Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2015; 6(10):1058-1065. doi:10.7150/jca.12519 This issue Cite
Review
Weaknesses and Pitfalls of Using Mice and Rats in Cancer Chemoprevention Studies
Yukui Ma1 ![]() , Yuping Jia1, Lichan Chen2, Lewis Ezeogu2, Baofa Yu3, Ningzhi Xu4
, Yuping Jia1, Lichan Chen2, Lewis Ezeogu2, Baofa Yu3, Ningzhi Xu4 ![]() , D. Joshua Liao2
, D. Joshua Liao2 ![]()
1. Shandong Academy of Pharmaceutical Sciences, Ji'nan, Shandong 250101, P.R. China
2. Hormel Institute, University of Minnesota, Austin, MN 55912, USA
3. Beijing Baofa Cancer Hospital, Shahe Wangzhuang Gong Ye Yuan, Chang Pin Qu, Beijing 102206, P.R. China
4. Laboratory of Cell and Molecular Biology, Cancer Institute, Chinese Academy of Medical Science, Beijing 100021, P.R. China.
Received 2015-4-27; Accepted 2015-7-19; Published 2015-9-1
Abstract
Many studies, using different chemical agents, have shown excellent cancer prevention efficacy in mice and rats. However, equivalent tests of cancer prevention in humans require decades of intake of the agents while the rodents' short lifespans cannot give us information of the long-term safety. Therefore, animals with a much longer lifespan should be used to bridge the lifespan gap between the rodents and humans. There are many transgenic mouse models of carcinogenesis available, in which DNA promoters are used to activate transgenes. One promoter may activate the transgene in multiple cell types while different promoters are activated at different ages of the mice. These spatial and temporal aspects of transgenes are often neglected and may be pitfalls or weaknesses in chemoprevention studies. The variation in the copy number of the transgene may widen data variation and requires use of more animals. Models of chemically-induced carcinogenesis do not have these transgene-related defects, but chemical carcinogens usually damage metabolic organs or tissues, thus affecting the metabolism of the chemopreventive agents. Moreover, many genetically edited and some chemically-induced carcinogenesis models produce tumors that exhibit cancerous histology but are not cancers because the tumor cells are still mortal, inducer-dependent, and unable to metastasize, and thus should be used with caution in chemoprevention studies. Lastly, since mice prefer an ambient temperature of 30-32°C, it should be debated whether future mouse studies should be performed at this temperature, but not at 21-23°C that cold-stresses the animals.
Keywords: mice and rats, carcinogenesis models
Introduction
Globally, cancer is becoming a greater and greater social and economic problem with the continuing expansion of the aging population [1], because cancer formation requires genetic mutation and cell proliferation, both incremental in many tissues, with a longer lifespan. While finding a cure for cancer is of importance, finding a way to prevent it is obviously an even-better solution. Actually, cancer prevention research has already achieved great progress in the scientific, social and economic aspects in recent decades. For example, decreases in gastric cancer incidences in some countries have been discerned after the refrigerator had come into common use in families decades ago, although inconsistent data still exist [2]. Use of refrigerators in daily life is thought to reduce stomach cancer risk by inhibiting bacterial growth in foods, by increasing fresh fruit and vegetable consumption, and by decreasing salt intake because a lot of pickled vegetable and cured or canned meat are replaced by frozen ones [3-5]. A nation-wide vaccination against hepatitis B virus since the late 1970s and early 1980s in some countries, such as in China, now is starting to result in a decrease in the incidences of not only hepatitis B but also liver cancer [6-8]. In some countries, decades of strict control of tobacco consumptions on both cigarette smoking and non-smoking usage have started to result in decreased lung cancer incidence in non-tobacco users, compared with the users, although globally this cancer is increasing due to complex reasons [9-12]. However, studies of cancer chemoprevention with a single chemical agent has basically not shown any practical progress, albeit there have been a plethora of research papers published with a tremendous financial outlay. For instance, albeit numerous research studies including animal experiments have been published, it still remains uncertain and thus requires further studies in humans whether or not green tea itself or its major components such as EGCG (epigallocatechin gallate) are effective on cancer prevention [13-16]. At least, none of the governmental agencies in the US or other countries have officially recommended green tea or EGCG as cancer preventive agent for all of us, albeit governmental agencies such as NIH have put a huge amount of money on the relevant studies. In our musing, the progress in chemoprevention research is greatly hampered by constraints from studies with mice or rats. In this primer, we describe several common pitfalls and weaknesses of the rodent models pertaining to cancer chemoprevention studies, while other defects that are not closely pertinent to chemoprevention or are common for all other biomedical issues are excluded.
