptional [88], translational, [89] and post-translational levels [90], often in a radiation dose and type dependent manner. While protein synthesis is regulated at the genetic, epigenetic, transciptomic, and translational levels [87, 91-94], protein degradation is mostly controlled via specific post-translational modifications such as phosphorylation, acetylation, glutathionylation, ubiquitinylation, and sumoylation at selected amino acid sites, and radiation is reported to influence these processes [25, 95-98]. Radiation exposure is also known to cause non-specific protein modifications such as oxidation, carbonylation and nitrosylation through overproduction of reactive oxygen (ROS) and nitrogen (RNS) species [99-101], resulting in loss of protein function [54, 102], accumulation of unfolded proteins, and initiation of the unfolded protein response (UPR) [103, 104]. Elimination of proteins modified post-translationally or oxidatively occurs either via lysosomal proteolysis or via ubiquitin-proteasome pathway, the two major pathways involved in the removal of cellular proteins [105], and radiation has been documented to modulate both the systems [25, 98, 106, 107].
The UPP, the focus of the current review, has been widely studied in relation to low-LET ionizing radiation [25, 98, 108]. Studies using purified proteasomes as well as various cell lines have demonstrated that radiation exposure is associated with inhibition of UPP [98, 109-113]. Importantly, impairment of radiation-induced proteasome function has in part been attributed to free-radical induced damage to ubiquitin-proteasome pathway-associated molecules [109, 110] and increased expression of endogenous proteasomal inhibitors Hsp90 and PI31 [114, 115]. Although it has yet to be explored, persistent alterations of the UPP, similar to stress response pathways observed by us in intestinal cells [54, 106, 116], are expected to occur long after radiation exposure especially after high-LET radiation exposures. Because UPP plays a major role in regulating levels of proteins involved in controlling growth and proliferation, radiation-induced long-term alterations of the UPP components will have major ramifications for cellular homeostasis with implications for carcinogenesis including colorectal carcinogenesis.
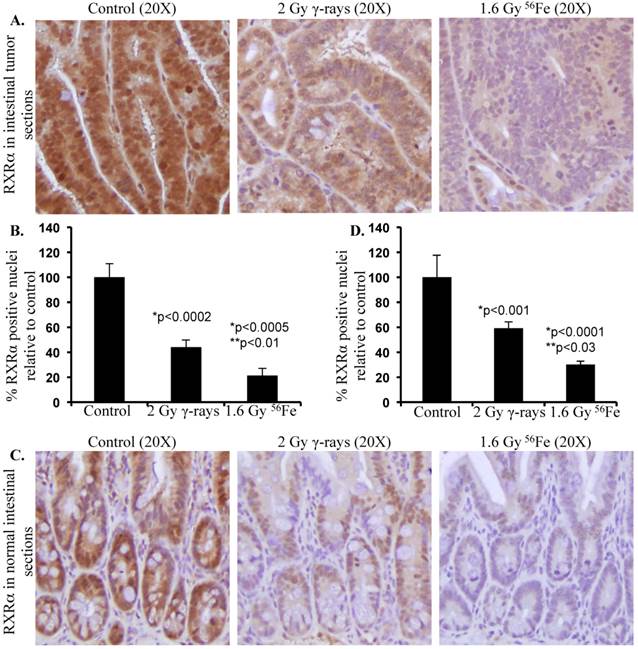
With increasing interest in space travel and search for life beyond earth, it is important to understand the effects of space radiation especially heavy ion radiation on cellular processes critical for maintenance of homeostasis, preservation of physiologic functioning, and prevention of initiation of pathologic processes. Although high-LET radiation including heavy ions has been demonstrated to cause greater protein modifications both via post-translational and oxidative processes relative to low-LET radiation [101, 106, 116-118], little progress has been made towards understanding its effects on UPP. We have demonstrated that exposure to high-LET heavy ion space radiation leads to increased risk of developing GI tumors in APC-mutant mouse models [10-12]. Exposure of APCMin/+ mice to high-LET 56Fe radiation led to higher intestinal tumor frequency relative to mice exposed to low-LET γ radiation. In APC1638N/+ mice, a greater increase in tumor frequency was observed throughout the GI tract after high-LET 56Fe radiation relative to low-LET γ radiation. Also, compared to low-LET tumors, significantly higher proportion of the high-LET heavy ion radiation-induced tumors was invasive adenocarcinoma. Therefore, we concluded from our previous studies that high-LET heavy ion radiation induced GI tumors were of higher frequency and grade relative to low-LET γ radiation [10-12, 119]. At the molecular level high-LET 56Fe radiation-induced tumors showed more pronounced oncogenic β-catenin accumulation relative to low-LET γ radiation [10]. Considering that APC is mutated in these mice, the APC-dependent Wnt/β-catenin degradation pathways are expected to be non-functional. Therefore, the cell may rely more on APC-independent pathways such as RXRα-mediated UPP for β-catenin degradation to provide some protection from tumorigenesis. Consequently, downregulation of the APC-independent RXRα pathway is expected to contribute to increased stabilization and thus accumulation of β-catenin leading to greater percentage of aggressive