Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2015; 6(12):1195-1205. doi:10.7150/jca.12452 This issue Cite
Research Paper
Vertical Targeting of AKT and mTOR as Well as Dual Targeting of AKT and MEK Signaling Is Synergistic in Hepatocellular Carcinoma
Florian Ewald1* ![]() , Dominik Nörz2*, Astrid Grottke2, Johanna Bach2, Christiane Herzberger2, Bianca T. Hofmann2,#, Björn Nashan1, Manfred Jücker2
, Dominik Nörz2*, Astrid Grottke2, Johanna Bach2, Christiane Herzberger2, Bianca T. Hofmann2,#, Björn Nashan1, Manfred Jücker2
1. Department of Hepatobiliary and Transplant Surgery, University Medical Center Hamburg-Eppendorf
2. Center of Experimental Medicine, Institute of Biochemistry and Signal Transduction, University Medical Center Hamburg-Eppendorf, Martinistr. 52, 20246 Hamburg, Germany.
* F.E. and D.N. contributed equally to this work
# Present address: Department of General, Visceral and Thoracic Surgery, University Medical Center Hamburg-Eppendorf, Martinistr. 52, 20246 Hamburg, Germany.
Received 2015-4-20; Accepted 2015-6-23; Published 2015-9-16
Abstract
Hepatocellular carcinoma (HCC) is the sixth most common cancer, and the third most common cause of cancer related death worldwide. The multi-kinase inhibitor Sorafenib represents the only systemic treatment option until today, and results from clinical trials with allosteric mTOR inhibitors were sobering. Since the PI3K/AKT/mTOR and RAF/MEK/ERK signaling pathways are frequently upregulated in HCC, we have analyzed the effects of AKT inhibitor MK-2206, MEK inhibitor AZD6244 (ARRY 142886) and mTOR kinase inhibitor AZD8055, given as single drugs or in combination, on proliferation and apoptosis of three HCC cell lines in vitro. We show that all three inhibitor combinations synergistically inhibit proliferation of the three HCC cell lines, with the strongest synergistic effect observed after vertical inhibition of AKT and mTORC1/2. We demonstrate that AKT kinase activity is restored 24h after blockade of mTORC1/2 by increased phosphorylation of T308, providing a rationale for combined targeting of AKT and mTOR inhibition in HCC. Our data suggest that a combination of inhibitors targeting those respective pathways may be a viable approach for future application in patients with hepatocellular carcinoma.
Keywords: Hepatocellular carcinoma, AKT, mTOR, MEK.
Introduction
Hepatocellular Carcinoma (HCC) represents the third most common cause of cancer related mortality, being the sixth most common cancer worldwide [1]. The greatest burden of the disease is concentrated in Asia and sub-Saharian Africa, where incidence rates are up to five times higher compared to Europe or North America [2], mostly due to the high prevalence of relevant risk factors for the development of HCC, including hepatitis B (HBV) and hepatitis C virus (HCV) infections in these countries [3]. However, incidence rates for HCC in North America have tripled since 1975 [4]. Treatment approaches depend on the stage of the tumor at diagnosis, and the only curative treatment options being surgical resection or liver transplantation [5]. There are no curative treatment approaches for advanced staged HCC, and the only FDA-approved systemic treatment available to date is Sorafenib, a multi-kinase inhibitor with modest efficacy in increasing quality-adjusted life-years [1, 6]. Therefore, new effective treatment strategies are urgently needed. In a study using immunohistochemical analysis of HCC tissue samples, activation of the PI3K/AKT/mTOR signaling pathway was frequently detected, i.e. activation of AKT was detected in 71,5%, and activation of mTOR in 47,5% of HCC samples analyzed [7]. AKT, also referred to as Protein kinase B, plays a pivotal role within the PI3K/AKT/mTOR pathway and numerous cellular functions, including proliferation, survival and migration [8]. Mammalian target of rapamycin (mTOR) is a downstream target of PI3K/AKT and acts as an integrator for a variety of stimuli, including mitogens as well as energy- and nutrient-levels, and takes influence on translation, proliferation and autophagy [9]. There is a complex interaction between AKT and mTOR, given that mTORC2 phosphorylates AKT within the carboxyterminus, which is required for full kinase activity of AKT, and AKT in turn controlls mTOR activity via regulation of the TSC1/2-complex [10-12]. Activation of the PI3K/AKT/mTOR pathway has been shown to be related to a poor overall prognosis in gastrointestinal and gynecological carcinoma [13]. Specifically in HCC, mTOR activation appears to be associated with less differentiated tumors, poor survival and early recurrence after resection [14]. Allosteric inhibitors of mTOR have been in the focus of oncological research for a long time [15]. However, recent results from the EVOLVE-1 trial using RAD001 as monotherapy in advanced HCC were desillusionating, since no significant difference in overall survival could be detected [16]. With an emerging understanding of the importance of mTORC2 signaling in tumorigenesis, compounds like the novel, highly selective, ATP competitive mTOR inhibitor AZD8055, that targets both mTORC1 and mTORC2, might therefore provide a therapeutic superiority compared to rapalogs, which primarily inhibit mTORC1 signaling [11, 17]. In this context, a feedback mechanism was demonstrated which restores a substantial part of AKT activity even after effective blockade of mTORC2 [18, 19]. To further address the functional role of AKT and mTOR in HCC cell lines, we analyzed the combined effects of AZD8055 and the allosteric AKT inhibitor MK-2206, that is currently being evaluated in numerous clinical trials [20].
The RAF/MEK/ERK signaling pathway plays a critical role in cancer development and progression, and was shown to be activated in up to 58% of all HCC samples analyzed [21-23]. Extracellular signal-regulated kinase (ERK) is a downstream kinase of many cell surface receptors, including EGFR, IGFR, MET and others [24], and has a wide range of substrates, which ultimately promote proliferation, cell survival, invasion and migration [25]. AZD6244 (ARRY-142886), also referred to as Selumetinib, is a selective allosteric inhibitor of the MEK1/2 kinases and can be used to disrupt downstream signaling to ERK. The efficacy of AZD6244 alone or combined with Sorafenib has already been demonstrated in a xenograft HCC model, and clinical trials have been initiated [26-28]. Both, the RAF/MEK/ERK and the PI3K/AKT/mTOR pathways play an important role in the control of cell proliferation and survival. There is evidence of a diligent cross-regulation between these two pathways and results from previous studies suggest a high level of functional redundancy between them [29]. Therefore, simultaneous inhibition of both pathways appears to be reasonable and has been shown to be effective in non-small-cell lung carcinoma (NSCLC) cell lines in both in vitro and in vivo experiments [30]. In this study, we demonstrate that combined targeting of AKT, mTOR and MEK/ERK using MK-2206, AZD6244 and AZD8055 is efficacious and synergistic in the inhibition of HCC cell proliferation. Our results suggest that dual targeting of AKT and mTOR, as well as AKT and MEK might be a promising therapeutic approach in the treatment of hepatocellular carcinoma.
