Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2015; 6(10):1049-1057. doi:10.7150/jca.12501 This issue Cite
Research Paper
Staurosporine Induced Apoptosis May Activate Cancer Stem-Like Cells (CD44+/CD24-) in MCF-7 by Upregulating Mucin1 and EpCAM
Na Zhou1, Rong Wang1, Yizhuang Zhang1, Zhen Lei2, Xuehui Zhang1, Ruobi Hu1, Hui Li1, Yiqing Mao1, Xi Wang1, David M. Irwin1,3 ![]() , Gang Niu2
, Gang Niu2 ![]() , Huanran Tan1
, Huanran Tan1 ![]()
1. Department of Pharmacology, Peking University, Health Science Center, Beijing 100191, China;
2. Beijing N&N Genetech Company, Beijing, 100082, China;
3. Department of Laboratory Medicine and Pathobiology, University of Toronto, Toronto, Ontario M5S 1A8, Canada
Received 2015-4-25; Accepted 2015-7-27; Published 2015-9-1
Abstract
Malignant tumors recur after chemotherapy. A small population of cancer stem-like cells within tumors is now generally considered the prime source of the recurrence. To better understand how cancer stem-like cells induce relapse after fractionated chemotherapy, we examined changes in the CD44+/CD24- cancer stem-like cells population and behavior using the breast cancer cell line MCF-7. Our results show that apart from an increase in the CD44+/CD24- population, proliferation and clone formation, but not migration, were enhanced after recovery from apoptosis induced by two pulses of staurosporine (STS). The distribution of cells in the cell cycle differed between acutely induced apoptosis and fractionated chemotherapy. Sorted CD44+/CD24- stem-like cells from MCF-7 cells recovered from STS treatment possessed greater proliferation abilities. We also observed that mucin1 (MUC1) and Epithelial cell adhesion molecule (EpCAM) were up-regulated in abundance coincidently with proliferation and clone formation enhancement. Our findings suggest that fractionated chemotherapy induced apoptosis could stimulate cancer stem-like cell to behave with a stronger malignant property than cancer cells themselves and MUC1 and EpCAM are important factors involving in this process. By demonstrating changes in cancer stem cell during chemotherapy and identifying the crucial factors, we potentially can target them, to eradicate tumors and overcome cancer relapse.
Keywords: cancer stem cells, apoptosis, chemotherapy, mucin1, EpCAM
Introduction
Although surgical ablation, chemotherapy and radiotherapy can diminish, or even eradicate, visible solid tumors, most patients with advanced cancers relapse, with new tumors developing that are frequently more aggressive and often fatal1-3. The cancer stem cells (CSCs) hypothesis has recently been proposed and is supported by a considerable amount of evidence. Tumor tissue is heterogeneous and contains cancer cells displaying differing levels of differentiation. CSCs are a small subset of cancer cells, which are undifferentiated and possess a series of “stem” properties such as self-renewal, differentiation potential and infinite propagation4. Clinical retrospective studies examining expression of CSCs markers have indicated that high levels of expression of CSCs molecular markers are directly correlated with poor prognosis in patients5-7. Classical therapeutic drugs are largely designed to target rapidly proliferating cells, such as highly differentiated cancer cells. The overwhelming majority of CSCs however, are arrested in the G0 phase of the cell cycle and express high levels of proteins related to drug resistance 4, 7, 8. Thus, shrinkage of tumor size that results from chemotherapy or radiotherapy is primarily due to the apoptosis or necrosis of highly differentiated cancer cells. Recurrence of cancer is due to the survival of the small population of non-dividing (or slowly dividing) CSCs. Since most cancers progress more rapidly after a traditional clinical treatment, this raises questions as to whether the cancer recurrence is just simply due to the residual CSCs or whether other changes also occur.
In humans, stem cells play crucial roles in replenishing mature cells damaged by injury or apoptosis. For example, hematopoietic stem cells (HSCs) are responsible for the generation and regeneration of circulating blood cells, including those involved in the immune system, and have been used in clinical therapy 4. Self-renewal is one of the crucial functions of stem cells, and this feature must persist for the lifetime of the animal. Interactions between apoptotic differentiated cells and stem cells must guide stem cells to self-renew and allow the replenishment of mature cells, but the precise mechanisms controlling these processes are not known. As both normal stem cell and cancer stem cell possess similar ”stem” properties, changes may occur to CSCs due to the apoptosis of highly differentiated cancer cells. CSCs are inconsistent with self-renewal, thus tumors are not controlled like normal injuries and remodeled. Therefore we suspect that these altered CSCs are not only the source of the original tumor, but might also be responsible for tumor progression, metastasis, and resistance to therapy, and subsequent tumor recurrence. Nevertheless, the specific mechanisms for this phenomenon remain obscure. As interactions may represent an important signaling mechanism for cancer recurrence and poor prognosis, we investigated the possible effects of cancer cell apoptosis on the number and “stem” properties of CSCs using an apoptotic MCF-7 cell model.
