Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2015; 6(9):901-912. doi:10.7150/jca.11910 This issue Cite
Research Paper
Initiate Tumors with Single Cell Spheres Formed in Serum-Containing Medium
Guozhou Zhang*, Kun Xiong*, Weiwei Ma, Wei Xu, Huihui Zeng ![]()
State Key Laboratory of Natural and Biomimetic Drugs, Beijing, P.R. China; Department of Chemical Biology, School of Pharmaceutical Sciences, Peking University, Beijing 100191, P.R. China
* Contributed equally to this work
Received 2015-2-16; Accepted 2015-6-12; Published 2015-7-28
Abstract
Background: It is difficult to prospectively identify and maintain putative tumor-initiating cells (TICs). Spheres that formed in serum-free media contained more TICs while spheres formed in serum-containing media were not used in tumor-initiating.
Methods: Soft-agar was used to isolate colonies. A continuous, static suspension culture using serum-containing media was modified from liquid overlay technique and tumor cell spheres could be maintained by this method for >90 days. Tumor-initiating capacity of these spheres was tested in nude mice. The nuclear staining of OCT4 in sphere cells and the expression profile of hepatic cell lineage related genes were examined.
Results: Soft-agar derived HepG2 colonies indicated different properties from their parental cells. The suspension cells of A549 and MCF7 could initiate tumors at 104 cells level. The growth proportions of individual A549, MCF7 and HepG2 spheres with diameter of 101-150 µm were significantly higher than smaller spheres. After suspension culture for 15-27 days, HepG2 and MCF7 spheres could initiate tumors with diameter up to 200 µm; the estimated TIC frequency was 1/1500-1/400. The HepG2 and MCF7 spheres retain tumor-initiating potential for at least 52 days.
Conclusion: After 15 days' serum-containing suspension culture small HepG2 and MCF7 cell spheres (diameter ~200 µm) could initiate tumors in nude mice.
Keywords: tumor-initiating, sphere, cell culture, xenograft
Introduction
Tumor-initiating process in human is largely deduced from xenografted models in which tumors are regenerated from prospectively isolated tumor-initiating cells (TICs). It is difficult to prospectively identify and maintain putative TICs because they only make up a fraction of tumor cells and the mechanism of TIC state control is largely unknown. Evidence indicates that tumor cell spheres formed in suspension culture harbored more TICs and these cells displayed different patterns of gene expression, phenotypes and subsequently their tumor initiating capacities [1-4]. These heterogeneous spheres can be used as starting materials for cancer stem cell study [2].
Most spheres hitherto are cultivated in serum-free media and some of them can be maintained for months [5] or years [6]; serum induces stem cells/progenitors differentiation [7]. The serum-free culture method is also consistent with the concepts of stem cell niche and inter-vascular tumor nodule, both lack direct blood supply. However, the serum-free media formulas are usually empirical and cell line-dependent, and formulas available limit to isolate TICs from dozens of heterogeneous tumor cells. On the other hand, tumor cells indicated limitless replicative potential and insensitivity to anti-grow signal [8] even in the presence of serum. It is quite possible to cultivate highly tumorigenic cells out of serum-containing media.
Transformed cells are able to switch their proliferation from as monolayer to in suspension in serum-containing media. Semisolid medium such as agar that can hold cells in suspension has been used to assay transformed cells for decades [9]. Recently, the suspension culture was postulated as a selective process favoring those de-differentiating cells in media with or without serum [10]. In suspension culture, single cells and cell aggregates coexist [10], and small aggregates originate from both clonal expansion and cell aggregating. A small cell aggregate or sphere is possible to grow into large multicellular tumor spheroids (MTSs). Both share the properties of cell-cell interactions within 3D structure despite other differences. The MTSs are regarded to mimic the avascular stage of tumor nodules, an intermediate between monolayer culture and tumor tissue [11]. As the spheres enlarged a core necrosis onsets where 50-300 µm from their peripheries under varying conditions [11], the inner MTS cells are deprived of media. It seems the sphere could shield its inner cells from surrounding media through structure assembled by cells. These inner cells could be different. If the inner sphere contains highly tumorigenic cells, it could initiate tumor with as few as possible cells.