Material and Methods
Chemicals and reagents
AZD8055 and AZD6244 were obtained from SelleckChem (Absource Diagnostics GmbH, Munich, Germany). MK-2206 was obtained from AbMole BioScience (Kowloon, Hongkong). Stock solutions with a concentration of 10 mM were prepared and stored at -80 °C. Antibodies against pan AKT, AKT1, AKT2, pAKT (S473), pAKT (T308), mTOR, pmTOR (S2448), pmTOR (S2481), pERK (T202/Y204), ERK, pMEK (S217/221), MEK 1/2, pGSK3-beta (S9), Cyclin D3, 4EBP-1, SKP-2 and pS6 (S240/244) were purchased from Cell Signaling Technology (Danvers, MA, USA). Antibodies against p27, PTEN and HSC-70 were purchased from Santa Cruz. Propidium Iodide (PI) was obtained from Sigma (Taufkirchen, Germany).
Cell culture
The three hepatocellular carcinoma cell lines Hep3B, HepG2 [31] and Huh-7 [32] were a kind gift from Prof. Dr. H. Will at the Heinrich-Pette institute in Hamburg, Germany. All cell lines were maintained in Dulbecco's Modified Eagle's Medium (DMEM), supplemented with 10% (v/v) fetal calf serum (FCS), and 1% (v/v) penicillin and streptomycin. Cells were cultured at 37°C in a humidified atmosphere containing 5% CO2. All cell lines were used at low passage number not exceeding 30 passages, except for a model of acquired MEK inhibitor resistance. For experiments including cells that underwent prolonged MEK-inhibitor treatment, HepG2 cells were cultured in standard DMEM medium with AZD6244 added to a final concentration of 5µM. Cells were maintained under these conditions for six months before experiments were carried out.
Western blot analysis
Western blot analysis was performed as described previously [33]. Protein expression was quantified using an LAS-3000 Imager from Fuji (Raytest, Straubenhardt, Germany).
Lentiviral knockdown of AKT isoforms
pLKO.1-puro vector encoding AKT1, AKT2 and non-target (scrambled, SCR) shRNA were purchased from Sigma-Aldrich (Taufkirchen, Germany). For double AKT isoform knockdown, puromycin resistance in the AKT2 and the control vector was exchanged for neomycin resistance (kind gift of Prof. Fehse, UKE Hamburg). Generation of pseudotyped lentiviruses and transduction were performed as previously described [33, 34]. Cells transduced with AKT1 shRNA containing vectors were selected by addition of puromycin (Sigma-Aldrich, Taufkirchen, Germany) to culture medium with a final concentration of 1.5 μg/ml for at least one week, followed by sequential transduction with an AKT2 shRNA containing vector and selection with puromycin (final concentration 1,5 µg/ml) and G418 (final concentration 800µg/ml) containing medium. Controls were transduced sequentially with the control shRNA vectors.
Proliferation, apoptosis, colony formation and cell cycle analysis
Proliferation was analyzed either by flow cytometry using the BrdU APC Flow Kit (BD, Pharmingen, CA, USA) or with the colorimetric BrdU ELISA Kit (Roche, Basel, CH) as indicated. For FACS-based assays, cells were seeded into 10 cm dishes and allowed to attach overnight. Then, medium was replaced by medium containing the respective inhibitor or inhibitor combination. Controls were treated with dimethyl sulfoxide (DMSO) only, and final DMSO concentration in culture medium was 0.1% (v/v) in all experiments. For cell labeling, BrdU was added to a final concentration of 10 µM, and cells were incubated for 12 to 16 h. For cell cycle analysis, cells were fixed in ice cold 70% ethanol for at least 6 h, washed and subsequently incubated with 5 µg PI and 5 µg RNAse A for one hour. Each experiment was performed in triplicates and has been repeated at least one time. Analysis was performed on BD Canto flow cytometer (BD Pharmingen, CA, USA). Cell cycle analysis was performed using FlowJo 7.6.5 software.
For BrdU ELISA assays, cells were seeded into 96-well plates and allowed to attach overnight. Cells were then incubated for 72 h with the respective compounds, and controls were treated with DMSO only. For apoptosis assays, cells were seeded into 96-well plates and grown in culture medium supplemented with 0.1 % FCS (v/v) before incubation with the different compounds for 24h. BrdU ELISA and Cell Death Detection ELISA plus (Roche, Basel, CH) were performed as described by the manufacturer. Each experiment was repeated at least three times in triplicates.
Immunoprecipitation and AKT isoform specific in vitro kinase assay
Immunoprecipitation of AKT using a pan AKT antibody and subsequent in vitro kinase assay was performed as described before [34, 35].
Statistical Analysis
Student's t-Test (unpaired, 2-tailed) or Kruskal-Wallis test was calculated based on the data of at least three independent experiments. Bonferroni correction for multiple testing was performed where applicable. Results were considered significant if p<0.05. All error bars represent SD, unless indicated otherwise. Drug interactions were analyzed based on the median effect method of Chou and Talalay [36]. CalcuSyn software (Biosoft, Cambridge, UK) was used to calculate a Combination Index (CI) for each combination point. CI values from 0.3 to 0.7 are considered to indicate synergism, CI values below 0.3 are considered to represent strong, and values below 0.1 very strong synergism. The CI values were used to draw a plot of CI values over a range of fractions affected as described [36]. IC50 values, i.e. the concentration of a compound that inhibits response by 50% corresponding to the Fraction affected (Fa) of 0.5, were calculated using CurveExpert Professional 1.3 software.