Methods
Cell culture and regents
MCF-7 was a gift from Professor Jing-Rong Cui (Peking University) and cultured in DMEM high glucose (Life Technologies, USA) supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin, and 10% heat inactivated FBS (Life Technologies, USA), and maintained at 37°C and 5% CO2. Staurosporine (STS) was purchased from Merck (Darmstadt, Germany).
Generation of acute apoptotic MCF-7, STS-1-R and STS-2-R
MCF-7 cells were treated for 12 h with 2 μM Staurosporine in full DMEM medium. Cell death was confirmed by flow cytometry, using Annexin V and propidium iodide (PI) co-staining (Biosea, Peking, China). To obtain cancer cells that recovered from apoptosis, we first used 2 μM STS to treat MCF-7 cells for 12 h, followed by a change to fresh medium to allow the cells recover. When cells reached 80% confluence they were harvested and named as STS-1-R. Subsequently, we treated STS-1-R with 2μM STS for 12 h, changed the media and waited for the cells to recover. When these cells reached 80% confluence they were harvested and named as STS-2-R.
Cell cycle assay
Cells were incubated in DMEM medium with or without apoptotic treatments. At appropriate intervals, 1 × 106 cells were collected and fixed in 70% ethanol (4°C, overnight), washed with 1ml PBS, treated with RNaseA (100 μg/ml) to eliminate interference of RNA and stained with PI (Sigma-Aldrich, Steinheim, Germany), and analyzed by a FACScan TM Calibur (Becton Dickinson, Franklin Lakes, NJ, USA). FCS Express version 3 (De Novo Software) was utilized to analyze the data.
Cell proliferation assay
Cell growth was examined by the MTS assay using CellTiter 96® AQueous One Solution (Promega Biotech Co, Ltd) according to the manufacturer's instructions. Briefly, cells were seeded at a density of 8000 cells/well into 96-well plate. At the time of assay, 20 μl MTS solution was added to each well and incubated for 1-4 h. Absorbance was measured at 490 nm using a Vmax microplate reader (Bio-Rad, Benchmark).
Flow cytometric analysis and Fluorescence-activated cell sorting (FACS)
Cells were collected and suspended in PBS, labeled with FITC mouse anti-human CD44 and PE mouse anti-human CD24 (BD Pharmingen TM), with 5 μl of antibody per one million cells in a final volume of 300 μl. A total of about 1.0× 106 cells were incubated with these two antibodies for 0.5 h at 4°C in the dark. Unbound antibody was washed off and the cells were analyzed on a BD FACS Calibur within 1 h of staining. Gating was established using the isotype controls FITC-labeled mouse IgG2a and PE-labeled mouse IgG2b (BD Pharmingen TM).
We used FACS to sort every passage of MCF-7 cells into two phenotypically distinct populations: CD44+/CD24- and all other phenotypes. The labeling method and condition were the same as described for the flow cytometric analysis.
Soft agar colony formation assay
1.2% agarose and 2×DMEM medium containing 20%FBS was mixed in equal volumes and 1ml was added per well of a 6-well tissue culture plate (Corning, NY, USA). This 0.6% matrigel was coagulated at room temperature for 0.5 h. 0.7% agarose and 2×DMEM medium containing 20% FBS was mixed with equal volumes. We used this mixture to suspend cells (10,000 cells /well) and lay them on the 0.6% matrigel gently. The cells were allowed to grow for about 21 days at 37°C and 5% CO2 before staining with 0.01% Crystal Violet and counting the number of colonies under a microscope.
Scratch assay
Cells were plated for 24 h before starving in FBS-free DMEM medium for 16 h prior to the beginning of the assay. A 10 μl pipette tip was used to create a “scratch'' in the cell monolayer. After removing debris and adding fresh media, cells were photographed at the 6 h, 12 h, 24 h, 36 h, 48 h, 60 h, 72 h, 84 h and 96 h time points. The leading edge of the migrating cells was approximated using Image J (National Institutes of Health) to generate a relative migration rate for each treatment condition.
Western blot analysis
Total proteins were extracted using a buffer containing 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 0.02% NaN3, 1% SDS, 1 mM EDTA, 0.5% Sodium deoxycholate, 100 mg/ml PMSF, 1 mg/ml leupeptin and 1% NP-40. For each sample, 60 mg of protein was separated by SDS-PAGE (with different concentrations appropriate for their differing molecular weight of the targeted proteins) at 80 V for 0.5 h and 120 V for 1 h using the Mini-PROTEAN 3 electrophoresis cell system (Bio-Rad). Proteins were then transferred to a PVDF membrane (Bio-Rad) by the semi-dry blotting method (Bio-Rad) and the Dunn carbonate transfer buffer that consisted of NaCHO3 (10 mM), Na2CO3 (3 mM), and 20% methanol. Membranes were blocked for 2 h by 5% w/v nonfat dry milk and then incubated overnight at 4°C with the primary antibodies, rabbit anti-EPCAM (1:1,000; Abcam, ab71916), mouse anti-MUC1 (1:500; CST, #4538), and rabbit anti-actin (1:1000; CST, #4970). Membranes were subsequently incubated with peroxidase-conjugated secondary antibodies. Antibody binding was detected after incubation with HRP-linked secondary antibodies, with the membrane-bound antibodies visualized by luminal chemiluminescence ChemiDoc XRS (BIO-RAD).