Beyond mice and rats, animals with much longer lifespans are needed for chemoprevention studies
Mice, rats and sometimes hamsters are the most commonly used experimental animals for cancer induction, mainly with radiation, chemical carcinogen and genetic modification. Mice and rats only have about 3 years of lifespan, which is about 1/30 of the human's. Therefore, treatment of mice or rats with an agent for one month may be calculated as an equivalence of 30 months, or 2.5 years, in humans. In most chemoprevention studies published, the tested agent is administered to the rodents for 3-6 months, with some reaching one year. Even if the rodent studies prove the safety and efficacy of one-year intake of an agent, in realty we cannot give it to humans at the same dose, calculated as per kilogram bodyweight, for 30 years, or over one-third of the lifespan, without much more convincing evidence for its long-term safety. Chemoprevention requires decades of intake of the chemical, as it is unlikely that a few months or years of intake of a compound can prevent a sporadic cancer for the rest of the subject's life. Moreover, chemopreventive agents should not have any acute or chronic toxicity, unlike chemotherapeutic drugs that are allowed to have some. Since a short-term safety shown in rodent studies does not guarantee a long-term safety in humans, the current practice is to gradually extend the duration of human trials, which is very wasteful in time, resources and efforts and is somewhat perilous to the subjects' health. In part because of this constraint, until now no any chemical has been approved by the US Food and Drug Administration (USFDA) for ordinary people to prevent cancers, albeit many preventive chemicals such as aspirin and EGCG are available on the shelves of drug and grocery stories. All agents that have been approved are for specific high-risk populations, such as tamoxifen approved for those women with a high risk to develop breast cancer. This constraint created by the lifespan-gap between the rodents and humans has seldom been emphasized and, in our cogitation, can only be solved by using those animal species that have a much longer lifespan, such as cats or dogs that have a lifespan of 15 years or longer, i.e. about 5 times longer than the rodents'.
Naked mole-rats have a lifespan of 30-33 years in the laboratory and thus are considered a better animal species for chemoprevention studies [17;18]. However, this species has unusual biological features, including refractoriness to cancer, diabetes, cardiovascular diseases, immunodeficiency, and atherosclerosis [19;20]. These animals live in colonies [21]; each colony contains a queen and several her “husbands” as breeding partners, and only the queen and the husbands breed [19;20]. Moreover, mole-rats in the wild actually live only about 3 years due to various extrinsic mortalities, mainly the death during fights with other colonies. We opine that these unusual biological and behavioral features [19;20], some of which may not be familiar to many non-experts on these animals, are weaknesses for chemoprevention studies.
The inserted element in genetically edited models may cause problems
Mice are much easier to genetically engineer and thus provide us with many more carcinogenesis models. Although these models are used mainly for studies on cancer mechanisms, they are sometimes used for chemoprevention studies as well. Traditionally, these gene-edited models include transgenic, knockout or knock-in mice in which the gene of interest is ectopically expressed, is inactivated, or is replaced with a mutant, respectively. In transgenic mouse, the ectopic gene, usually just the open reading frame (ORF) of its cDNA, is linked to a DNA regulatory element called promoter (Fig 1). Sometimes the transgenic animal is created to serve as a tool, in which the gene of interest is an enzyme or a regulatory DNA itself. Crossing one tool animal with another allows activate, repress or exchange a gene of interest at a specific cell type of the animal. The so-called Cre/LoxP is a typical of such too systems but, unfortunately, itself can cause toxicity and affect the development [22] or growth [23] of the tumor. Recently, a new genetic tool called CRISPR/Cas9 emerges for inactivating genes in animals. Since this new technique still has off-target problem [24-26], it should be used with caution in chemoprevention study, because some mistakenly targeted genes may affect activation or inactivation of the preventive agent. Besides all man-created animals described above, there are some spontaneously occurring genetic models, as best exemplified by the APC(Min/+) mouse that is derived from a spontaneous mutation in the APC gene for colorectal cancer research [27;28]. These naturally occurring animals do not have the promoter-derived weaknesses described in this paper.
Illustration of transgene construct and copy number. A: The target gene, usually its ORF, is linked to a promoter sequence that helps in targeting the transgene to the tissue or organ of interest. A promoter contains different transcription factor (TF) binding sites (TFB) and is activated upon binding by these TFs that usually act as homo- or hetero-dimers (indicated by the same or different shapes). Often a promoter is not very specific because the TFs bind to and regulate other genes as well. The TFs may also be expressed at different time points of the development, thus regulating the transgene at different ages of the life. B: In traditional technique, the transgene (dot) is randomly inserted into the genome, thus often being in different sites of a chromosome and in multiple chromosomes. If the mouse used as a breeder has two copies of the transgene on chromosome 2, one copy on chromosome 4 and another on chromosome 6 of either the paternal (dark bar) or the maternal (light grey bar) origin, during meiosis one of the two sister chromosomes of each chromosomal pair is randomly assigned to a sperm or egg, making different sperms or eggs having different copy numbers of the transgene (seven different genotypes are illustrated as examples). In reality the number of transgene-carrying pups may not meet the Mendelian inheritance because of natural selection for the healthier genotype.
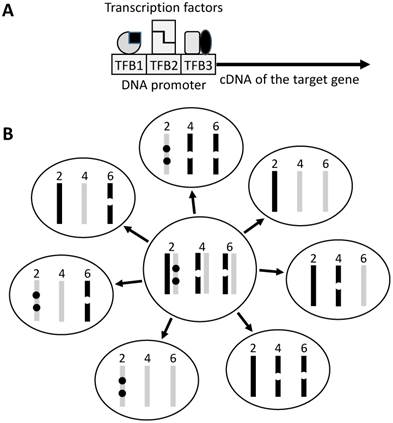
The impact of copy number of the transgene is often forgotten
A founder of a transgenic strain usually contains multiple copies of the transgene that are inserted into different chromosomes or different sites of a chromosome (Fig 1). For instance, an interleukin-5 transgenic mouse line carries 49 copies of the transgene [29], whereas a tumor necrosis factor transgenic mouse line carries 50 copies of the transgene [30]. Both the copy number and the insertion sites at the chromosomes have impacts on the functions of not only the transgene but also the inserted mouse chromosomes. However, of many transgenic models, how many copies of the transgene as well as which chromosomes and where of the chromosomes the transgenes are inserted, are not clear to the animal users.