Results
Combined inhibition of AKT and MEK or mTOR is synergistic in HCC cell lines
We first analyzed the activity of the PI3K/AKT/mTOR and RAF/MEK/ERK signaling pathways in the three HCC cell lines Hep3B, HepG2 and Huh-7. Constitutive activation of both pathways was detected by Western blot analysis, as previously described (Figure S1 and [37]). We then analyzed the efficacy of the MEK inhibitor AZD6244 and the mTOR kinase inhibitor AZD8055 in suppressing the activity of their corresponding downstream targets ERK and S6, as shown in Figure S1. Of note, AZD6244 was unable to suppress phosphorylation of ERK at T202/Y204 even at 1000nM, probably due to a relief of the feedback inhibition of BRAF, as indicated by the concomitant increase in pMEK [38]. MK-2206 has been evaluated in the three HCC cell lines before as published by our group in [37]. To assess the impact of AZD6244 and AZD8055 on proliferation, the three HCC cell lines were treated with a broad range of concentrations over 72h, and proliferation was measured by BrdU incorporation (Figure S2). AZD8055 caused a strong reduction in growth of Hep3B and Huh-7, with IC50 values of 47.9 nM and 24.5 nM respectively. However, AZD8055 failed to induce a fifty percent inhibition of proliferation in HepG2 cells even at a concentration of 1000 nM. Conversely, HepG2 cells were highly susceptible for the MEK-Inhibitor AZD6244, with an IC50 value of 27.8 nM, whereas IC50 value was 3031 nM for Huh7 cells. Hep3B appeared to be intrinsically resistant to AZD6244 with an IC50 value of greater than 10 µM and minimal influence on proliferation even at high concentrations. The effects of MK-2206 on these HCC cell lines were described before by our group [37]. Next, we analyzed the efficacy of combining AKT and mTOR, AKT and MEK, or mTOR and MEK inhibition in these cell lines (Figure 1).
Combined treatment of HCC cell lines with combinations of MK-2206, AZD6244 and AZD8055 synergistically suppresses proliferation. HCC cells were seeded into 96-well plates and incubated with increasing concentrations of AKT inhibitor MK-2206, MEK inhibitor AZD6244 or mTOR inhibitor AZD8055 or a combination of two inhibitors, as indicated over 72 h. Controls were treated with DMSO only. Proliferation was analyzed after 72h by BrdU incorporation. Each data point represents mean of three independent experiments measured in triplicated, normalized to controls; bars, SD. *, p < 0.05
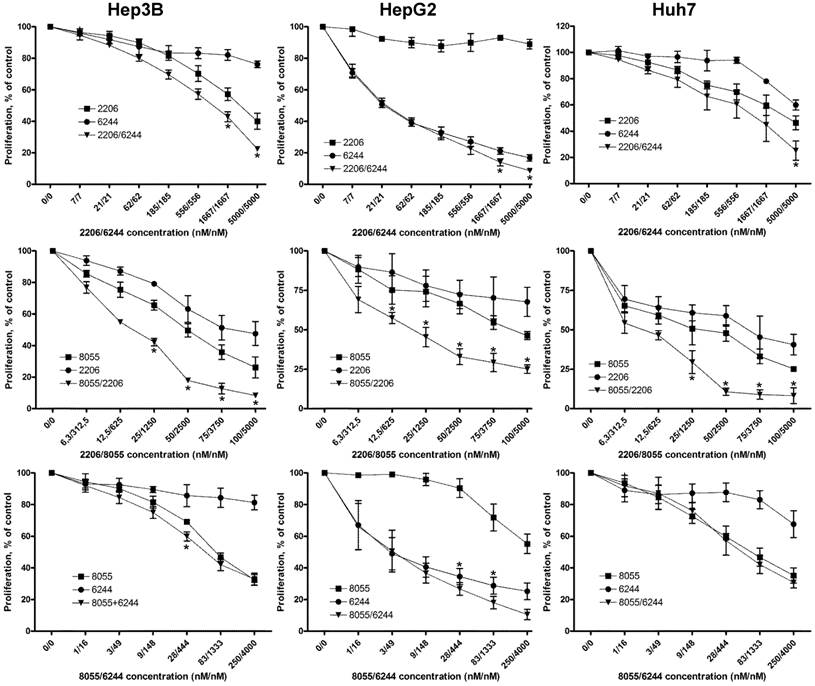
Combined targeting of AKT and MEK synergistically inhibited proliferation of all HCC cell lines, although the synergistic effect appears less prominent in HepG2 cell line because of its high susceptibility to AZD6244. Fractional effect blots for all cell lines and combinations treatments can be found in Figure S3. Similar results have been reported previously in NSCLC, thyroid cancer and cholangicarcinoma cell lines [19, 30, 39].
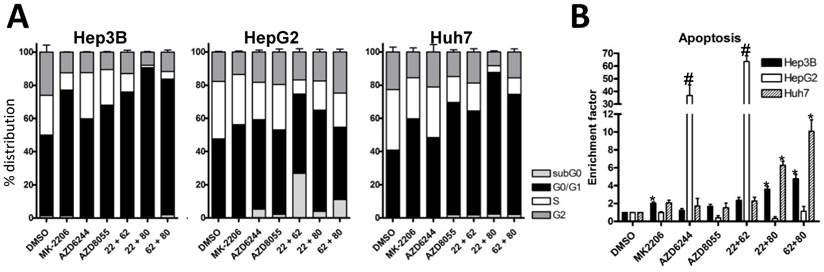
Vertical inhibition of the AKT-mTOR pathway by combining MK-2206 and AZD8055 yielded robust synergistic effects in the three cell lines, even at low concentrations. Finally, combining MEK inhibitor AZD6244 with the mTOR inhibitor AZD8055 resulted in weak synergistic or mere additive effects in Hep3B and Huh7 cells, whereas the combination was strongly synergistic in HepG2 cells. Similar results were observed when we performed cell cycle analysis of HCC cell lines treated with the afore-mentioned compounds and combinations, as seen in Fig. 2A. The combination of MK-2206 and AZD8055, which has shown the most robust impact on proliferation, caused an almost complete reduction of cells in S and G2 phase in Hep3B and Huh-7. Finally, we analyzed the induction of apoptosis in HCC cell lines after treatment with these inhibitors, either alone or in combination (Figure 2B). We observed a strong induction of apoptosis only in HepG2 cells when treated with AZD6244 alone or in combination with 2206.