Statistical analysis
All experiments were conducted in at least 3 independent cultures. All data were expressed as means ± standard error of mean and analyzed with one-way ANOVA followed by the Tukey's post hoc test for multiple comparisons. P values less than 0.05 were considered significant.
Results
STS could induce apoptosis of MCF-7 cells, which was intensified in a dose dependent manner
The effect of Staurosporine (STS) on cell proliferation was assessed with the MTS assay in cells treated with differing concentrations of STS for 12 h. No significant difference in cell proliferation was detected between untreated cells and those treated with 0.05 to 0.3 μM STS, however, proliferation was significantly inhibited by concentrations of 0.5 μM to 2 μM STS (Fig. 1A). Assessment with the apoptotic assay for PI-Annexin V co-staining in cells treated with STS at various concentrations of STS for 12h showed that apoptosis was induced in MCF-7 at a dose of 0.25 μM and above, with the effect increasing with higher concentrations of STS (Fig. 1C). The number of apoptotic cells increased from 10% for the control (untreated cells) to a maximum of 40% at a STS concentration of 2 μM (Fig. 1B and 1C). These results indicate that STS has anti-proliferative and apoptotic effects on human MCF-7 cell line and that 2 μM STS could be used to generate an apoptosis model in MCF-7 cells.
STS induces apoptosis in MCF-7 cells in a dose dependent manner (A) The effect of STS on cell proliferation by MTS assay. Data represent means ± S.E. (n=3). *** p<0.001 vs. control group. (B) Changes in the levels of apoptosis in STS treated MCF-7 cells. Annexin V/Propidium Iodide staining and flow cytometry were used to quantify apoptosis proportion. The percentage of apoptotic cells is the sum of the events in the upper right and lower right quadrants. (C) Quantification analysis of flow cytometry results. These results are typical for three independent experiments. Data represent means ± S.E. (n=3). ** p<0.01, *** p<0.001 vs. control group.

Proliferation and clone formation but not migration was enhanced after recovery from apoptosis induced by two pulses of STS
To mimic clinic chemotherapy, we first used 2 μM STS to treat MCF-7 cells for 12 h, followed by a change to fresh media to allow cells to recover. When these cells grew to 80% confluence, the cells were harvested and named as STS-1-R. Subsequently, we treated the STS-1-R with 2 μM STS for 12 h, changed the media and waited for the cells to recover. When these cells grew to 80% confluence, they were harvested and named as STS-2-R. Using the MTS proliferation assay, no significant difference in cell proliferation ability was detected between STS-1-R and untreated cells at 24 h, whereas STS-2-R showed a significant increase in cell numbers compared to control and STS-1-R as soon as 24 h, however this difference was not seen at 48 h (Fig. 2A).
Analysis of cell proliferation, migration and colony formation abilities in control and STS treated MCF-7 cells. (A) Analysis of cell proliferation among control group, recovery from first STS treated group (STS-1-R) and recovery from second STS treated group (STS-2-R). Results are typical of the three independent experiments. Data represent means ± S.E. (n=3). * p<0.05vs control, ### p<0.001vs STS-1-R. (B) Phase-contrast microscopy images and number analysis of clones in control and STS treated MCF-7 cells. Data represent means ± S.E. (n=3). ** p<0.01vs control, ## p<0.01vs STS-1-R. (C) Scratch assay to compare migration rates among control, STS-1-R and STS-2-R cells. Scratches were made in the monolayer with a 10 μl pipette tip. Distance cells migrated was quantified using Image J. Experiment was repeated three times in triplicates and data denote the mean ± S.E (n=3).