tumors in response to 56Fe radiation. While we already demonstrated that the high-LET 56Fe radiation exposure caused greater β-catenin and higher percent of invasive carcinoma in APCMin/+ mice [10], recently we have reported for the first time that 56Fe also led to downregulation of RXRα in the tumor-bearing as well as in the tumor-free areas of the intestine in these mice [120]. Conversely, differential effects of low- and high-LET radiation on UPP pathway have not yet been clearly delineated. Our initial results presented in this review demonstrate that exposure to 1.6 Gy high-LET 56Fe radiation caused higher downregulation of RXRα in tumor as well as in normal intestinal tissues (Figure 2) suggesting greater inhibitory effects of 56Fe on UPP relative to an equitoxic (calculated and published previously in [121]) 2 Gy dose of γ radiation. While further studies into the effects of high-LET radiation on UPP in relation to low-LET radiation at the molecular level are ongoing, our results suggest for the first time that radiation quality can differentially impact UPP with implications for overall cellular protein turnover and thus for high-LET radiation carcinogenesis including GI carcinogenesis.
Differential effects of low- and high-LET radiation on RXRα expression in intestine. Sections from control and irradiated (2 Gy γ-rays or 1.6 Gy 56Fe) mice were fixed in buffered formalin, paraffin embedded, and sectioned for immunohistochemistry. Detail of mice, irradiation, and sample collection were described previously [10, 106]. Immunohistochemistry for RXRα (Santa Cruz Biotechnology, Dallas, TX) was performed and quantified according to procedures described previously [101, 120]. Staining in control samples were considered 100% and quantification data from irradiated samples are presented graphically as percent change relative to controls. Data from five mice were used for statistical analysis and p<0.05 was considered as significant. Error bars represent mean ± standard error of mean (SEM). A) Representative images of RXRα staining in intestinal tumors from female APCMin/+ mice 100 to 110 d after radiation exposure. B) Quantification of RXRα immunohistochemistry staining in intestinal tumors. C) Representative images of RXRα staining in normal intestinal sections from female C57BL/6J mice two months after radiation exposure. D) Quantification of RXRα immunohistochemistry staining in normal intestinal sections. Significance - *relative to control and **relative to γ-radiation.
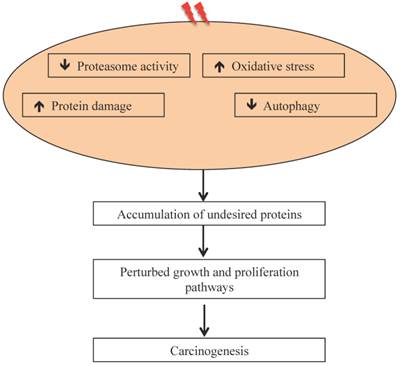
Our results on intestinal tumorigenesis in APCMin/+ with Wip1 (wild-type p53 induced phosphatase-1) knockout (APCMin/+; Wip1-/-) mice showed that Wip1 abrogation abolishes radiation-induced intestinal tumorigenesis in these mice [122]. Since, Wip1, a phosphatase, is a radiation responsive protein known to regulate the p53-MDM2 auto-regulatory loop through dephosphorylation of p53 and dephosphorylated p53 is subjected to MDM2 mediated ubiquitination and its subsequent degradation via proteasome [123], abrogation of Wip1 is believed to allow p53 accumulation and thus block tumorigenesis. We also reported persistent downregulation of autophagy in mouse intestine long-term after radiation exposure [106], which could also be associated as reported earlier with decreased UPP flux [124, 125]. It has been postulated that decreased UPP flux in autophagy-inhibited cells is caused due to accumulation of p62 [126] observed in radiation-exposed mouse intestine [106]. While these studies suggest effects of heavy ion radiation on UPP, there are considerable uncertainties in understanding how qualitatively different types of radiation affects UPP and how UPP modulates GI cancer risks after long duration space missions such as mission to Mars. Key to the risk estimates is to acquire in vivo mechanistic data on GI carcinogenesis using surrogate biological endpoints. The UPP is linked to a number of carcinogenic precursor events such as oxidative stress and inflammation, which we demonstrated to occur persistently in response to high-LET 56Fe radiation and UPP inhibition itself has the potential to promote carcinogenesis (Figure 3). Additional studies focused on proteasome structure and function will be required to fully delineate the link between UPP and GI cancer risk after energetic heavy ion space radiation exposures.