The initiating techniques, growth kinetics and morphology of MTSs were well studied. The cell-cell interactions, extracellular matrix components within MTS were explored [11-14]. Few studies investigated tumor-initiating of MTSs because pure cell was a prerequisite of cell biological study. In contrast to using large, uniform tumor spheroids in pharmaceutical assays and tumor biological study, the cell spheres used in tumor-initiating should be as small as possible, ideally, merely containing single TIC. To minimize the size of cell sphere in tumor-initiating, only those cells displayed constant dividing capacity should be preserved.
In this study, the liquid overlay technique [15] was modified to a continuous, static suspension culture in which no dissociation of sphere was required. Tumor cell spheres could be maintained in serum-containing media for >90 days. Randomly selected individual spheres could initiate tumor after 15-27 days in suspension; this period could be extended to 52 days for HepG2 and MCF7 cell spheres. Considering the successful rate of tumor initiation, the estimated TIC frequency in spheres was 1/1500-1/400. Further data are required to identify the very cell that initiated tumor and the underlying mechanisms.
Materials and Methods
Cell maintenance and soft-agar cloning
Cell lines of A549, HepG2, MCF7 and U87-MG were introduced from Chinese Academy of Medical Science and maintained as monolayer in RPMI 1640 (Invitrogen, Carlsbad, CA) supplemented with 10% FCS (ExCell, China) and 50 µg/ml gentamycin sulfate (Amresco #0304, Solon, OH). Soft-agar assay was used to isolate the cell colonies of HepG2 and to test the secondary colony forming capability. The agar (Sigma, St. Louis, MO) was dissolved in complete media to 0.6% and 0.3% as the under- and upper-layer respectively. A549 encoding mitochondrial thioredoxin reductase-eGFP (A549_TR) was transfected by using Lipofectamine 2000 (Invitrogen) and isolated by cell sorting. This fusion protein in A549_TR did not show enzymatic activity. In order to trace the cell origin of spheres, the geneticin (G418) was not used when culturing A549_TR cells in suspension.
Continuous static suspension culture
The plate (Corning, Cambridge, MA) was coated with 1.2% agar dissolved in pure water 30 min prior to cell seeding. Trypsinized adherent cells were transferred into coated plate to initiate suspension culture. Up to 5×105 cells (in 5 ml) could be seeded per well in the six-well plate. Suspension cells were passaged every 3-7 days in accordance with their growth state. The passaging process facilitated direct cell-cell contact. To minimize the cell re-aggregating during incubation, the plate was left undisturbed.
No mechanical or enzymatic dissociation was required to break the spheres. By slightly extending the interval of media renewal, pH of media dropped to 6.8-7.0, a portion of cells died and the majority of spheres were easily broken by gentle pipetting. The spheres were sedimented for 15 min and 4/5 of used media was removed; or the cells were pelleted at 80 ×g for 3 min and removed all supernatant.
Single cell and sphere (~100 sphere/ml) in cool RPMI 1640 media were picked randomly under dissecting microscope and traced individually in agar-coated 96-well plate (200 µl of complete media). Smallest diameters of the spheres were measured by using a calibrated minuscule scale. After two weeks, the spheres with ≥2 folds smallest diameter increase, presumed 8 folds cell number increase, were used to estimate the growth proportion.
Cytotoxic assay
The cells (2-10×103 cells/200 µl) were allowed to attach for 24 h in 96-well plate. The medium then was changed to 180 µl complete medium plus 20 µl chemicals. The chemicals of cisplatin (USP32 99%; Italo bioloigical techanology, China), 5-fluorouracil (Sigma), Ethaselen, Butaselen and Pentaselen (investigational drugs synthesized in our lab) or selenite (Zhonglian, China) were assayed. After designated time of exposure, the cytotoxicity was estimated by MTT (Sigma) staining. Drug concentration of 50% inhibition (IC50) was calculated by using four-parameter Logistic formula. Data were reported as IC50±SE.