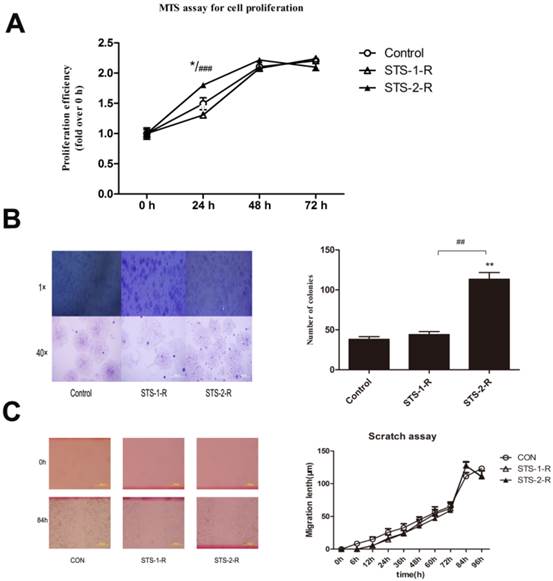
Stem-like cells can grow under anchorage-independent conditions in soft agarose and form single-cell colonies. Therefore, in vitro foci formation assays potentially reveal the in vivo tumorigenic ability of cells, reflecting stem cell properties and possibly forecast the degree of malignancy. There was little difference in the number of foci obtained between the control and the STS-1-R cells. STS-2-R cells, however, produced a significantly increased number of foci compared to both control and STS-1-R cells. Phase-contrast microscopy revealed that the colonies generated from the STS-2-R cells were larger than the other two groups (Fig. 2B). According to above data, we presume that when the breast cell line MCF-7 cells were first treated with STS, cellular functions became impaired, and could not rapidly adapt. This may explain why STS-1-R cells are similar to the control cells. In contrast, after two pulses of STS treatment, STS-2-R behaved more like cancer cells and appears more malignant.
To further investigate the specific role of STS-induced apoptosis in wound closure and cell migration, we performed the scratch assay with STS-1-R and STS-2-R cells, but found no significant difference in their migration rates compared to control cells (Fig. 2C). These results clearly suggest that proliferation and clone formation ability but not migration was enhanced after recovery from apoptosis induced by two pulses of STS.
Acute induced apoptosis contribute to changes in the cell cycle and the proportion of CD44+/CD24- cancer stem cells
We then examined the cell cycle to determine possible mechanisms for the apoptosis-induced enhanced proliferation. MCF-7 cells were treated with 2 μM STS for 12 h and then the DNA content of the cells was analyzed. Flow cytometry analysis revealed a decrease in the proportion of cells in the G1 phase, from 51.14% to 42.80% and an increase of cells in S phase, from 31.00% to 42.07%, in the STS treated group, with no significant differences observed for the G2/M phase (Fig. 3A). These results demonstrated that acute treatment of MCF-7 cells with STS leads to a change in the cell cycle.
Apoptosis contributed to changes in the cell cycle and increases in the proportion of CD44+/CD24- cancer stem cells. (A) Quantification of the percentages of cells in different phases of the cell cycle, determined by flow cytometry. Data represent means ± S.E. (n=3). * p<0.05, **p<0.01. (B) Flow cytometry analysis of the percentage of the CD44+/CD24- subpopulation in MCF-7 cells treated with STS at different concentrations. (C) Quantification of the CD44+/CD24- subpopulation. Results are typical of three independent experiments. Data represent means ± S.E. (n=3). * p<0.05, ** p<0.01.
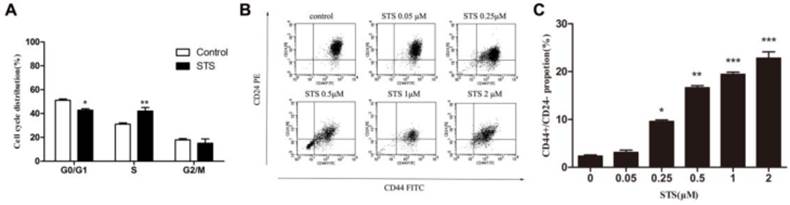
Cancer stem-like cells are a small subpopulation of cells in various types of cancer, including breast cancer, which have been shown to be highly tumorigenic and exhibits some stem cell characteristics such as self-renewal and the ability to form tumor spheres 4. CD44+/CD24-cells were considered to be the cancer stem-like cells in breast cancer9, 10. To examine whether changes in the cancer behaviors recognized above was related with CSCs, we quantified the difference in CD44+/CD24- cell proportions in population of cells treated with STS for 12 h using flow cytometry. Untreated MCF-7 cells contain a subpopulation of 2.95±0.75% of the cells that are CD44+/CD24-. The percentage of the CD44+/CD24- cells was found to significantly increase, compared to control MCF-7 cells, when the cells were treated with STS at a concentration of 0.25 μM or above, with 2 μM STS yielding a CD44+/CD24- cell proportion of 20.72±3.45% (Fig. 3B,3C).