Radiation-induced oxidative stress, protein damage, and altered protein degradation pathways are implicated in carcinogenesis.
Acknowledgements
This study is supported in part by NASA grants NNX13AD58G and NNX09AU95G. We are very much thankful to the members of the NASA Space Radiation Laboratory (NSRL) especially to Drs. Peter Guida and Adam Rusek form Brookhaven National Laboratory for their excellent support in conducting heavy ion radiation exposures. We are also thankful to Mr. Steven J. Strawn and Ms. Pelagie Ake for administrative and mice breeding support.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Durante M, Cucinotta FA. Heavy ion carcinogenesis and human space exploration. Nat Rev Cancer. 2008;8:465-472
2. Jakel O. The relative biological effectiveness of proton and ion beams. Z Med Phys. 2008;18:276-285
3. Setlow RB. The U.S. National Research Council's views of the radiation hazards in space. Mutat Res. 1999;430:169-175
4. Suman S, Datta K, Trani D, Laiakis EC, Strawn SJ, Fornace AJJ. Relative biological effectiveness of (12)C and (28)Si radiation in C57BL/6J mice. Radiat Environ Biophys. 2012;51:303-309
5. Cucinotta FA. Space radiation risks for astronauts on multiple International Space Station missions. PLoS One. 2014;9:e96099
6. Cucinotta FA, Kim MH, Chappell LJ, Huff JL. How safe is safe enough? Radiation risk for a human mission to Mars. PLoS One. 2013;8:e74988
7. Okada T, Kamada T, Tsuji H, Mizoe JE, Baba M, Kato S. et al. Carbon ion radiotherapy: clinical experiences at National Institute of Radiological Science (NIRS). J Radiat Res. 2010;51:355-364
8. Schulz-Ertner D, Jakel O, Schlegel W. Radiation therapy with charged particles. Semin Radiat Oncol. 2006;16:249-259
9. Cucinotta FA, Durante M. Cancer risk from exposure to galactic cosmic rays: implications for space exploration by human beings. Lancet Oncol. 2006;7:431-435
10. Datta K, Suman S, Kallakury BV, Fornace AJJ. Heavy ion radiation exposure triggered higher intestinal tumor frequency and greater beta-catenin activation than gamma radiation in APC(Min/+) mice. PLoS One. 2013;8:e59295
11. Trani D, Datta K, Doiron K, Kallakury B, Fornace AJJ. Enhanced intestinal tumor multiplicity and grade in vivo after HZE exposure: mouse models for space radiation risk estimates. Radiat Environ Biophys. 2010;49:389-396
12. Trani D, Nelson SA, Moon BH, Swedlow JJ, Williams EM, Strawn SJ. et al. High-energy particle-induced tumorigenesis throughout the gastrointestinal tract. Radiat Res. 2014;181:162-171
13. Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M. et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007;449:1003-1007
14. Barker N, van de Wetering M, Clevers H. The intestinal stem cell. Genes Dev. 2008;22:1856-1864
15. Barker N, van Es JH, Jaks V, Kasper M, Snippert H, Toftgard R. et al. Very long-term self-renewal of small intestine, colon, and hair follicles from cycling Lgr5+ve stem cells. Cold Spring Harb Symp Quant Biol. 2008;73:351-356
16. Barker N. Adult intestinal stem cells: critical drivers of epithelial homeostasis and regeneration. Nat Rev Mol Cell Biol. 2014;15:19-33
17. Henderson BR, Fagotto F. The ins and outs of APC and beta-catenin nuclear transport. EMBO Rep. 2002;3:834-839
18. Kolligs FT, Bommer G, Goke B. Wnt/beta-catenin/tcf signaling: a critical pathway in gastrointestinal tumorigenesis. Digestion. 2002;66:131-144