DNA content analysis
The trypsinized cells were pelleted at 200 ×g for 2 min, followed by two D-PBSA washes. Adjusted the cell number to <106 cells/500 µl and fixed with 5 ml cold (-20°C) ethanol, added drop wise with mixing, at 4 °C overnight. The cells were pelleted at 200 ×g for 5 min, followed by two washes with D-PBSA+1% BSA (106 cells/3 ml). The cells were resuspended in 500 µl D-PBSA+1% BSA, treated with 50 µl 10×PI (Sigma) and 50 µl 10×RNase A (Amresco) at 37 °C for 30 min, and filtered through 40 µm mesh cell strainer (BD Biosciences, Bedford, MA). Guava Easycyte Plus System (Millipore, Billerica, MA) was used to analyze 5000 cells per sample. The cell cycle distribution was fitted by using ModFit LT 3.2 software.
Immunofluorescence Assay
The spheres were allowed to attach to 25×25 mm glass coverslips for 2 h, fixed with cool 4% paraformaldehyde for 30 min, permeated in PBST (0.1% Triton X-100 in D-PBSA) for 30 min. The sphere sample was blocked by 3% BSA for 1 h, incubated in anti-OCT3/4 (1:500) (rabbit polyclonal antibody; Biorbyt, UK) at 4°C overnight, and then incubated in FITC- or TRITC-conjugated antibody (1:500) (Santa Cruz, CA) for 1 h. The nuclei were stained with 10 µg/ml Hochest 33342 for 10 min. The coverslips were mounted with sphere towards slides by using Prolong Antifade Reagent (Invitrogen).
The spheres were examined by using Leica TCS SP2 confocal system (Leica Microsystems, Heidelberg GmBH, Germany). Full-frame x-y plane image (512×512 pixels) was recorded along the z-axis by 2 µm stepping. The nuclear staining of OCT4 was colocalized by using ImageJ 1.46r (http://rsb.nih.gov/ij) with “colocalization” plugin. Only those colocalized signals appeared in three consecutive sections of nuclei were recorded as OCT4 positive.
Western blot
Cells were lysed with RIPA (150 mM NaCl, 1.5% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris-HCl, pH 8.0; supplemented with 1 mM PMSF just before use) for 30 min (100 mm dish per ml lysis). The lysate was centrifuged at 12000 ×g for 30 min and the supernatant was collected (above, performed at 4°C). After the protein quantifying (BCA reagent, Applygen, China), the protein samples were diluted with 5×Laemmli buffer (10% SDS, 25% 2-mercaptoethanol, 50% glycerol, 0.01% bromophenol blue, 312.5 mM Tris-HCl, pH 6.8) and boiled for 5 min. The proteins were separated with polyacrylamide gel (20 µg/lane) and blotted onto PVDF membrane. The membranes were blocked with 5% skimmed milk, and incubated with the antibodies of mAb cyclin B1 (Santa Cruz, #sc-7393, 1:5000), mouse anti-cyclin D1 (Santa Cruz, #sc-8396, 1:5000), rabbit anti-cdk2 (Santa Cruz, #sc-163, 1:5000) and mAb β-actin (ZSGB-BIO, #TA-09, 1:5000) in TBST (137 mM NaCl, 20 mM Tris-HCl, pH 7.6, 0.5% Tween 20)+1% BSA at 4°C overnight, followed by incubating with secondary antibodies (1:5000) for 1 h at RT. The bands were developed with Amersham ECL Reagent (GE Healthcare Life Sciences, UK) and recorded with X-ray film (Kodak, Cat#6535876).
RT-PCR
The cells cultured in suspension for >12 days were used in this procedure. Total RNA was extracted with TRIzol Reagent (Invitrogen) according to the instructions. Reverse transcription, in duplicate, was conducted by following the instructions of Oligo d(T)15 (TIANGEN, China) and TIANScript M-MLV kit (TIANGEN). The 2×Taq PCR Mix (Aidlab, China) was used in PCR amplification. Each PCR run included non-template control. The primers for fetal liver cell markers (CD34, CD90/Thy-1, CD117/c-Kit, CD133/prominin-1), early hepatic lineage (biliary) markers (AFP, CK19) and late hepatic lineage (hepatocyte) markers (ALB, CK18) were used [16]. The primers for the markers that associated with pluripotency including Oct4, Nanog, SMO, Bmi1, Notch1, β-catenin were used (the primers and cycling conditions refer Supplementary Table 1).