Effects of combined treatment on cell cycle distribution and apoptosis in three HCC cell lines. (A) Cell cycle analysis of HCC cell lines after 24h treatment with 2 µM MK-2206, 1 µM AZD6244, 100 nM AZD8055, or the combination of two inhibitors, as indicated. Control cells were treated with DMSO only. Columns: mean of one representative experiment performed in triplicates; bars, SD. (B) HCC cells were seeded into 96-well plates and treated with afore mentioned compounds and concentrations for 24 hours, before apoptosis was measured by Cell Death Detection ELISA. Controls were treated with DMSO. Each data point represents three independent experiments, performed in triplicates; bars: SD. *, p <0.05.
Analysis of intracellular signalling by Western blot
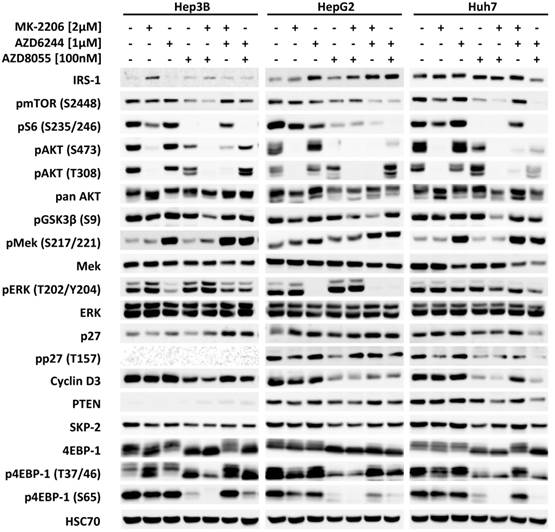
Next, the effects of MK-2206, AZD6244, AZD8055 and their combinations on intracellular signaling in HCC cells were analyzed by western blot, as seen in Figure 3. All inhibitors effectively suppressed the phosphorylation of their respective downstream targets, i.e. AKT, ERK and S6, with the exception of AZD6244 in Huh-7, as described above. Correspondingly, proliferation of Huh-7 cells was almost unaffected at low doses of AZD6244. Figure S4 demonstrates that although pERK is initially suppressed by addition of AZD6244 within the first 6 hours, it then quickly recovers and restores its former level within 12h following treatment onset. Neither the AKT inhibitor MK-2206, nor the mTOR inhibitor AZD8055 alone were able to significantly reduce the level of pGSK3β (S9) in any of the cell lines analyzed, even though both had a distinct impact on pAKT (S473). However, the combination of both compounds resulted in a synergistic inhibition of pGSK3β levels. This combination also resulted in a more thorough reduction of phosphorylation of mTOR substrate 4EBP-1 at S65, compared to AZD8055 alone (Figure 3).
Effects of combined treatment on intracellular signalling in the three HCC cell lines. HCC cell lines were treated with 2 µM MK-2206, 1 µM AZD6244, 75 nM AZD8055, or a combination of two of compounds, as indicated. PI3K-AKT-mTOR and RAF-MEK-ERK signaling pathway activity was analyzed by western blot with antibodies directed against the indicated targets. HSC70 served as loading control.
Inhibition of mTORC2 causes elevated phosphorylation of AKT at threonine residue 308 (T308), accompanied by increased residual AKT activity
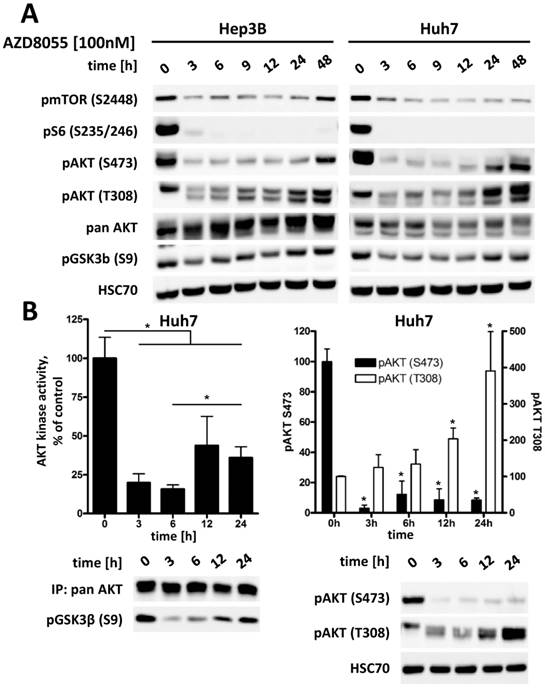
Treatment with mTOR inhibitor AZD8055 results in an effective suppression of phosphorylation of AKT at S473 in all three HCC cell lines, as seen in Figure S1. However, phosphorylation at T308 shows a rapid recovery to its original or even higher level within 24 to 48 hours (Figure 4A). To further investigate the significance of this interaction between AKT and mTOR after mTORC1/2 inhibition in HCC cells, the kinase activity of AKT was measured directly by an in vitro kinase assay. As demonstrated in Figure 4B, addition of AZD8055 reduces AKT activity to approximately 15% compared to baseline after 3 h, but is then restored to about 35% within 24h following treatment. This increase of AKT activity correlates to an increase in pAKT T308 of up to four times compared to baseline, whereas pAKT S473 remains strongly suppressed. We also observed a concomitant increase in pGSK3β S9 over time as a readout of the restoration of AKT activity, as seen in Figure 4A. This is in line with results published by Rodrik-Outmezguine et al [18] and our own findings in cholangicarcinoma cell lines [19], suggesting that the increased phosphorylation of AKT at T308 is sufficient for a residual enzymatic activity of AKT. These results also illustrate the rationale for vertical targeting of AKT and mTOR for cancer therapy.