19. Hildesheim J, Salvador JM, Hollander MC, Fornace AJJ. Casein kinase 2- and protein kinase A-regulated adenomatous polyposis coli and beta-catenin cellular localization is dependent on p38 MAPK. J Biol Chem. 2005;280:17221-17226
20. Segditsas S, Tomlinson I. Colorectal cancer and genetic alterations in the Wnt pathway. Oncogene. 2006;25:7531-7537
21. Fu H, Lin YL, Fatimababy AS. Proteasomal recognition of ubiquitylated substrates. Trends Plant Sci. 2010;15:375-386
22. Karpov D, Glickman MH, Bar-Nun S, Coffino P. Ubiquitin proteasome system in stress and disease. Biochem Res Int. 2012;2012:454796
23. Kish-Trier E, Hill CP. Structural biology of the proteasome. Annu Rev Biophys. 2013;42:29-49
24. Wolf DH, Hilt W. The proteasome: a proteolytic nanomachine of cell regulation and waste disposal. Biochim Biophys Acta. 2004;1695:19-31
25. McBride WH, Iwamoto K, Pervan M. The proteasome and radiation. Radiat Res. 2005;163:706-707
26. Voutsadakis IA. The ubiquitin-proteasome system in colorectal cancer. Biochim Biophys Acta. 2008;1782:800-808
27. http://www.nrc.gov/reading-rm/doc-collections/fact-sheets/bio-effects-radiation.html
28. Krewski D, Lubin JH, Zielinski JM, Alavanja M, Catalan VS, Field RW. et al. A combined analysis of North American case-control studies of residential radon and lung cancer. J Toxicol Environ Health A. 2006;69:533-597
29. Brenner DJ, Hricak H. Radiation exposure from medical imaging: time to regulate? JAMA. 2010;304:208-209
30. Cucinotta FA, Schimmerling W, Wilson JW, Peterson LE, Badhwar GD, Saganti PB. et al. Space radiation cancer risks and uncertainties for Mars missions. Radiat Res. 2001;156:682-688
31. Chancellor JC, Scott GB, Sutton JP. Space Radiation: The Number One Risk to Astronaut Health beyond Low Earth Orbit. Life (Basel). 2014;4:491-510
32. Cucinotta FA, Wu H, Shavers MR, George K. Radiation dosimetry and biophysical models of space radiation effects. Gravit Space Biol Bull. 2003;16:11-18
33. Hamilton SA, Pecaut MJ, Gridley DS, Travis ND, Bandstra ER, Willey JS. et al. A murine model for bone loss from therapeutic and space-relevant sources of radiation. J Appl Physiol. 2006;101:789-793
34. Zeitlin C, Hassler DM, Cucinotta FA, Ehresmann B, Wimmer-Schweingruber RF, Brinza DE. et al. Measurements of energetic particle radiation in transit to Mars on the Mars Science Laboratory. Science. 2013;340:1080-1084
35. Steele CB, Rim SH, Joseph DA, King JB, Seeff LC. Colorectal cancer incidence and screening - United States, 2008 and 2010. MMWR Surveill Summ. 2013;62(Suppl 3):53-60
36. Haggar FA, Boushey RP. Colorectal cancer epidemiology: incidence, mortality, survival, and risk factors. Clin Colon Rectal Surg. 2009;22:191-197
37. Cragle DL, McLain RW, Qualters JR, Hickey JL, Wilkinson GS, Tankersley WG. et al. Mortality among workers at a nuclear fuels production facility. Am J Ind Med. 1988;14:379-401
38. Dupree-Ellis E, Watkins J, Ingle JN, Phillips J. External radiation exposure and mortality in a cohort of uranium processing workers. Am J Epidemiol. 2000;152:91-95
39. Preston DL, Shimizu Y, Pierce DA, Suyama A, Mabuchi K. Studies of mortality of atomic bomb survivors. Report 13: Solid cancer and noncancer disease mortality: 1950-1997. Radiat Res. 2003;160:381-407
40. Reynolds P, Austin DF. Cancer incidence among employees of the Lawrence Livermore National Laboratory, 1969-1980. West J Med. 1985;142:214-218
41. Nottage K, McFarlane J, Krasin MJ, Li C, Srivastava D, Robison LL. et al. Secondary colorectal carcinoma after childhood cancer. J Clin Oncol. 2012;30:2552-2558