A somatic mutation carried by a parent has a 50% chance of being passed to an offspring, whereas multiple mutations in different chromosomes are randomly combined and are reduced by different numbers in different sperms or eggs because meiosis reduces the chromosome number by half (Fig 1). Multiple transgene copies resemble multiple mutations in different chromosomes. Therefore, when two heterozygous single-transgene breeders are crossed to engender double-transgenic pups that are heterozygous for each transgene, the copy number of each transgene may be reduced by different numbers in the pups. Polymerase chain reaction (PCR) technique is the most common method used to genotype the pups but it is not sensitive enough to detect the difference in copy number among the pups. Therefore, it is difficult to show, but is very likely, that the heterozygous paternal and maternal breeders have a larger copy-number of the transgene than, and thus are not valid controls for comparison with, the double-transgenic pups. Unfortunately, such comparison is often made and this potential pitfall has hardly been discussed in the literature. A related concern is the heterozygous or homozygous state, since a homozygote has a doubled copy-number. With regard to chemoprevention studies, the difference in the copy number among animals may widen data variation. Theoretically, this weakness, if realized, can be overcome by using more animals, which, however, is sometimes undoable due to the Darwinian selection against the detrimental transgene, as explained below.
In those animal models that contain two, three or even more different transgenes driven by different promoters, such as in the MMTV-myc/MT-tgfα double transgenic [31;32] and the LSL-KrasG12D/pdx-cre/Arf-/- triple transgenic mice [33], producing pups that bear all different transgenes is highly costly and laborious, because only a low percentage of the pups are the positive ones. Even worse, in many occasions, the percentage is lower than the Mendelian inheritance because one or more of the transgenes are detrimental and the natural selection favors the healthier embryos with the healthier genotypes while prevents the full gestation of the embryos with more detrimental genotypes, as mentioned before [34]. This constraint often prevents researchers from using more animals to overcome the abovementioned weakness of copy number variation.
Influence from minor target-cell-types in transgenic mice is often neglected
The DNA promoter in many transgenic models is activated in different cell types and thus has multiple targeted-cell-types. For example, MMTV-c-myc and MT-c-myc transgenic mice use the MMTV (the long terminal repeat of mouse mammary tumor virus) and the metallothionein-1 (MT) promoter, respectively, as the transactivator for the c-myc oncogene. Although the MMTV-promoted transgenic mice are mostly used in the studies of the mammary gland, mainly its development and carcinogenesis, the MMTV promoter actually targets a wide spectrum of organs or tissues, mainly secretory ones and the hematopoietic systems [35] as well as the lung [36-38]. Of the hematopoietic cell lineages, lymphocytes are probably the main targets [39], as evolution has assigned lymphocytes to be frontier fighters against viruses such as MMTV. The best-studied activators of the MMTV promoter are steroid hormones, including glucocorticoids, progesterone and androgens [40-44] but, peculiarly, not estrogens [43;44]. Similarly, the three well-studied targeted-cell-types of the MT promoter are the mammary gland epithelium, the pancreatic ductal epithelium, and the liver hepatocytes [31;34;45-47]. However, when MMTV or MT is used to prod an oncogene (e.g. c-myc) to induce carcinogenesis in the mammary gland, it is often neglected that other targeted-cell-types, even if they might only be affected mildly, may still have impacts on the mammary carcinogenesis. For example, the potential alteration in immune functions due to the MMTV-activation in lymphocytes, or the potential abnormal liver and pancreatic functions due to the MT-activation in the liver and pancreas, may influence the mammary carcinogenesis. Unfortunately, such secondary influences from other targeted organs or tissues have seldom been addressed in the literature about the effect of transgene on its major targeted organs. For instance, the hematopoietic cells and the lung as targets of the MMTV promoter have hardly been mentioned and thus, to our knowledge, are not known to many peers who use MMTV transgenic mice to study mammary carcinogenesis.
Many DNA promoter sequences used in genetic engineering are actually not well characterized for their activators and suppressors as well as for the full spectrum of their targeted-cell-types. Since, as abovementioned, the MMTV promoter can be activated by glucocorticoids, one can imagine that it will, to some extent, have impacts on all glucocorticoid-responsive cell types, which are huge in number. Similarly, since metallothionein-1 is the key enzyme metabolizing heavy metals, heavy metals are main activators of the MT-1 promoter [45], making it perceivable that a variety of tissues and organs, especially those metabolic ones, are targeted by the MT-1 at different extents. Pertaining to chemoprevention studies, various minor targeted organs or tissues may intervene with the metabolism, and in turn the activity and toxicity, of the compound in question.