Sorted CD44+/CD24- stem cells from recovered MCF-7 possess greater proliferation ability
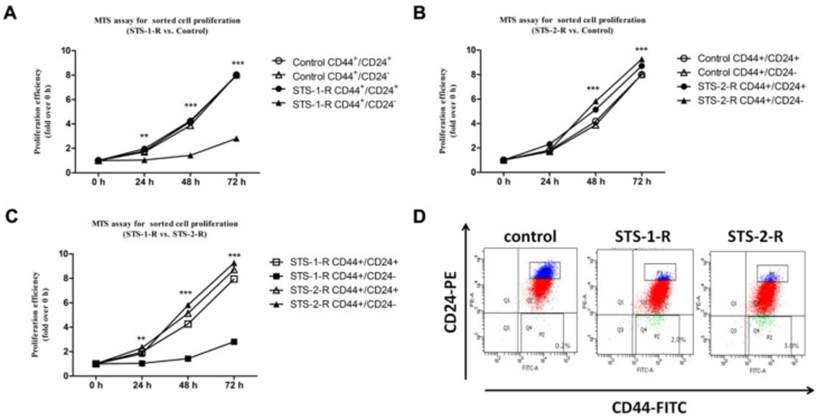
Accumulating evidence has proven that chemotherapy helps enrich cancer stem-like cells11. Our data similarly show an increase in the CD44+/CD24- subpopulation. These observations suggest that the changes in the behavior of cancer cell were related to characteristics of the CSCs. To address this conjecture, we sorted CD44+/CD24- and CD44+/CD24+ subpopulations as CSCs and non-CSCs, respectively, from control MCF-7, STS-1-R and STS-2-R cells using FACS, followed by cultivating and the MTS proliferation assay. Compared to the control group, non-CSCs from STS-1-R cells did not show any change in proliferation at any time point, whereas significant decreases in proliferative ability of CSCs sorted from the STS-1-R were observed from 24 h to 72 h (Fig. 4A). As to STS-2-R, both CSCs and non-CSCs sorted from the STS-2-R had significantly increased proliferative ability, compared to control and STS-1-R. In addition, CSCs of STS-2-R showed higher proliferative ability than non-CSCs of STS-2-R (Fig. 4B, 4C). These data may indicate that after two pulses of STS, the recovered cancer cells contained a larger amount of cancer stem-like cells (Fig. 4D) that act as the origins of differentiated cancer cells, and after 24 h of adjustment and replenishment, possess more vigorous proliferation ability.
Analysis of the proliferative abilities of CD44+CD24- and non CD44+CD24- cells in control and STS treated MCF-7 cells assayed by the MTS assay. (A, B, C) Proliferative ability is measured over time. Results are typical of three independent experiments. Data represent means ± S.E. ** p<0.01, *** p<0.001. (D) Proportion of CSCs proportion sorted from the three groups.
MUC1 and EpCAM may act in a crucial role in apoptosis induced CSCs activation
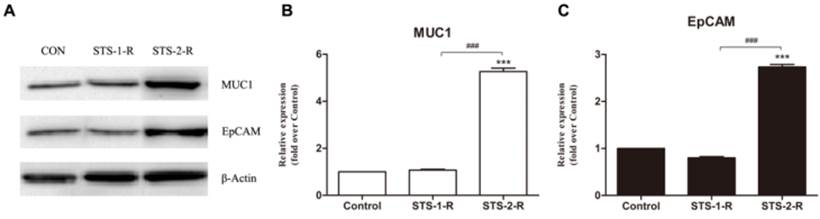
To further explore the mechanism of apoptosis induced CSCs activation, we investigated several candidate molecules related to tumorigenesis and progression, including ALDH1, Her2, and cyclinD1, CXCR4, P65, P50, Bcl2, Bax, EpCAM and MUC1. Protein from control, STS-1-R and STS-2-R cells were extracted and Western blotted (Fig. 5A). Apart from EpCAM and MUC1, the others did not change significantly among the different groups (data not shown). As shown in Fig. 5, when cells were treated with two pulses of STS, the recovery cells had significantly higher levels of MUC1 and EpCAM compared to the control group, where the abundance of MUC1 and EpCAM differed markedly between the STS-1-R and STS-2-R cells (Fig. 5B, 5C). MUC1 in STS-2-R cells was significantly more abundant than in the control or STS-1-R, as was EpCAM, concordant with our cancer cell behavior experiments, such as the cell proliferation and colony forming assays. These results suggest that MUC1 and EpCAM may have crucial roles in apoptosis induced CSCs activation.
Changes in the abundance of MUC1 and EpCAM in control and STS treated MCF-7 cells. (A) Western blot analysis of MUC1 and EpCAM. Bands were analyzed using Quantity One software and β-actin used as a loading control. (B) Quantification of the Western blot analysis. Protein levels are compared to the control group. Results are typical of three independent experiments. Data represent means ± S.E. (n=3). *** p<0.001 vs control, ### p<0.001 vs. STS-1-R.
Discussion
Despite developments in surgical methods, chemotherapy, radiotherapy and agents targeted against some tumor markers (e.g., estrogen receptor ER, HER2 for breast cancer and EGFR for non-small-cell lung carcinoma), a considerable number of patients with advanced stage carcinoma experience tumor recurrence or metastasis a few years after treatment 1, 2. These disappointing facts have led investigators to attribute this to cancer stem cells. The cancer stem cell hypothesis was proposed to explain cancer heterogeneity and the risk of cancer recurrence, where researchers presumed that cell subpopulations may contribute to drug resistance induced by over expression of both drug resistance related genes and anti-apoptosis genes that drive tumor recurrence or metastasis 4. Furthermore, as conventional chemotherapeutic strategies are directed against rapidly cycling tumor cell fractions 12, the CSCs population would be up-regulated after chemotherapy. Consistently, our data showed that when MCF-7 endured the protein kinase inhibitor STS, a substantial portion of them became apoptotic, but the size of the CD44+/CD24- CSCs population increased (Fig. 1 and 3). Yu et al. demonstrated that large numbers of BT-IC (breast tumor-initiating cells) enriched cells could be generated by in vivo passage of breast cancer cells in NOD/SCID mice treated with chemotherapeutic agents, with some of their tumorigenic properties enhanced11. Above all, it is generally believed that chemotherapy induced tumor cell death not only enriches cancer stem cells, but also augments their malignant properties, which consequently leads to recurrence.