42. Rapiti E, Fioretta G, Verkooijen HM, Zanetti R, Schmidlin F, Shubert H. et al. Increased risk of colon cancer after external radiation therapy for prostate cancer. Int J Cancer. 2008;123:1141-1145
43. Hall EJ, Brenner DJ. Cancer risks from diagnostic radiology. Br J Radiol. 2008;81:362-378
44. Brenner DJ, Hall EJ. Computed tomography-an increasing source of radiation exposure. N Engl J Med. 2007;357:2277-2284
45. Jacob P, Meckbach R, Kaiser JC, Sokolnikov M. Possible expressions of radiation-induced genomic instability, bystander effects or low-dose hypersensitivity in cancer epidemiology. Mutat Res. 2010;687:34-39
46. Kaiser JC, Meckbach R, Jacob P. Genomic instability and radiation risk in molecular pathways to colon cancer. PLoS One. 2014;9:e111024
47. Ohno T. Particle radiotherapy with carbon ion beams. EPMA J. 2013;4:9
48. Tsujii H, Kamada T. A review of update clinical results of carbon ion radiotherapy. Jpn J Clin Oncol. 2012;42:670-685
49. Cheema AK, Suman S, Kaur P, Singh R, Fornace AJJ, Datta K. Long-term differential changes in mouse intestinal metabolomics after gamma and heavy ion radiation exposure. PLoS One. 2014;9:e87079
50. Denisova NA, Shukitt-Hale B, Rabin BM, Joseph JA. Brain signaling and behavioral responses induced by exposure to (56)Fe-particle radiation. Radiat Res. 2002;158:725-734
51. Hall EJ, Brenner DJ, Worgul B, Smilenov L. Genetic susceptibility to radiation. Adv Space Res. 2005;35:249-253
52. Manda K, Ueno M, Anzai K. Memory impairment, oxidative damage and apoptosis induced by space radiation: ameliorative potential of alpha-lipoic acid. Behav Brain Res. 2008;187:387-395
53. Manda K, Ueno M, Anzai K. Space radiation-induced inhibition of neurogenesis in the hippocampal dentate gyrus and memory impairment in mice: ameliorative potential of the melatonin metabolite, AFMK. J Pineal Res. 2008;45:430-438
54. Datta K, Suman S, Kallakury BV, Fornace AJJ. Exposure to heavy ion radiation induces persistent oxidative stress in mouse intestine. PLoS One. 2012;7:e42224
55. Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol. 2004;20:781-810
56. Rao TP, Kuhl M. An updated overview on Wnt signaling pathways: a prelude for more. Circ Res. 2010;106:1798-1806
57. Bienz M, Clevers H. Linking colorectal cancer to Wnt signaling. Cell. 2000;103:311-320
58. Eletr ZM, Wilkinson KD. Regulation of proteolysis by human deubiquitinating enzymes. Biochim Biophys Acta. 2014;1843:114-128
59. Rubinsztein DC. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature. 2006;443:780-786
60. Mani A, Gelmann EP. The ubiquitin-proteasome pathway and its role in cancer. J Clin Oncol. 2005;23:4776-4789
61. Hicke L. Protein regulation by monoubiquitin. Nat Rev Mol Cell Biol. 2001;2:195-201
62. Forster F, Unverdorben P, Sledz P, Baumeister W. Unveiling the long-held secrets of the 26S proteasome. Structure. 2013;21:1551-1562
63. Bedford L, Paine S, Sheppard PW, Mayer RJ, Roelofs J. Assembly, structure, and function of the 26S proteasome. Trends Cell Biol. 2010;20:391-401
64. Pickart CM, Cohen RE. Proteasomes and their kin: proteases in the machine age. Nat Rev Mol Cell Biol. 2004;5:177-187
65. Zhou P. Targeted protein degradation. Curr Opin Chem Biol. 2005;9:51-55
66. Kitagawa K, Kotake Y, Kitagawa M. Ubiquitin-mediated control of oncogene and tumor suppressor gene products. Cancer Sci. 2009;100:1374-1381