Inhibition of mTORC1/2 causes upregulation of phospho-AKT at T308, resulting in increased residual AKT activity over time in HCC cell lines. (A) Hep3B and Huh7 cells were treated with either DMSO or 100 nM AZD8055 for up to 48 h and cell lysates were prepared at the indicated time points. PI3K-AKT-mTOR pathway signaling was analyzed by western blot. HSC70 was used as loading control. (B) Huh7 cells were treated with 100 nM AZD8055 for 0, 3, 6, 12 and 24 hours. AKT in vitro kinase assays were performed after quantitative pan AKT immunoprecipitation in triplicates per timepoint. GSK3α/β fusion protein was used as AKT substrate and phosphorylation at S9/21 detected by western blot. pAKT S473 and T308 levels were analyzed by western blot. Bars: SD. *, p < 0.05; #, p < 0.01
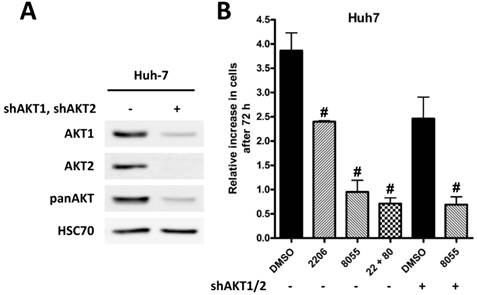
Knockdown of AKT1 and AKT2 in Huh-7 cells is synergistic with inhibition of mTOR
To underline the importance of AKT signaling after mTOR inhibition, we generated Huh-7 AKT1/AKT2 double knockdown cells (Figure 5A). Proliferation of control cells treated with DMSO, MK-2206, AZD8055, or the combination of both, and AKT1/2 double knockdown cells treated with DMSO or AZD8055 was measured by cell counting after 72h. As shown in Figure 5B, combined MK2206 or knockdown of AKT1/2 with AZD8055 results in an additional inhibition of proliferation compared to inhibition of mTOR or AKT alone.
Knock-down of AKT is synergistic with mTORC1/2 inhibition. (A) Knockdown of AKT1 and AKT2 in Huh7 cells was confirmed by Western blot, HSC70 served as loading control. (B) Huh7 SCR and AKT1/2 knockdown cells were incubated with DMSO, 2 µM MK-2206, 100 nM AZD8055, or a combination of both over 72h. After 72h cells were trypsinized and counted, and the relative increase in cell number in given in the graph. The Experiments were performed in triplicates. Bars: SD; #, p < 0.01
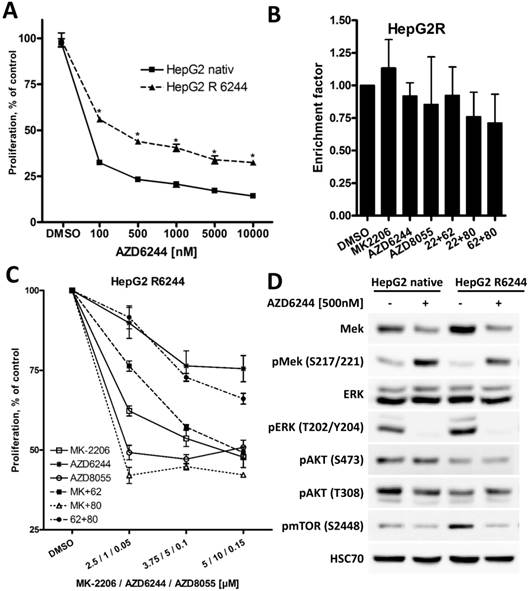
HepG2 cells overcome high apoptosis induction of AZD6244 after prolonged treatment
Among the three HCC cell lines analyzed, HepG2 cells were most susceptible to MEK inhibitor AZD6244, and a strong induction of apoptosis was only observed in these cells, reflecting the importance of ERK1/2 signaling in HepG2 cells harboring a NRAS mutation [40]. To investigate the impact of prolonged exposure to AZD6244, HepG2 cells were cultivated in medium containing 5 µM AZD6244 for six months (these cells will be referred to as HepG2R). As seen in Figure 6A, proliferation of HepG2R cells was still affected by AZD6244, but they were significantly more resistant compared to parental HepG2 cells. Furthermore, AZD6244, alone or in combination with MK-2206 or AZD8055, failed to induce apoptosis in HepG2R cells (Figure 6B). This is in contrast to the strong induction of apoptosis observed in parental HepG2 cells after treatment with these drugs as shown in Figure 2. Rapid development of MEKi resistance has been well documented in other cell lines as well, and reflects a not yet overcome challenge in the clinical application of BRAF and MEK inhibitors [41]. Of note, HepG2R cells were also resistant to MEK inhibitor PD0325901, but HepG2R cells were still susceptible to multi-tyrosine kinase inhibitor Sorafenib, as seen in Figure S5. HepG2R cells were then treated with AZD6244 either in combination with MK-2206 or AZD8055, as shown in Figure 6C. However, in HepG2R cells only the combination of MK-2206 and AZD8055 resulted in a stronger inhibition of proliferation, whereas the combination of AZD6244 with MK-2206, or AZD8055, even had strong antagonistic effects compared to MK-2206 or AZD8055 alone.
Long term exposure of HepG2 cell to AZD6244 results in acquired resistance. (A) HepG2 and AZD6244 resistant HepG2R cells were seeded into 96-well plates and treated with increasing concentrations of AZD6244, controls were treated with DMSO. Proliferation was analyzed after 72h by BrdU incorporation. Each data point represents three independent experiments, each performed in triplicates. Bars: SD; *, p < 0.05. (B) HepG2R cells were treated with 2 µM MK-2206, 1 µM AZD6244, 75 nM AZD8055, or a combination of two of compounds as indicated for 24h, and induction of apoptosis compared to untreated cells was determined using the Cell Death Detection ELISA kit. No significant induction of apoptosis was observed in any experimental condition in HepG2R cells. (C) HepG2R cells were seeded into 96-well plates and treated with the indicated compounds or their combinations. Proliferation was analyzed after 72h by BrdU incorporation. Each data point represents mean of three independent experiments. Bars: SD. (D) HepG2R cells were treated with 500nM AZD6244 or DMSO. PI3K/AKT/mTOR and RAF/MEK/ERK signaling pathway activity was analyzed by Western blot. HSC70 served as loading control.