Most animals have some ability to regenerate damaged cells and tissues 13. Research in several model organisms has revealed that apoptosis may be the driving force for cell proliferation during tissue regeneration, which was proposed in 198814-16. Stem cells are typically quiescent or passing slowly through the cell cycle in adult tissues, but can be activated in response to cell loss and wounding. Therefore, stem cells and undifferentiated progenitor cells play an important role in both tissue homeostasis and tissue regeneration 17. Hristov et al. demonstrated that apoptotic bodies from endothelial cells enhance the number and initiate the differentiation of human endothelial progenitor cells in vitro18. We presume that crosstalk occurs between apoptotic cells and stem cells, which regulates stem cells differentiation and the replacement of dead cells as part of tissue homeostasis. These events could mirror the relationship between highly differentiated cancer cells and CSCs, as CSCs have similar properties with normal stem or progenitor cells 4.
In our research, we found that when MCF-7 was under acute treatment by STS, CSCs were enriched. To our knowledge, CSCs are generally in G0 arrest, but interestingly, the cell cycle assay showed the opposite phenomenon, a decrease in the proportion of G1 phase cells and an increase of S phase cells (Fig. 3A). The anti-proliferative activity of staurosporine is due to its dramatic effect on the cell cycle, by interfering with multiple components of the CDK system. The interference results in the arrest of cells at G2/M or G0/G1, depending on cell type19,20. Developing resistance to STS could allow CSCs to quickly differentiate, with the highly differentiated cancer cells rapidly propagating.
We mimicked clinical fractionated chemotherapy by giving two pulses of STS treatment to MCF-7 cells, with the STS-2-R cells yielding higher proliferation ability and could form more colonies than the other cells. In addition, CD44+/CD24- CSCs sorted from the STS-2-R showed significantly higher proliferation ability (Figs. 2 and 4). Apart from indefinite proliferation, another key malignant characteristic is invasion. We used the scratch assay to estimate whether recovery of MCF-7 cells from two pulses of STS treatment affected invasion and repair abilities. The answer was no. Neither STS-1-R nor STS-2-R cells had stronger invasion abilities compared to control cells. Many previous breast cancer studies have established the invasive nature of various malignant breast tumor cells such as MDA-MB-231, and MCF-721, 22. These studies show that MDA-MB-231 cells are highly invasive, whereas MCF-7 cells are weakly invasive. Invasion was only observed after 48 h for the MCF-7 cells, thus we conclude that invasion is not a principal malignant property of MCF-7, and only proliferation ability was changed.
MUC1 is a transmembrane glycoprotein, which is aberrantly glycosylated and over expressed in a variety of epithelial cancers and has also been reported to be associated with proliferation, metabolism, invasion and metastasis, angiogenesis, chemoresistance and resistance to apoptosis 23-25. Although the relationship between MUC1 and cancer stem cells remains unexplored, a considerable number of recent studies have revealed that MUC1 is associated with cancer stem cells. MUC1 is expressed on human embryonic pluripotent stem cells and functions as a growth factor receptor to boost the proliferation of hESCs and prevents them from differentiating 26. MUC1 is also involved in increasing the proportion of SP cells in the breast cancer cell line MCF-7 27.
EpCAM is increasingly being recognized as a specific cancer stem cell marker for a variety of tumors including liver, colon, breast, cholangiocarcinomas and pancreatic cancer28-30. Increased EpCAM expression indicates a poor prognosis, as shown for example, in breast and gallbladder carcinomas30-32. In addition, metastatic and recurrent cancers were found to express significantly higher levels of EpCAM compared with primary carcinomas33. Rather than antagonizing apoptosis, EpCAM exerts a pro-tumor effect by inducing proliferation with a direct impact on cell cycle control, upregulating the proto-oncogene c-myc and Cyclins A and E, and transducing signals into the cell nucleus through the Wnt signaling pathway34-36. In our research, MCF-7 cells that recovered from two pulses of STS showed significantly elevated MUC1 and EpCAM abundance compared to the other two groups (Fig. 5). STS-2-R has a high proportion of CSCs and is direct products of chemotherapy. We conclude that MUC1 and EpCAM may be an important factor mediating apoptosis induced CSCs activation.