67. Chen D, Kon N, Li M, Zhang W, Qin J, Gu W. ARF-BP1/Mule is a critical mediator of the ARF tumor suppressor. Cell. 2005;121:1071-1083
68. Ramadan K, Meerang M. Degradation-linked ubiquitin signal and proteasome are integral components of DNA double strand break repair: New perspectives for anti-cancer therapy. FEBS Lett. 2011;585:2868-2875
69. Harhaj EW, Dixit VM. Deubiquitinases in the regulation of NF-kappaB signaling. Cell Res. 2011;21:22-39
70. Tanaka T. Colorectal carcinogenesis: Review of human and experimental animal studies. J Carcinog. 2009;8:5
71. Fodde R. The APC gene in colorectal cancer. Eur J Cancer. 2002;38:867-871
72. Armaghany T, Wilson JD, Chu Q, Mills G. Genetic alterations in colorectal cancer. Gastrointest Cancer Res. 2012;5:19-27
73. Voutsadakis IA. The ubiquitin-proteasome system and signal transduction pathways regulating Epithelial Mesenchymal transition of cancer. J Biomed Sci. 2012;19:67
74. Salvat C, Jariel-Encontre I, Acquaviva C, Omura S, Piechaczyk M. Differential directing of c-Fos and c-Jun proteins to the proteasome in serum-stimulated mouse embryo fibroblasts. Oncogene. 1998;17:327-337
75. Cirit M, Grant KG, Haugh JM. Systemic perturbation of the ERK signaling pathway by the proteasome inhibitor, MG132. PLoS One. 2012;7:e50975
76. Wu YT, Ouyang W, Lazorchak AS, Liu D, Shen HM, Su B. mTOR complex 2 targets Akt for proteasomal degradation via phosphorylation at the hydrophobic motif. J Biol Chem. 2011;286:14190-14198
77. Wong NA, Pignatelli M. Beta-catenin-a linchpin in colorectal carcinogenesis? Am J Pathol. 2002;160:389-401
78. Lemieux E, Cagnol S, Beaudry K, Carrier J, Rivard N. Oncogenic KRAS signalling promotes the Wnt/beta-catenin pathway through LRP6 in colorectal cancer. Oncogene. 2014
79. Liu J, Stevens J, Rote CA, Yost HJ, Hu Y, Neufeld KL. et al. Siah-1 mediates a novel beta-catenin degradation pathway linking p53 to the adenomatous polyposis coli protein. Mol Cell. 2001;7:927-936
80. Neufeld KL, Zhang F, Cullen BR, White RL. APC-mediated downregulation of beta-catenin activity involves nuclear sequestration and nuclear export. EMBO Rep. 2000;1:519-523
81. Shah S, Hecht A, Pestell R, Byers SW. Trans-repression of beta-catenin activity by nuclear receptors. J Biol Chem. 2003;278:48137-48145
82. Xiao JH, Ghosn C, Hinchman C, Forbes C, Wang J, Snider N. et al. Adenomatous polyposis coli (APC)-independent regulation of beta-catenin degradation via a retinoid X receptor-mediated pathway. J Biol Chem. 2003;278:29954-29962
83. Polakis P. More than one way to skin a catenin. Cell. 2001;105:563-566
84. Matsuzawa SI, Reed JC. Siah-1, SIP, and Ebi collaborate in a novel pathway for beta-catenin degradation linked to p53 responses. Mol Cell. 2001;7:915-926
85. Zhang XK, Su Y, Chen L, Chen F, Liu J, Zhou H. Regulation of the nongenomic actions of retinoid X receptor-alpha by targeting the coregulator-binding sites. Acta Pharmacol Sin. 2015;36:102-112
86. Wu Y, Cai Y, Aguilo J, Dai T, Ao Y, Wan YJ. RXRalpha mRNA expression is associated with cell proliferation and cell cycle regulation in Hep3B cell. Exp Mol Pathol. 2004;76:24-28
87. Aypar U, Morgan WF, Baulch JE. Radiation-induced epigenetic alterations after low and high LET irradiations. Mutat Res. 2011;707:24-33
88. Woloschak GE, Chang-Liu CM. Differential modulation of specific gene expression following high- and low-LET radiations. Radiat Res. 1990;124:183-187