Discussion
In this study, we aimed to investigate the effects of combined targeting of the PI3K/AKT/mTOR pathway and the RAS/MEK/ERK pathway, as well as vertical targeting of AKT and mTOR in HCC cell lines. Over the past years, preclinical and clinical evidence has accumulated that pharmacological inhibition of single targets will induce clinically relevant responses only in a minority of cancer patients [42]. This might be due to intrinsic resistance of cancer cells [43], as well as feedback mechanisms and redundancy among signaling pathways, alleviating the drug effect [25, 29, 44]. For example, RAD001 was shown to be only effective in KRAS wild type breast cancer cell lines harboring PI3KCA mutations [43], whereas activating mutations in KRAS results in intrinsic resistance. Furthermore, inhibition of AKT using MK-2206 or mTOR using AZD8055 was shown to result in increased phosphorylation of ERK1/2, indicative of a compensatory activation of this signaling pathway [45, 46]. Therefore, inhibitor combinations could be used to disrupt critical feedback mechanisms, to overcome intrinsic drug resistance and therefore induce more profound clinical responses [47, 48]. Combining the AKT inhibitor MK-2206 with the allosteric MEK inhibitor AZD6244 was synergistic in all three HCC cell lines, in line with previous results in another HCC cell line as well as NSCLC cell lines [30, 45]. Of note, HepG2 was the only HCC cell line tested that exhibited a strong induction of apoptosis after treatment with AZD6244. Induction of apoptosis is supposed to be an important effect of this inhibitor [49], and the effect was augmented by the addition of MK-2206. The high susceptibility of HepG2 cells for AZD6244 demonstrates the potential use of NRAS/BRAF mutation status as a biomarker to predict responsiveness to treatment with RAF/MEK inhibitors in HCC patients [50], although these mutations are detected in only 1-2% of all HCC (http://cancer.sanger.ac.uk/cosmic; 03/2015). A list of mutated cancer genes as reported in the COSMIC Cell Lines (http://cancer.sanger.ac.uk/cell_lines) database can be found in Table S1. For HepG2, only CTNNB1 (β-catenin) and NRAS-mutations have been reported [40, 51]. NRASQ61L mutation is the only present aberration known to predict sensitivity to MEK-inhibitor AZD6244 [50]. No correlation between sensitivity to MK-2206, or AZD8055 has been reported for any other mutation listed in Table S1. Copy number alterations of the three HCC cell lines were analyzed by Chen et al. [52], reporting an EGFR-amplification in Hep3B and Huh7 cells. Amplification of EGFR might result in an activation of downstream PI3K/AKT/mTOR signaling. In preclinical models, activation of PI3K/AKT/mTOR signaling correlated with sensitivity to drugs targeting this pathway [53]. However, there is only limited validation for this association in the clinical setting [54]. Taken together, the possibility to predicting the sensitivity of cell lines to inhibitors targeting AKT and mTOR is limited, and reliable biomarkers still need to be identified. We have further demonstrated that AZD6244 was unable to achieve a suppression of pERK at low to moderate doses in the less responsive cell lines Hep3B and Huh7 due to a rapid rebound activation upstream of pERK, restoring pERK levels to its original level. A similar effect was observed in melanoma cells harboring KRAS mutations, correlating with a poor response to MEK inhibition [55]. These finding underline the importance of appropriate patient selection before therapy initiation using molecular targeted inhibitors. We also investigated the effect of long-term exposure of HepG2 cells to AZD6244, demonstrating that HepG2 cells become resistant to the antiproliferative and proapoptotic effect of AZD6244 within a few months. Combining AZD6244 with either MK-2206 or AZD8055 was unable to reverse the acquired resistance against AZD6244, and we observed even an antagonistic interaction in these combinations. However, HepG2R cells appeared to be more susceptible to treatment with the AKT-inhibitor MK-2206 alone, compared to parental HepG2 cells, and the combination of AZD8055 and MK-2206 still had a weak, synergistic inhibitory effect of HepG2R proliferation. We found the combination of AKT inhibitor MK-2206 and mTOR inhibitor AZD8055 to be most effective in all three HCC cell lines analyzed, resulting in a synergistic inhibition of proliferation even at low concentrations. This is in line with previous results from our group as well as others demonstrating the efficacy of vertical targeting [19, 56, 57]. Here, we have shown that AKT activity is significantly reduced after the initial administration of AZD8055, but quickly recovers to around 35% of the baseline value within the first 24 hours of treatment. This process is accompanied by an increase in pAKT T308, although pAKT S473, which is considered to be most important for full AKT activity and is generated by mTOR complex 2, was thoroughly suppressed throughout the whole time course [58]. The increased phosphorylation at T308 is caused by the relief of feedback inhibition on RTK signaling, as previously demonstrated [18]. We assume that this feedback mechanism plays an important role for the synergistic effects of vertical AKT/mTOR inhibition, and we confirmed this interaction using a dual knockdown of AKT1 and AKT2 in Huh7 cells. Combining the MEK inhibitor AZD6244 and the mTOR inhibitor AZD8055 resulted in only weak synergistic effects in the three HCC cell lines tested. The same agents were used in rhabdomyosarcoma cell lines, showing strong synergistic effects due to the suppression of a feedback loop between ERK, mTOR and AKT, which was able to maintain tumor cell viability as long as only one signaling pathway was targeted [59]. A study on uveal melanoma cells demonstrated that the susceptibility for this inhibitor combination depends on further genetic alterations, especially the presence of guanine nucleotide-binding protein, q polypeptide (GNAQ) mutations, rendering cells less sensitive to MEK inhibitors [60]. However, GNAQ-mutations appear to be extremely rare in HCC (http://cancer.sanger.ac.uk/cosmic; 03/2015).
In conclusion, we have demonstrated the efficacy of inhibitors targeting the PI3K/AKT/mTOR-, and the RAS/MEK/ERK pathways in HCC cell lines. Combining the AKT inhibitor MK-2206 with either the MEK inhibitor AZD6244, or the mTOR kinase inhibitor AZD8055 resulted in synergistic effects in all three HCC cell lines. We observed the strongest antitumor potential after combined inhibition of AKT and mTOR in Hep3B and Huh7 cells, whereas HepG2 cells were most susceptible to combined inhibition of MEK and AKT/mTOR. Generally, exploiting synergistic effects by combining two or more targeted therapies might allow treatment protocols with lower inhibitor doses, causing less side effects, while still sustaining sufficient impact on cancer cells. Therefore, our results might be used to guide future preclinical and clinical trials to evaluate new, effective treatment regimens.
Abbreviations
BrdU: 5-bromo-2'-deoxyuridine; CI: combination index; DMSO: dimethyl sulfoxide; HCC: hepatocellular carcinoma; mTOR: mammalian target of rapamycin; PBS: phosphate buffered saline; PI3K: phosphatidylinositol 3-kinase.