There are some limitations of our study. First, breast cancer stem cells have not yet been identified, although they can be enriched and propagated in vitro with the techniques we used. Second, using cell lines and the protein kinase C inhibitor Staurosporine (STS) to mimic clinical chemotherapy in vitro may not represent the in vivo microenvironment of breast cancer completely. Thus, our data need validation with tissues derived directly from patient breast cancer specimens.
In conclusion, our data indicated that cancer cell apoptosis induced by two pulses of STS treatment not only enrich the CSCs population, but also enhance the proliferation ability of sorted CD44+/CD24- CSCs and the clone forming ability of the cancer cell recovered from STS. It is presumed that cancer cell death induced by chemotherapy or radiotherapy will not eliminate all cancer stem cells; to the contrary, it may directly or indirectly stimulate concealed characteristics of CSCs and aggravate their malignancy, which consequently results in poor prognosis and high mortality. Moreover, MUC1 and EpCAM is an important factor involved in this crosstalk. Thus, the exact signaling pathways involved in this apoptosis induced CSCs proliferation needs to be clarified so that we can target it to realize tumor eradication.
Abbreviations
CSCs: cancer stem cells; STS: staurosporine; MUC1: mucin1; EpCAM: Epithelial cell adhesion molecule.
Acknowledgements
This study was supported by grants from the National Natural Science Foundation of China (NSFC) Grant Number 81102484 and 30772603, National Key Technologies R&D Program (Grant Numbers 2012BAK25B01), and a grant from the National Science Foundation of China - Canadian Institutes of Health Research (NSFC-CIHR) China-Canada Joint Health Research Initiative (Grant Numbers 81061120525 and CCI-109605).
Competing Interests
The authors have declared that no competing interest exists.
References
1. da Silva SD, Hier M, Mlynarek A. et al. Recurrent oral cancer: current and emerging therapeutic approaches. Front Pharmacol. 2012;3:149
2. Locke JA, Zafarana G, Ishkanian AS. et al. NKX3.1 haploinsufficiency is prognostic for prostate cancer relapse following surgery or image-guided radiotherapy. Clin Cancer Res. 2012;18:308-16
3. Kerbel RS. Reappraising antiangiogenic therapy for breast cancer. The Breast. 2011;20:S56-S60
4. Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105-11
5. Abubaker K, Latifi A, Luwor R. et al. Short-term single treatment of chemotherapy results in the enrichment of ovarian cancer stem cell-like cells leading to an increased tumor burden. Mol Cancer. 2013;12:24
6. Okudela K, Woo T, Mitsui H. et al. Expression of the potential cancer stem cell markers, CD133, CD44, ALDH1, and beta-catenin, in primary lung adenocarcinoma--their prognostic significance. Pathol Int. 2012;62:792-801
7. Oktem G, Sanci M, Bilir A. et al. Cancer stem cell and embryonic development-associated molecules contribute to prognostic significance in ovarian cancer. Int J Gynecol Cancer. 2012;22:23-9
8. Kondo T, Setoguchi T, Taga T. Persistence of a small subpopulation of cancer stem-like cells in the C6 glioma cell line. Proc Natl Acad Sci U S A. 2004;101:781-6
9. Hughes L, Malone C, Chumsri S. et al. Characterisation of breast cancer cell lines and establishment of a novel isogenic subclone to study migration, invasion and tumourigenicity. Clin Exp Metastasis. 2008;25:549-57
10. Dontu G, Liu S, Wicha MS. Stem cells in mammary development and carcinogenesis: implications for prevention and treatment. Stem Cell Rev. 2005;1:207-13
11. Yu F, Yao H, Zhu P. et al. let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell. 2007;131:1109-23
12. Brunner TB, Kunz-Schughart LA, Grosse-Gehling P. et al. Cancer stem cells as a predictive factor in radiotherapy. Semin Radiat Oncol. 2012;22:151-74
13. Steller H. Mechanisms and genes of cellular suicide. Science. 1995;267:1445-9
14. Chera S, Ghila L, Dobretz K. et al. Apoptotic cells provide an unexpected source of Wnt3 signaling to drive hydra head regeneration. Dev Cell. 2009;17:279-89
15. Li F, Huang Q, Chen J. et al. Apoptotic cells activate the "phoenix rising" pathway to promote wound healing and tissue regeneration. Sci Signal. 2010;3:ra13
16. Kondo S. Altruistic cell suicide in relation to radiation hormesis. Int J Radiat Biol Relat Stud Phys Chem Med. 1988;53:95-102