89. Stahl S, Fung E, Adams C, Lengqvist J, Mork B, Stenerlow B. et al. Proteomics and pathway analysis identifies JNK signaling as critical for high linear energy transfer radiation-induced apoptosis in non-small lung cancer cells. Mol Cell Proteomics. 2009;8:1117-1129
90. Whalen MK, Gurai SK, Zahed-Kargaran H, Pluth JM. Specific ATM-mediated phosphorylation dependent on radiation quality. Radiat Res. 2008;170:353-364
91. Maroschik B, Gurtler A, Kramer A, Rossler U, Gomolka M, Hornhardt S. et al. Radiation-induced alterations of histone post-translational modification levels in lymphoblastoid cell lines. Radiat Oncol. 2014;9:15
92. Dent P, Yacoub A, Contessa J, Caron R, Amorino G, Valerie K. et al. Stress and radiation-induced activation of multiple intracellular signaling pathways. Radiat Res. 2003;159:283-300
93. Braunstein S, Badura ML, Xi Q, Formenti SC, Schneider RJ. Regulation of protein synthesis by ionizing radiation. Mol Cell Biol. 2009;29:5645-5656
94. Rothman S. How is the balance between protein synthesis and degradation achieved? Theor Biol Med Model. 2010;7:25
95. Zhao Y, Brickner JR, Majid MC, Mosammaparast N. Crosstalk between ubiquitin and other post-translational modifications on chromatin during double-strand break repair. Trends Cell Biol. 2014;24:426-434
96. Xie A, Odate S, Chandramouly G, Scully R. H2AX post-translational modifications in the ionizing radiation response and homologous recombination. Cell Cycle. 2010;9:3602-3610
97. Abraham J, Kelly J, Thibault P, Benchimol S. Post-translational modification of p53 protein in response to ionizing radiation analyzed by mass spectrometry. J Mol Biol. 2000;295:853-864
98. McBride WH, Iwamoto KS, Syljuasen R, Pervan M, Pajonk F. The role of the ubiquitin/proteasome system in cellular responses to radiation. Oncogene. 2003;22:5755-5773
99. Davies MJ. The oxidative environment and protein damage. Biochim Biophys Acta. 2005;1703:93-109
100. Gould N, Doulias PT, Tenopoulou M, Raju K, Ischiropoulos H. Regulation of protein function and signaling by reversible cysteine S-nitrosylation. J Biol Chem. 2013;288:26473-26479
101. Suman S, Rodriguez OC, Winters TA, Fornace AJJ, Albanese C, Datta K. Therapeutic and space radiation exposure of mouse brain causes impaired DNA repair response and premature senescence by chronic oxidant production. Aging (Albany NY). 2013;5:607-622
102. Machado A, Ayala A, Gordillo E, Revilla E, Santa Maria C. Relationship between enzymatic activity loss and post-translational protein modification in aging. Arch Gerontol Geriatr. 1991;12:187-197
103. Zhang B, Wang Y, Pang X, Su Y, Ai G, Wang T. ER stress induced by ionising radiation in IEC-6 cells. Int J Radiat Biol. 2010;86:429-435
104. Schroder M, Kaufman RJ. ER stress and the unfolded protein response. Mutat Res. 2005;569:29-63
105. Mehlhase J, Grune T. Proteolytic response to oxidative stress in mammalian cells. Biol Chem. 2002;383:559-567
106. Datta K, Suman S, Fornace AJJ. Radiation persistently promoted oxidative stress, activated mTOR via PI3K/Akt, and downregulated autophagy pathway in mouse intestine. Int J Biochem Cell Biol. 2014;57:167-176
107. Somosy Z, Takats A, Bognar G, Kovacs AL, Telbisz A, Racz A. et al. X-irradiation-induced changes of the prelysosomal and lysosomal compartments and proteolysis in HT-29 cells. Scanning Microsc. 1996;10:1079-90 discussion 1090
108. Nizard C, Bulteau AL, Moreau M, Saunois A, Friguet B. Proteasome: a new target of UV radiations. Eur J Dermatol. 2002;12:XXVII-XXVIII