Animal transplanting
The use of animals was approved by Laboratory Animal Welfare Sub-Committee of Biomedical Ethics Council, Peking University. The nude mice were provided by Laboratory Animal Department of Peking University Health Science Center. Five to seven female mice, four weeks of age, were maintained in an individually ventilated cage. The cells or spheres were dispersed in cool RMPI 1640 media for <2 h before injection. In sphere transplanting, a small subcutaneous cavity was enlarged by using small tweezers through a small skin cut (~2 mm). Single sphere in 5 µl media (or mixed with equal volume of Matrigel) (BD Biosciences) was injected into this small cavity. The skin cut recovered in 2-3 days without specific treatment. After transplanting, the mice were examined daily for the first two weeks and twice per week for the subsequent ten weeks. At end point, the mice were sacrificed by cervical dislocation. In serial passaging, tumors were removed and the connective tissue was trimmed. The tumors were placed in cool RPMI 1640 containing 2×penicillin/streptomycin and dissected into small dices (≈1mm3). One dice was embedded into each subcutaneous cavity. For histological examination, the tumors were sliced (5 µm) and stained with hematoxylin and eosin (H&E).
Statistics
Proportions were compared by Chi-square or Fisher exact test. TIC frequency was estimated through limit dilution analysis (LDA). The Poisson statistics of one-hit model in LDA was estimated by using the “limdil” function of the “StatMod” package (Smyth GK, http://bioinf.wehi.edu.au/software/limdil) included in the R-project (http://www.r-project.org).
Results
Suspension cells differ from their parental cell
Three sub-populations of HepG2 could proliferate from single colony to >105 cells in adherent culture and were designated as SM01, SM02 and SM03. Though maintained in adherent culture for three weeks post isolation, these sub-populations increased in forming secondary colony (Figure 1A). Three sub-populations were not resistant to 5-FU, CDDP, Ethaselen, Butaselen or selenite. The SM02 was more sensitive to these chemicals than its parental cells (Figure 1B).
Suspension cells differ from their parental cells. (A) Three HepG2 subpopulations increased in forming secondary colony ≤3 weeks following isolation (mean±SD, student t test); continuous data of >3 weeks were not available, but latter data indicated the reduction of colony-forming capacity in SM01, SM02 and SM03. (B) Adherent SM02 was more sensitive to chemicals than its parental, HepG2, cell (IC50±SE). Eight concentrations were used for each chemical in curve fitting. 5FU: 5-fluorouracil; CDDP: cisplatin; recov: recovery time after removal of chemical, otherwise no recovery time. Wilcoxon matched-pairs signed rank test is employed in comparison between IC50 of SM01, SM02, SM03 and HepG2. (C) The proportion of G1/S/G2 phase was estimated via DNA content. The chi-square test was used in proportion comparison. (D) The cyclin D1 in HepG2 and its tumor were lower than SM01, SM02 and SM03 at 24 h after seeding. (E) The A549_TR, MCF7 and U87MG cells grown in suspension could initiate tumor at 104 cells level or less in nude mice (H&E, ×100). Arrow beside tumor indicates the double tumors at single injection site. Arrows in image indicate the alveolus-like structure in A549_TR tumor and glandular structure in MCF7 tumor. Note: *, P<0.05; **, P<0.01.