Temporal aspect of transgene activation is often neglected as well
Not only is the spatial aspect of transgene promoters a concern as described above, but so also is their temporal aspect [34;48]. Different organs or tissues start to develop and differentiate at different embryonic time points or different ages after birth; even different cell types in the same organ or tissue may show such temporal difference. Mechanistically, these developmental differences are attributed to the temporal differences in the expression of cell- or tissue-specific genes, which in turn is due to the temporal differences in the activation or suppression of these genes' DNA regulatory elements. For instance, the Mist-1 promoter is activated in multiple tissues of embryos [49;50]. On the other hand, since the level of progesterone, a potent activator of the MMTV promoter, and its interactions with androgens, glucocorticoids, estrogens and prolactin start to increase at puberty [51;52], the activity of the MMTV promoter starts to increase at puberty in the mammary gland of females. The progesterone level peaks at pregnancy, which is why mammary carcinogenesis in many MMTV transgenic females is enhanced by pregnancy [31]. Unfortunately, when selecting a DNA promoter as a transgene driver, most attention has been focused on the spatial aspect, i.e. the cell-, tissue- or organ-specificity, and much less on the temporal aspect, i.e. the time point during the murine development and growth at which the promoter is turned on and/or turned off. Indeed, we have asked many peers whether they know when the transgenes in the transgenic mice they use (but which they have not created) start to be active and inactive, many cannot give us a clear answer, although they know which organ or tissue should display a phenotype. Neglect of the temporal aspect may lead to data misinterpretation. Some transgenic mouse models develop cancers at a very early age, in contrast to their human counterparts that usually occur in the elder group. For instance, the Mist-1-KrasG12D knock-in mice develop both pancreatic and liver cancers at an early age [49], albeit most sporadic pancreatic and liver cancers in humans occur after mid age, because the Mist-1 promoter starts to be active at the embryonic stage when the liver and pancreas are developing.
One can imagine that the earlier at an embryonic life a DNA promoter is activated, the less differentiated tumors the animal may engender, because the transgene activation retains the targeted cells at an earlier embryonic, i.e. less differentiated, states, although it is not always the case because the functions of the transgenes influence the tumor differentiation as well. One has to bear it in mind that the carcinogenesis in these animal models occurs via refraining the targeted cells from differentiation, i.e. via a stop-differentiation mechanism, whereas many cases of the same type of cancer in humans may occur via de-differentiation of already mature cells [48]. In other words, the animal models with embryonically activated promoters and sporadic human cancers may be opposite in the carcinogenic mechanism. Researchers often claim that the cancer histology in the animal model they made or used resembles its human counterpart. Such description hints that the animal model recapitulates the human carcinogenesis and thus is a good one. However, it may be a pitfall and a great misleading if the mechanism is opposite, i.e. if the animal tumor derives from stop of differentiation of embryonic cells while the human counterpart comes from de-differentiation of adult mature cells. Therefore, transgenic models using embryonic DNA promoters may be useful in the investigation of pediatric cancers that may indeed be initiated during an embryonic stage, but may not suit the research of sporadic cancers in adulthood. Pertaining to the chemoprevention studies, preventing an antithetical carcinogenic mechanism or process has little human relevance.
The malignant nature of many animal tumors is actually questionable
Cancers developed from many transgenic models are dependent on the transgene until a very late stage, manifested as regression of the tumors upon the inactivation of the transgenes [48]. For example, cancers induced in some strains of c-myc and/or k-ras transgenic mice regress after inactivation of the transgene, although reactivation of the transgene can quickly induce the tumors again [53-55]. Some, but not most, chemical-induced cancers show a similar inducer-addiction as well. For instance, it has been known for 80 years that sex steroids can induce cancers in multiple organs of mice, rats and hamsters but the overt tumors, before they reach the terminal stage, regress completely after withdrawal of the steroid [51;56-62]. These spontaneous regressions indicate that most tumor cells are still mortal and are not autonomous, and thus should not even be classified as benign, and certainly not malignant, ones, according to the definition of tumor in pathology textbooks. Indeed, in some studies the tumor cells have been put in culture dishes wherein the cells are proved to remain mortal [63], as explained before [64]. Using these animal models to demonstrate oncogenicity of a chemical or a gene actually goes into a circular demonstration: The appearance of the cancer is used to “prove” the oncogenicity of the inducer and, in the same time, the inducer is used to sustain the presence of the cancer. In other words, these animal cancers, as long as they have not yet reached a very advanced stage, do not need to be prevented by any agent or cured by any drug, because withdrawal of the inducer is the best and simplest preventive or therapeutic approach, which contrasts human cancers that rarely regress spontaneously. Therefore, before a chemoprevention study is initiated, it needs to be known whether the to-be-used animal model is inducer dependent. If the study has been done, it needs to be determined whether the tested agent inhibits the carcinogenesis by depowering the cancer-inducer, i.e. by inactivating the transgene or metabolically cleaning up the chemical carcinogen. Unfortunately, these two possibilities, especially the inducer-addiction, have not yet been fully discussed in many animal studies of chemoprevention.
In human pathology, a malignant histology can well predict a malignant nature of the tumor, owing to several hundred years of experience in the anatomic pathology practice. Therefore, diagnosis can be made based on the histology of surgical or biopsy samples without a need to wait for years to see whether the patients eventually show or do not show metastases, which is the only reliable criterion for malignancy [65]. However, a large percentage, probably the majority, of the animal models of carcinogenesis do not show metastasis in the whole life time of the animals, although the tumors exhibit malignant histology [48;65]. If we consider these tumors cancerous, we actually think that most overt cancers can be cured simply by surgical removal, which usually is not the case in humans and thus makes the animal models poorly relevant to humans [48]. Likely, the malignant histology is barely a manifestation of a function of the inducer (e.g. an oncogene), and thus usually is inducer-dependent, but does not really mean that the cells are truly malignant, as inferred before [48]. In short, the histology of the tumors from a large number of animal models is disassociated from later metastasis and thus is not a reliable indicator for malignancy as seen in human pathology. Using these animal models in chemoprevention studies may lead to misinterpretation of the tested agents.