109. Pajonk F, McBride WH. Ionizing radiation affects 26s proteasome function and associated molecular responses, even at low doses. Radiother Oncol. 2001;59:203-212
110. Pajonk F, McBride WH. The proteasome in cancer biology and treatment. Radiat Res. 2001;156:447-459
111. McBride WH, Pajonk F, Chiang CS, Sun JR. NF-kappa B, cytokines, proteasomes, and low-dose radiation exposure. Mil Med. 2002;167:66-67
112. Bulteau AL, Moreau M, Nizard C, Friguet B. Impairment of proteasome function upon UVA- and UVB-irradiation of human keratinocytes. Free Radic Biol Med. 2002;32:1157-1170
113. Bulteau AL, Szweda LI, Friguet B. Age-dependent declines in proteasome activity in the heart. Arch Biochem Biophys. 2002;397:298-304
114. Conconi M, Friguet B. Proteasome inactivation upon aging and on oxidation-effect of HSP 90. Mol Biol Rep. 1997;24:45-50
115. Zaiss DM, Standera S, Kloetzel PM, Sijts AJ. PI31 is a modulator of proteasome formation and antigen processing. Proc Natl Acad Sci U S A. 2002;99:14344-14349
116. Suman S, Kallakury BV, Fornace AJJ, Datta K. Protracted Upregulation of Leptin and IGF1 is Associated with Activation of PI3K/Akt and JAK2 Pathway in Mouse Intestine after Ionizing Radiation Exposure. Int J Biol Sci. 2015;11:274-283
117. Eguchi K, Inada T, Yaguchi M, Satoh S, Kaneko I. Induction and repair of DNA lesions in cultured human melanoma cells exposed to a nitrogen-ion beam. Int J Radiat Biol Relat Stud Phys Chem Med. 1987;52:115-123
118. Spotheim-Maurizot M, Davidkova M. Radiation damage to DNA in DNA-protein complexes. Mutat Res. 2011;711:41-48
119. Trani D, Moon BH, Kallakury B, Hartmann DP, Datta K, Fornace AJJ. Sex-dependent differences in intestinal tumorigenesis induced in Apc1638N/+ mice by exposure to gamma rays. Int J Radiat Oncol Biol Phys. 2013;85:223-229
120. Suman S, Kumar S, Fornace AJJ, Datta K. Decreased RXRα is associated with increased β-catenin/TCF4 in 56-Fe-induced intestinal tumors. Front. Oncol. 2015;5:218. doi: 10.3389/fonc.2015.00218
121. Datta K, Suman S, Trani D, Doiron K, Rotolo JA, Kallakury BV. et al. Accelerated hematopoietic toxicity by high energy (56)Fe radiation. Int J Radiat Biol. 2012;88:213-222
122. Suman S, Moon BH, Thakor H, Fornace AJJ, Datta K. Wip1 abrogation decreases intestinal tumor frequency in APC(Min/+) mice irrespective of radiation quality. Radiat Res. 2014;182:345-349
123. Lu X, Nguyen TA, Zhang X, Donehower LA. The Wip1 phosphatase and Mdm2: cracking the “Wip” on p53 stability. Cell Cycle. 2008;7:164-168
124. Korolchuk VI, Mansilla A, Menzies FM, Rubinsztein DC. Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol Cell. 2009;33:517-527
125. Korolchuk VI, Menzies FM, Rubinsztein DC. A novel link between autophagy and the ubiquitin-proteasome system. Autophagy. 2009;5:862-863
126. Korolchuk VI, Menzies FM, Rubinsztein DC. Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett. 2010;584:1393-1398
Author contact
![]() Corresponding author: Kamal Datta, M.D., Associate Professor, Department of Biochemistry and Molecular & Cellular Biology, Georgetown University, Research Building, Room E518, 3970 Reservoir Rd., NW, Washington, DC 20057, USA. Email: kd257edu
Corresponding author: Kamal Datta, M.D., Associate Professor, Department of Biochemistry and Molecular & Cellular Biology, Georgetown University, Research Building, Room E518, 3970 Reservoir Rd., NW, Washington, DC 20057, USA. Email: kd257edu