Supplementary Material
Table S1, Figures S1-S5.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ferenci P, Fried M, Labrecque D, Bruix J, Sherman M, Omata M. et al. Hepatocellular carcinoma (HCC): a global perspective. Journal of clinical gastroenterology. 2010;44:239-45
2. Venook AP, Papandreou C, Furuse J, de Guevara LL. The incidence and epidemiology of hepatocellular carcinoma: a global and regional perspective. Oncologist. 2010;15(Suppl 4):5-13
3. El-Serag HB. Hepatocellular carcinoma. N Engl J Med. 2011;365:1118-27
4. Altekruse SF, McGlynn KA, Reichman ME. Hepatocellular carcinoma incidence, mortality, and survival trends in the United States from 1975 to 2005. J Clin Oncol. 2009;27:1485-91
5. Forner A, Llovet JM, Bruix J. Hepatocellular carcinoma. Lancet. 2012;379:1245-55
6. Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF. et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378-90
7. Chen JS, Wang Q, Fu XH, Huang XH, Chen XL, Cao LQ. et al. Involvement of PI3K/PTEN/AKT/mTOR pathway in invasion and metastasis in hepatocellular carcinoma: Association with MMP-9. Hepatol Res. 2009;39:177-86
8. Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9:550-62
9. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274-93
10. Inoki K, Corradetti MN, Guan KL. Dysregulation of the TSC-mTOR pathway in human disease. Nat Genet. 2005;37:19-24
11. Sparks CA, Guertin DA. Targeting mTOR: prospects for mTOR complex 2 inhibitors in cancer therapy. Oncogene. 2011;29:3733-44
12. Liu P, Gan W, Inuzuka H, Lazorchak AS, Gao D, Arojo O. et al. Sin1 phosphorylation impairs mTORC2 complex integrity and inhibits downstream Akt signalling to suppress tumorigenesis. Nat Cell Biol. 2013;15:1340-50
13. Ocana A, Vera-Badillo F, Al-Mubarak M, Templeton AJ, Corrales-Sanchez V, Diez-Gonzalez L. et al. Activation of the PI3K/mTOR/AKT pathway and survival in solid tumors: systematic review and meta-analysis. PLoS One. 2014;9:e95219
14. Matter MS, Decaens T, Andersen JB, Thorgeirsson SS. Targeting the mTOR pathway in hepatocellular carcinoma: current state and future trends. J Hepatol. 2014;60:855-65
15. Markman B, Dienstmann R, Tabernero J. Targeting the PI3K/Akt/mTOR pathway--beyond rapalogs. Oncotarget. 2010;1:530-43
16. Zhu AX, Kudo M, Assenat E, Cattan S, Kang YK, Lim HY. et al. Effect of everolimus on survival in advanced hepatocellular carcinoma after failure of sorafenib: the EVOLVE-1 randomized clinical trial. JAMA. 2014;312:57-67
17. Lv X, Ma X, Hu Y. Furthering the design and the discovery of small molecule ATP-competitive mTOR inhibitors as an effective cancer treatment. Expert Opin Drug Discov. 2013;8:991-1012
18. Rodrik-Outmezguine VS, Chandarlapaty S, Pagano NC, Poulikakos PI, Scaltriti M, Moskatel E. et al. mTOR kinase inhibition causes feedback-dependent biphasic regulation of AKT signaling. Cancer Discov. 2011;1:248-59
19. Ewald F, Norz D, Grottke A, Hofmann BT, Nashan B, Jucker M. Dual Inhibition of PI3K-AKT-mTOR- and RAF-MEK-ERK-signaling is synergistic in cholangiocarcinoma and reverses acquired resistance to MEK-inhibitors. Invest New Drugs. 2014
20. Pal SK, Reckamp K, Yu H, Figlin RA. Akt inhibitors in clinical development for the treatment of cancer. Expert Opin Investig Drugs. 2010;19:1355-66
21. Dhillon AS, Hagan S, Rath O, Kolch W. MAP kinase signalling pathways in cancer. Oncogene. 2007;26:3279-90
22. Schmidt CM, McKillop IH, Cahill PA, Sitzmann JV. Increased MAPK expression and activity in primary human hepatocellular carcinoma. Biochem Biophys Res Commun. 1997;236:54-8
23. Ito Y, Sasaki Y, Horimoto M, Wada S, Tanaka Y, Kasahara A. et al. Activation of mitogen-activated protein kinases/extracellular signal-regulated kinases in human hepatocellular carcinoma. Hepatology. 1998;27:951-8
24. Yoon JH, Gwak GY, Lee HS, Bronk SF, Werneburg NW, Gores GJ. Enhanced epidermal growth factor receptor activation in human cholangiocarcinoma cells. J Hepatol. 2004;41:808-14
25. McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Wong EW, Chang F. et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta. 2007;1773:1263-84
26. Huynh H, Soo KC, Chow PK, Tran E. Targeted inhibition of the extracellular signal-regulated kinase kinase pathway with AZD6244 (ARRY-142886) in the treatment of hepatocellular carcinoma. Mol Cancer Ther. 2007;6:138-46
27. Huynh H, Ngo VC, Koong HN, Poon D, Choo SP, Toh HC. et al. AZD6244 enhances the anti-tumor activity of sorafenib in ectopic and orthotopic models of human hepatocellular carcinoma (HCC). J Hepatol. 2009;52:79-87
28. Shen YC, Lin ZZ, Hsu CH, Hsu C, Shao YY, Cheng AL. Clinical trials in hepatocellular carcinoma: an update. Liver Cancer. 2013;2:345-64
29. Mendoza MC, Er EE, Blenis J. The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends Biochem Sci. 2011;36:320-8
30. Meng J, Dai B, Fang B, Bekele BN, Bornmann WG, Sun D. et al. Combination treatment with MEK and AKT inhibitors is more effective than each drug alone in human non-small cell lung cancer in vitro and in vivo. PLoS One. 2010;5:e14124
31. Aden DP, Fogel A, Plotkin S, Damjanov I, Knowles BB. Controlled synthesis of HBsAg in a differentiated human liver carcinoma-derived cell line. Nature. 1979;282:615-6
32. Nakabayashi H, Taketa K, Miyano K, Yamane T, Sato J. Growth of human hepatoma cells lines with differentiated functions in chemically defined medium. Cancer Res. 1982;42:3858-63
33. Grabinski N, Bartkowiak K, Grupp K, Brandt B, Pantel K, Jucker M. Distinct functional roles of Akt isoforms for proliferation, survival, migration and EGF-mediated signalling in lung cancer derived disseminated tumor cells. Cell Signal. 2011;23:1952-60