In a nutshell, the mortal nature, the inducer-dependency, and the metastasis-incompetence of the tumor cells seen in a large number of animal models challenge the malignant nature of the tumors, albeit they are histologically malignant and probably even morphologically resemble their human counterparts. For these tumors, the best preventive and therapeutic approach is the withdrawal of the inducer. Cancers induced by many chemical carcinogens may be less dependent on the carcinogens and thus are better models in this regard. However, they suffer from other faults such as the damage of metabolic organs or tissues, typically the liver and kidneys, by the chronic toxicity of the chemical carcinogens, which in turn influences the metabolism of the preventive agents in question and leads to biased conclusions.
Unfortunately, almost all mouse studies are conducted at a wrong ambient temperature
The core body temperature of mice and rats varies much more easily and greatly, compared with that of humans, because the rodents have a larger ratio of surface area to body mass and are so-called “metabolic strategists” that maintain the body temperature more by generating heat via metabolism than by the peripheral vasomotor control of the skin temperature [66;67]. Whereas rats and mice prefer 30-32°C [68], at least 28°C [69], globally the ambient temperature for these experimental animals is set at 21-23°C as required by the guideline of NIH and similar governmental agency of other countries. The 21-23°C is cold-stressing to the animals and is the lower limit of the so-called thermoneutral zone that allows the animals to maintain a thermostasis [66;68;70;71]. The constant cold-stress at 21-23°C has been shown to make xenograft tumors in mice grow much faster than at an ambient temperature of 30-31°C [72]. Restated, raising the ambient temperature to 30-31°C can significantly inhibit xenograft tumor growth in mice, which is elicited mechanistically via stimulating the innate immunity [69;72] and affecting heat shock protein expression [73]. Moreover, the cold-stressed situation affects significantly the clearance and toxicity of xenobiotics in a complex manner [74;75], which should be taken into account when interpreting the data from chemoprevention studies with mice housed at 21-23°C. Pertaining to cancer prevention in mice, is there any other approach cheaper and easier than tuning-up the thermo control of the room?
Conclusions
Mice and rats have about three years of lifespan, whereas further trials in humans require a much stronger guarantee that decades of intake of a cancer-preventive compound will not cause any chronic toxicity. In our rumination, after chemoprevention studies have been performed with the rodents, which are important, necessary and more cost-effective, larger animals with much longer lifespans should be involved to bridge the lifespan gap between humans and the rodents, although this may encounter social resistance and requires amendment of governmental guidelines. A huge number of transgenic mouse models of carcinogenesis have been available, which are used mainly for mechanistic research but sometimes also for chemoprevention studies. The copy number of the transgene, which usually cannot be determined in routine genotyping with PCR, may vary among the pups and broaden data variation, which should be compensated for by using more animals. The spatial and temporal aspects of the DNA promoters used to drive the transgenes require special attentions when selecting these murine models for chemoprevention studies, otherwise they may be potential pitfalls and weaknesses, especially for those promoters that are still poorly known about their spatial and temporal features. Models of chemically induced carcinogenesis do not have these transgene-related defects, but chemical carcinogens damage major metabolic organs or tissues and alter their functions, in turn affecting the metabolism of the to-be-tested chemopreventive agents. Moreover, cancers developed in many genetically edited and in some chemically induced carcinogenesis models may not really be malignant if judged by their behaviors, and not just by their histology, and thus should be used with caution in chemoprevention studies. Lastly, probably all published studies using mice have been performed at the ambient temperature of 21-23°C that is cold-stressing to the animals. Probably it needs to continue the already-started debate whether governmental guidelines should be amended to suggest that future mouse studies should be performed at the ambient temperature preferred by mice, which is about 30-32°C, to avoid cold-stress [76-78].