17. Bergmann A, Steller H. Apoptosis, stem cells, and tissue regeneration. Sci Signal. 2010;3:re8
18. Hristov M, Erl W, Linder S, Weber PC. Apoptotic bodies from endothelial cells enhance the number and initiate the differentiation of human endothelial progenitor cells in vitro. Blood. 2004;104:2761-6
19. Gescher A. Staurosporine analogues - pharmacological toys or useful antitumour agents? Crit Rev Oncol Hematol. 2000;34:127-35
20. Johnson LN, De Moliner E, Brown NR. et al. Structural studies with inhibitors of the cell cycle regulatory kinase cyclin-dependent protein kinase 2. Pharmacol Ther. 2002;93:113-24
21. Chen H, Zhu G, Li Y. et al. Extracellular signal-regulated kinase signaling pathway regulates breast cancer cell migration by maintaining slug expression. Cancer Res. 2009;69:9228-35
22. Zhang L, Sullivan PS, Goodman JC. et al. MicroRNA-1258 suppresses breast cancer brain metastasis by targeting heparanase. Cancer Res. 2011;71:645-54
23. Besmer DM, Curry JM, Roy LD. et al. Pancreatic ductal adenocarcinoma mice lacking mucin 1 have a profound defect in tumor growth and metastasis. Cancer Res. 2011;71:4432-42
24. Chaika NV, Gebregiworgis T, Lewallen ME. et al. MUC1 mucin stabilizes and activates hypoxia-inducible factor 1 alpha to regulate metabolism in pancreatic cancer. Proc Natl Acad Sci U S A. 2012;109:13787-92
25. Kitamoto S, Yokoyama S, Higashi M. et al. MUC1 enhances hypoxia-driven angiogenesis through the regulation of multiple proangiogenic factors. Oncogene. 2013;32:4614-21
26. Sherry T. Hikita KSK, Dennis O. Clegg, Cynthia Bamdad. MUC1* Mediates the Growth of Human Pluripotent Stem Cells. PloS one. 2008:3
27. Engelmann K, Shen H, Finn OJ. MCF7 side population cells with characteristics of cancer stem/progenitor cells express the tumor antigen MUC1. Cancer Res. 2008;68:2419-26
28. Yamashita T, Ji J, Budhu A. et al. EpCAM-positive hepatocellular carcinoma cells are tumor-initiating cells with stem/progenitor cell features. Gastroenterology. 2009;136:1012-24
29. Kokuryo T, Yokoyama Y, Nagino M. Recent advances in cancer stem cell research for cholangiocarcinoma. J Hepatobiliary Pancreat Sci. 2012;19:606-13
30. Osta WA, Chen Y, Mikhitarian K. et al. EpCAM is overexpressed in breast cancer and is a potential target for breast cancer gene therapy. Cancer Res. 2004;64:5818-24
31. Gastl G, Spizzo G, Obrist P. et al. Ep-CAM overexpression in breast cancer as a predictor of survival. The Lancet. 2000;356:1981-2
32. Varga M, Obrist P, Schneeberger S. et al. Overexpression of epithelial cell adhesion molecule antigen in gallbladder carcinoma is an independent marker for poor survival. Clin Cancer Res. 2004;10:3131-6
33. Bellone S, Siegel ER, Cocco E. et al. Overexpression of epithelial cell adhesion molecule in primary, metastatic, and recurrent/chemotherapy-resistant epithelial ovarian cancer: implications for epithelial cell adhesion molecule-specific immunotherapy. Int J Gynecol Cancer. 2009;19:860-6
34. Munz M, Baeuerle PA, Gires O. The emerging role of EpCAM in cancer and stem cell signaling. Cancer Res. 2009;69:5627-9
35. Maaser K, Borlak J. A genome-wide expression analysis identifies a network of EpCAM-induced cell cycle regulators. Br J Cancer. 2008;99:1635-43
36. Maetzel D, Denzel S, Mack B. et al. Nuclear signalling by tumour-associated antigen EpCAM. Nat Cell Biol. 2009;11:162-71
Author contact
![]() Corresponding authors: Prof. Huanran Tan, Department of Pharmacology Peking University, Health Science Center Physiology Building#232 38 Xue Yuan Road Beijing 100191, China. Tel: +86 10 82802004; Fax: +86 10 82802004; E-mail: tanlabedu.cn, Prof. David M. Irwin, Department of Laboratory Medicine and Pathobiology, University of Toronto, Toronto, Ontario M5S 1A8, Canada. Tel.: +1-416-978-0519; E-Mail: david.irwinca, Mr Gang Niu, Beijing N&N Genetech Company, Beijing, 100082, China. E-Mail: nngenecom; Tel.: +86-10-6404-7343 (G.N.)
Corresponding authors: Prof. Huanran Tan, Department of Pharmacology Peking University, Health Science Center Physiology Building#232 38 Xue Yuan Road Beijing 100191, China. Tel: +86 10 82802004; Fax: +86 10 82802004; E-mail: tanlabedu.cn, Prof. David M. Irwin, Department of Laboratory Medicine and Pathobiology, University of Toronto, Toronto, Ontario M5S 1A8, Canada. Tel.: +1-416-978-0519; E-Mail: david.irwinca, Mr Gang Niu, Beijing N&N Genetech Company, Beijing, 100082, China. E-Mail: nngenecom; Tel.: +86-10-6404-7343 (G.N.)