34. Ewald F, Grabinski N, Grottke A, Windhorst S, Norz D, Carstensen L. et al. Combined targeting of AKT and mTOR using MK-2206 and RAD001 is synergistic in the treatment of cholangiocarcinoma. Int J Cancer. 2013
35. Grabinski N, Bartkowiak K, Grupp K, Brandt B, Pantel K, Jucker M. Distinct functional roles of Akt isoforms for proliferation, survival, migration and EGF-mediated signalling in lung cancer derived disseminated tumor cells. Cell Signal. 2011;23:1952-60
36. Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27-55
37. Grabinski N, Ewald F, Hofmann BT, Staufer K, Schumacher U, Nashan B. et al. Combined targeting of AKT and mTOR synergistically inhibits proliferation of hepatocellular carcinoma cells. Mol Cancer. 2012;11:85
38. Fritsche-Guenther R, Witzel F, Sieber A, Herr R, Schmidt N, Braun S. et al. Strong negative feedback from Erk to Raf confers robustness to MAPK signalling. Mol Syst Biol. 2011;7:489
39. Kandil E, Tsumagari K, Ma J, Abd Elmageed ZY, Li X, Slakey D. et al. Synergistic inhibition of thyroid cancer by suppressing MAPK/PI3K/AKT pathways. J Surg Res. 2013;184:898-906
40. Omerovic J, Hammond DE, Clague MJ, Prior IA. Ras isoform abundance and signalling in human cancer cell lines. Oncogene. 2008;27:2754-62
41. Moriceau G, Hugo W, Hong A, Shi H, Kong X, Yu CC. et al. Tunable-Combinatorial Mechanisms of Acquired Resistance Limit the Efficacy of BRAF/MEK Cotargeting but Result in Melanoma Drug Addiction. Cancer Cell. 2015;27:240-56
42. Wu M, Sirota M, Butte AJ, Chen B. Characteristics of drug combination therapy in oncology by analyzing clinical trial data on ClinicalTrials.gov. Pac Symp Biocomput. 2015:68-79
43. Di Nicolantonio F, Arena S, Tabernero J, Grosso S, Molinari F, Macarulla T. et al. Deregulation of the PI3K and KRAS signaling pathways in human cancer cells determines their response to everolimus. J Clin Invest. 2010;120:2858-66
44. Yamanaka K, Petrulionis M, Lin S, Gao C, Galli U, Richter S. et al. Therapeutic potential and adverse events of everolimus for treatment of hepatocellular carcinoma - systematic review and meta-analysis. Cancer Med. 2013;2:862-71
45. Simioni C, Martelli AM, Cani A, Cetin-Atalay R, McCubrey JA, Capitani S. et al. The AKT inhibitor MK-2206 is cytotoxic in hepatocarcinoma cells displaying hyperphosphorylated AKT-1 and synergizes with conventional chemotherapy. Oncotarget. 2013;4:1496-506
46. Wei F, Zhang Y, Geng L, Zhang P, Wang G, Liu Y. mTOR inhibition induces EGFR feedback activation in association with its resistance to human pancreatic cancer. Int J Mol Sci. 2015;16:3267-82
47. Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, Upadhyay R. et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14:1351-6
48. Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J. et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367:1694-703
49. Meng J, Fang B, Liao Y, Chresta CM, Smith PD, Roth JA. Apoptosis induction by MEK inhibition in human lung cancer cells is mediated by Bim. PLoS One. 2010;5:e13026
50. Thumar J, Shahbazian D, Aziz SA, Jilaveanu LB, Kluger HM. MEK targeting in N-RAS mutated metastatic melanoma. Mol Cancer. 2014;13:45
51. de La Coste A, Romagnolo B, Billuart P, Renard CA, Buendia MA, Soubrane O. et al. Somatic mutations of the beta-catenin gene are frequent in mouse and human hepatocellular carcinomas. Proc Natl Acad Sci U S A. 1998;95:8847-51
52. Chen CF, Hsu EC, Lin KT, Tu PH, Chang HW, Lin CH. et al. Overlapping high-resolution copy number alterations in cancer genomes identified putative cancer genes in hepatocellular carcinoma. Hepatology. 2010;52:1690-701
53. Owonikoko TK, Khuri FR. Targeting the PI3K/AKT/mTOR pathway: biomarkers of success and tribulation. Am Soc Clin Oncol Educ Book. 2013
54. Gonzalez-Angulo AM, Blumenschein GR Jr. Defining biomarkers to predict sensitivity to PI3K/Akt/mTOR pathway inhibitors in breast cancer. Cancer Treat Rev. 2013;39:313-20
55. Lito P, Saborowski A, Yue J, Solomon M, Joseph E, Gadal S. et al. Disruption of CRAF-mediated MEK activation is required for effective MEK inhibition in KRAS mutant tumors. Cancer Cell. 2014;25:697-710
56. Rebecca VW, Alicea GM, Paraiso KH, Lawrence H, Gibney GT, Smalley KS. Vertical inhibition of the MAPK pathway enhances therapeutic responses in NRAS-mutant melanoma. Pigment Cell Melanoma Res. 2014
57. Werzowa J, Koehrer S, Strommer S, Cejka D, Fuereder T, Zebedin E. et al. Vertical inhibition of the mTORC1/mTORC2/PI3K pathway shows synergistic effects against melanoma in vitro and in vivo. J Invest Dermatol. 2010;131:495-503
58. Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098-101
59. Renshaw J, Taylor KR, Bishop R, Valenti M, De Haven Brandon A, Gowan S. et al. Dual blockade of the PI3K/AKT/mTOR (AZD8055) and RAS/MEK/ERK (AZD6244) pathways synergistically inhibits rhabdomyosarcoma cell growth in vitro and in vivo. Clin Cancer Res. 2013;19:5940-51
60. Ho AL, Musi E, Ambrosini G, Nair JS, Deraje Vasudeva S, de Stanchina E. et al. Impact of combined mTOR and MEK inhibition in uveal melanoma is driven by tumor genotype. PLoS One. 2012;7:e40439
61. Futreal PA, Coin L, Marshall M, Down T, Hubbard T, Wooster R. et al. A census of human cancer genes. Nat Rev Cancer. 2004;4:177-83
Author contact
![]() Corresponding author: Florian Ewald, MD, Department of Hepatobiliary and Transplant Surgery, University Medical Center Hamburg-Eppendorf, Martinistr.52, 20246 Hamburg, Germany. f.ewaldde
Corresponding author: Florian Ewald, MD, Department of Hepatobiliary and Transplant Surgery, University Medical Center Hamburg-Eppendorf, Martinistr.52, 20246 Hamburg, Germany. f.ewaldde