Acknowledgements
We want to give special thanks to Dr. Fred Bogott at the Medical Center, Austin of Minnesota, for his valuable comments and excellent English editing of this manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Gompel A, Baber RJ, de Villiers TJ, Huang KE, Santen RJ, Shah D. et al. Oncology in midlife and beyond. Climacteric. 2013;16(5):522-535
2. Park B, Shin A, Park SK, Ko KP, Ma SH, Lee EH. et al. Ecological study for refrigerator use, salt, vegetable, and fruit intakes, and gastric cancer. Cancer Causes Control. 2011;22(11):1497-1502
3. van den Brandt PA, Botterweck AA, Goldbohm RA. Salt intake, cured meat consumption, refrigerator use and stomach cancer incidence: a prospective cohort study (Netherlands). Cancer Causes Control. 2003;14(5):427-438
4. Pakseresht M, Forman D, Malekzadeh R, Yazdanbod A, West RM, Greenwood DC. et al. Dietary habits and gastric cancer risk in north-west Iran. Cancer Causes Control. 2011;22(5):725-736
5. Peleteiro B, Lopes C, Figueiredo C, Lunet N. Salt intake and gastric cancer risk according to Helicobacter pylori infection, smoking, tumour site and histological type. Br J Cancer. 2011;104(1):198-207
6. Kane MA, Hadler SC, Lee L, Shapiro CN, Cui F, Wang X. et al. The inception, achievements, and implications of the China GAVI Alliance Project on Hepatitis B Immunization. Vaccine. 2013;31(Suppl 9):J15-J20
7. Chiang CJ, Yang YW, You SL, Lai MS, Chen CJ. Thirty-year outcomes of the national hepatitis B immunization program in Taiwan. JAMA. 2013;310(9):974-976
8. Sun Z, Chen T, Thorgeirsson SS, Zhan Q, Chen J, Park JH. et al. Dramatic reduction of liver cancer incidence in young adults: 28 year follow-up of etiological interventions in an endemic area of China. Carcinogenesis. 2013;34(8):1800-1805
9. Tota JE, Ramanakumar AV, Franco EL. Lung cancer screening: review and performance comparison under different risk scenarios. Lung. 2014;192(1):55-63
10. Schivo M, Avdalovic MV, Murin S. Non-cigarette tobacco and the lung. Clin Rev Allergy Immunol. 2014;46(1):34-53
11. Alberg AJ, Brock MV, Ford JG, Samet JM, Spivack SD. Epidemiology of lung cancer: Diagnosis and management of lung cancer, 3rd ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest. 2013;143(5 Suppl):e1S-29S
12. Samet JM. Tobacco smoking: the leading cause of preventable disease worldwide. Thorac Surg Clin. 2013;23(2):103-112
13. Butt MS, Ahmad RS, Sultan MT, Qayyum MM, Naz A. Green tea and anticancer perspectives: updates from last decade. Crit Rev Food Sci Nutr. 2015;55(6):792-805
14. Hayat K, Iqbal H, Malik U, Bilal U, Mushtaq S. Tea and its consumption: benefits and risks. Crit Rev Food Sci Nutr. 2015;55(7):939-954
15. Lambert JD. Does tea prevent cancer? Evidence from laboratory and human intervention studies. Am J Clin Nutr. 2013;98(6 Suppl):1667S-1675S
16. Sturgeon JL, Williams M, van SG. Efficacy of green tea in the prevention of cancers. Nurs Health Sci. 2009;11(4):436-446
17. Deweerdt S. Comparative biology: Naked ambition. Nature. 2014;509(7502):S60-S61
18. Gorbunova V, Seluanov A, Zhang Z, Gladyshev VN, Vijg J. Comparative genetics of longevity and cancer: insights from long-lived rodents. Nat Rev Genet. 2014;15(8):531-540
19. Skulachev VP. What is "phenoptosis" and how to fight it? Biochemistry (Mosc). 2012;77(7):689-706
20. Skulachev MV, Skulachev VP. New data on programmed aging - slow phenoptosis. Biochemistry (Mosc ). 2014;79(10):977-993
21. Williams SA, Shattuck MR. Ecology, longevity and naked mole-rats: confounding effects of sociality? Proc Biol Sci. 2015;282(1802):pii 20141664. doi: 10.1098/rspb.2014.1664
22. Li Y, Choi PS, Casey SC, Felsher DW. Activation of Cre recombinase alone can induce complete tumor regression. PLoS One. 2014;9(9):e107589. doi: 10.1371/journal.pone.0107589
23. Schmidt-Supprian M, Rajewsky K. Vagaries of conditional gene targeting. Nat Immunol. 2007;8(7):665-668
24. Wu X, Kriz AJ, Sharp PA. Target specificity of the CRISPR-Cas9 system. Quant Biol. 2014;2(2):59-70
25. Hendel A, Fine EJ, Bao G, Porteus MH. Quantifying on- and off-target genome editing. Trends Biotechnol. 2015;33(2):132-140
26. Wade M. High-Throughput Silencing Using the CRISPR-Cas9 System: A Review of the Benefits and Challenges. J Biomol Screen. 2015 pii: 1087057115587916
27. McCart AE, Vickaryous NK, Silver A. Apc mice: models, modifiers and mutants. Pathol Res Pract. 2008;204(7):479-490
28. Yamada Y, Mori H. Multistep carcinogenesis of the colon in Apc(Min/+) mouse. Cancer Sci. 2007;98(1):6-10
29. Dent LA, Strath M, Mellor AL, Sanderson CJ. Eosinophilia in transgenic mice expressing interleukin 5. J Exp Med. 1990;172(5):1425-1431
30. Keffer J, Probert L, Cazlaris H, Georgopoulos S, Kaslaris E, Kioussis D. et al. Transgenic mice expressing human tumour necrosis factor: a predictive genetic model of arthritis. EMBO J. 1991;10(13):4025-4031
31. Liao DJ, Dickson RB. c-Myc in breast cancer. Endocr Relat Cancer. 2000;7(3):143-164
32. Liao DJ, Natarajan G, Deming SL, Jamerson MH, Johnson M, Chepko G. et al. Cell cycle basis for the onset and progression of c-Myc-induced, TGFalpha-enhanced mouse mammary gland carcinogenesis. Oncogene. 2000;19(10):1307-1317
33. Aguirre AJ, Bardeesy N, Sinha M, Lopez L, Tuveson DA, Horner J. et al. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev. 2003;17(24):3112-3126
34. Liao JD, Adsay NV, Khannani F, Grignon D, Thakur A, Sarkar FH. Histological complexities of pancreatic lesions from transgenic mouse models are consistent with biological and morphological heterogeneity of human pancreatic cancer. Histol Histopathol. 2007;22(6):661-676
35. Wagner KU, McAllister K, Ward T, Davis B, Wiseman R, Hennighausen L. Spatial and temporal expression of the Cre gene under the control of the MMTV-LTR in different lines of transgenic mice. Transgenic Res. 2001;10(6):545-553
36. Linnerth NM, Sirbovan K, Moorehead RA. Use of a transgenic mouse model to identify markers of human lung tumors. Int J Cancer. 2005;114(6):977-982
37. Moorehead RA, Sanchez OH, Baldwin RM, Khokha R. Transgenic overexpression of IGF-II induces spontaneous lung tumors: a model for human lung adenocarcinoma. Oncogene. 2003;22(6):853-857
38. Moorehead RA, Fata JE, Johnson MB, Khokha R. Inhibition of mammary epithelial apoptosis and sustained phosphorylation of Akt/PKB in MMTV-IGF-II transgenic mice. Cell Death Differ. 2001;8(1):16-29
39. Danke C, Grunz X, Wittmann J, Schmidt A, Agha-Mohammadi S, Kutsch O. et al. Adjusting transgene expression levels in lymphocytes with a set of inducible promoters. J Gene Med. 2010;12(6):501-515
40. Aurrekoetxea-Hernandez K, Buetti E. Transforming growth factor beta enhances the glucocorticoid response of the mouse mammary tumor virus promoter through Smad and GA-binding proteins. J Virol. 2004;78(5):2201-2211
41. Truss M, Bartsch J, Schelbert A, Hache RJ, Beato M. Hormone induces binding of receptors and transcription factors to a rearranged nucleosome on the MMTV promoter in vivo. EMBO J. 1995;14(8):1737-1751
42. Beato M, Arnemann J, Chalepakis G, Slater E, Willmann T. Gene regulation by steroid hormones. J Steroid Biochem. 1987;27(1-3):9-14
43. Bradham BM, Bolander FF Jr. The role of sex steroids in the expression of MMTV in the normal mouse mammary gland. Biochem Biophys Res Commun. 1989;159(3):1020-1025
44. Glover JF, Darbre PD. Multihormone regulation of MMTV-LTR in transfected T-47-D human breast cancer cells. J Steroid Biochem. 1989;32(3):357-363
45. Gulick J, Robbins J. Cell-type-specific transgenesis in the mouse. Methods Mol Biol. 2009;561:91-104
46. Ohgaki H, Sanderson ND, Ton P, Thorgeirsson SS. Molecular analyses of liver tumors in c-myc transgenic mice and c-myc and TGF-alpha double transgenic mice. Cancer Lett. 1996;106(1):43-49
47. Liao DJ, Wang Y, Wu J, Adsay NV, Grignon D, Khanani F. et al. Characterization of pancreatic lesions from MT-tgfalpha, Ela-myc and MT-tgfalpha/Ela-myc single and double transgenic mice. J Carcinog. 2006:5 DOI: 10.1186/1477-3163-5-19
48. Zhang J, Lou XM, Zellmer L, Liu SQ, Xu NZ, Liao DJ. Just like the rest of evolution in Mother Nature, the evolution of cancers may be driven by natural selection, and not by haphazard mutations. Oncoscience. 2014;1(9):580-590
49. Tuveson DA, Zhu L, Gopinathan A, Willis NA, Kachatrian L, Grochow R. et al. Mist1-KrasG12D knock-in mice develop mixed differentiation metastatic exocrine pancreatic carcinoma and hepatocellular carcinoma. Cancer Res. 2006;66(1):242-247
50. Lemercier C, To RQ, Swanson BJ, Lyons GE, Konieczny SF. Mist1: a novel basic helix-loop-helix transcription factor exhibits a developmentally regulated expression pattern. Dev Biol. 1997;182(1):101-113
51. Liao DJ, Dickson RB. Roles of androgens in the development, growth, and carcinogenesis of the mammary gland. J Steroid Biochem Mol Biol. 2002;80(2):175-189
52. Yart L, Lollivier V, Marnet PG, Dessauge F. Role of ovarian secretions in mammary gland development and function in ruminants. Animal. 2014;8(1):72-85
53. Wang C, Tai Y, Lisanti MP, Liao DJ. c-Myc induction of programmed cell death may contribute to carcinogenesis: a perspective inspired by several concepts of chemical carcinogenesis. Cancer Biol Ther. 2011;11(7):615-626
54. Wu CH, van RJ, Yetil A, Fan AC, Bachireddy P, Felsher DW. Cellular senescence is an important mechanism of tumor regression upon c-Myc inactivation. Proc Natl Acad Sci U S A. 2007;104(32):13028-13033
55. Tran PT, Fan AC, Bendapudi PK, Koh S, Komatsubara K, Chen J. et al. Combined Inactivation of MYC and K-Ras oncogenes reverses tumorigenesis in lung adenocarcinomas and lymphomas. PLoS One. 2008;3(5):e2125. doi: 10.1371/journal.pone.0002125
56. Noble RL, Hochachka BC, King D. Spontaneous and estrogen-produced tumors in Nb rats and their behavior after transplantation. Cancer Res. 1975;35(3):766-780
57. Noble RL. Sex steroids as a cause of adenocarcinoma of the dorsal prostate in Nb rats, and their influence on the growth of transplants. Oncology. 1977;34(3):138-141
58. Noble RL. Production of Nb rat carcinoma of the dorsal prostate and response of estrogen-dependent transplants to sex hormones and tamoxifen. Cancer Res. 1980;40(10):3547-3550
59. Noble RL, Cutts JH. Mammary tumors of the rat: a review. Cancer Res. 1959;19:1125-1139
60. Burrows H. Carcinoma mammae occurring in a male mouse under continued treatment with oestrin. Am J Cancer. 1935;24(3):613-616
61. Geschickter CF, Lewis D, Hartman CG. Tumors of the breast related to the oestrin hormone. Am J Cancer. 1934;21:828-859
62. Mceuen CS. Occurrence of cancer in rats treated with oestrone. Am J Cancer. 1938;34:184-195
63. Lazarov M, Kubo Y, Cai T, Dajee M, Tarutani M, Lin Q. et al. CDK4 coexpression with Ras generates malignant human epidermal tumorigenesis. Nat Med. 2002;8(10):1105-1114
64. Wang C, Lisanti MP, Liao DJ. Reviewing once more the c-myc and Ras collaboration: converging at the cyclin D1-CDK4 complex and challenging basic concepts of cancer biology. Cell Cycle. 2011;10(1):57-67
65. Lazebnik Y. What are the hallmarks of cancer? Nat Rev Cancer. 2010;10(4):232-233
66. Gordon C.J. The mouse: An "average" homoetherm. J Therm Biol. 2012;37:286-290
67. Povinelli BJ, Kokolus KM, Eng JW, Dougher CW, Curtin L, Capitano ML. et al. Standard sub-thermoneutral caging temperature influences radiosensitivity of hematopoietic stem and progenitor cells. PLoS One. 2015;10(3):e0120078
68. Gordon CJ, Aydin C, Repasky EA, Kokolus KM, Dheyongera G, Johnstone AF. Behaviorally mediated, warm adaptation: a physiological strategy when mice behaviorally thermoregulate. J Therm Biol. 2014;44:41-46
69. Messmer MN, Kokolus KM, Eng JW, Abrams SI, Repasky EA. Mild cold-stress depresses immune responses: Implications for cancer models involving laboratory mice. Bioessays. 2014;36(9):884-891
70. Gaskill BN, Gordon CJ, Pajor EA, Lucas JR, Davis JK, Garner JP. Heat or insulation: behavioral titration of mouse preference for warmth or access to a nest. PLoS One. 2012;7(3):e32799
71. Gaskill BN, Gordon CJ, Pajor EA, Lucas JR, Davis JK, Garner JP. Impact of nesting material on mouse body temperature and physiology. Physiol Behav. 2013;110-111:87-95
72. Kokolus KM, Capitano ML, Lee CT, Eng JW, Waight JD, Hylander BL. et al. Baseline tumor growth and immune control in laboratory mice are significantly influenced by subthermoneutral housing temperature. Proc Natl Acad Sci U S A. 2013;110(50):20176-20181
73. Eng JW, Reed CB, Kokolus KM, Repasky EA. Housing temperature influences the pattern of heat shock protein induction in mice following mild whole body hyperthermia. Int J Hyperthermia. 2014;30(8):540-546
74. Gordon CJ. Response of the thermoregulatory system to toxic insults. Front Biosci (Elite Ed). 2010;2:293-311
75. Gordon CJ, Johnstone AF, Aydin C. Thermal stress and toxicity. Compr Physiol. 2014;4(3):995-1016
76. Gaskill BN, Garner JP. Letter-to-the-editor on "Not so hot: Optimal housing temperatures for mice to mimic the thermal environment of humans". Mol Metab. 2014;3(4):335-336
77. Speakman JR, Keijer J. Not so hot: Optimal housing temperatures for mice to mimic the thermal environment of humans. Mol Metab. 2012;2(1):5-9
78. Speakman JR, Keijer J. Not so nuanced: Reply to the comments of Gaskill and Garner on 'Not so hot: Optimal housing temperatures for mice to mimic the environment of humans'. Mol Metab. 2014;3(4):337
Author contact
![]() Corresponding authors: Yukui Ma, Shandong Academy of Pharmaceutical Sciences, Ji'nan, Shandong 250101, P.R. China. Email: yukuimacom. Ningzhi Xu, Laboratory of Cell and Molecular Biology Cancer Institute, Chinese Academy of Medical Science, Beijing 100021, P.R. China. Email: xningzhibta.net.cn. D. Joshua Liao, Hormel Institute, University of Minnesota, Austin, MN 55912, USA. Email: djliaoumn.edu
Corresponding authors: Yukui Ma, Shandong Academy of Pharmaceutical Sciences, Ji'nan, Shandong 250101, P.R. China. Email: yukuimacom. Ningzhi Xu, Laboratory of Cell and Molecular Biology Cancer Institute, Chinese Academy of Medical Science, Beijing 100021, P.R. China. Email: xningzhibta.net.cn. D. Joshua Liao, Hormel Institute, University of Minnesota, Austin, MN 55912, USA. Email: djliaoumn.edu