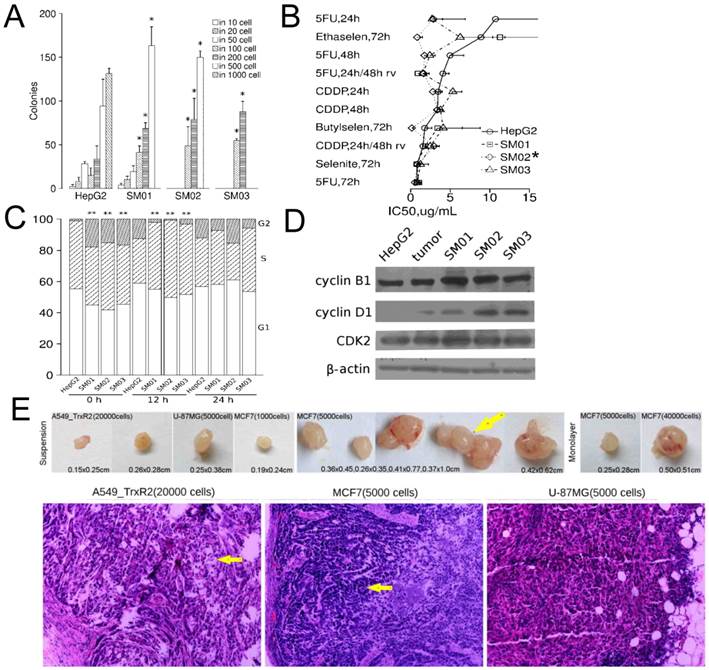
Three sub-populations of HepG2 showed a rapid G1/S phase transition at 12 h following seeding (Figure 1C). After 24 h, the G1/S/G2 phase distributions of HepG2 and its sub-populations were not different, still the relative protein levels of cyclin B1, D1 in HepG2 cells and HepG2 tumor were lower than those in SM01, SM02 and SM03 cells (Figure 1D).
The secondary colony cells of SM01 and SM02, collected directly from soft-agar, could regenerate tumors at 104 cells level in six weeks (1/1 tumor/mouse each). The adherent HepG2 and SM03 cells failed at 104 cells level though the adherent HepG2 cells could regenerate tumors at 106 and 105 cells levels, 3/5 and 1/1 tumor/mice, respectively. The colony cell collecting was inefficient; the xenografted animal number was insufficient for statistical inference.
Suspension cells are slightly more tumorigenic
It was unknown whether these suspension cells collected directly from soft-agar were more tumorigenic. To provide cells for constant analysis, the liquid overlay technique was modified to a continuous, static suspension culture. A less cell level, 104 cells per injection [17], for tumor initiating was examined. The suspension cells of A549_TR, MCF7 and U87-MG regenerated tumors at 104 cells level or less but in most cases the tumors appeared in <1/4 mice (Table 1). A549_TR cells produced tumors at 2×104 cells level, the tumor exhibited alveolus-like structure. U87-MG cells produced tumor at 5×103 cells level, and the tumor cells and nuclei displayed pleomorphism. In MCF7 tumors, ill-organized glandular structure could be seen (Figure 1E). The TIC frequency of MCF7 cells, compared with its monolayer counterpart, increased >6-fold (P=0.005) and the latency for suspension cells was shortened. Two MCF7 tumors out of single injection site (5×103 cells/site, two sites in total) suggested the cell number to initiate tumor was sufficient. Suspension SM01 and SM02 cells failed to initiate tumor at 104 cells level which was inconsistent with their colony cells.
Cells in suspension initiate tumor in nude mice
| cell line | cells per injection # of tumor/# of injection | latency (day) | TIC frequency (95% C.I.) | |||
|---|---|---|---|---|---|---|
| 106 | 105 | 104 | 103 | |||
| A549_TR | 2/8a | 0/4c | 42-70 | 1/71535(17852-286654) | ||
| HepG2 | 3/6 | 3/6 | 0/6 | 0/4 | 14-34 | 1/782616(313163-1955810) |
| SM01 | 0/9 | 0/7 | NA | NA | ||
| SM02 | 0/9 | 0/7 | NA | NA | ||
| SM03 | 0/5 | 0/11 | NA | NA | ||
| MCF7 | 5/6b | 3/4c, 1/2 | 15-19 | 1/11681(4332-31494)* | ||
| MCF7(adh) | 1/4b | 1/4d | 19-27 | 1/77884(18220-332921) | ||
| U-87MG | 1/6d | 70 | 1/27424(3852-195215) | |||
Note: cells were transplanted s.c. in nude mice of four weeks old; a: 20000 cells, b: 40000 cells, c: 2000 cells, d: 5000 cells per injection; adh: adherent; TIC: tumor-initiating cell; *: P=0.008 compared with MCF7 adherent culture.
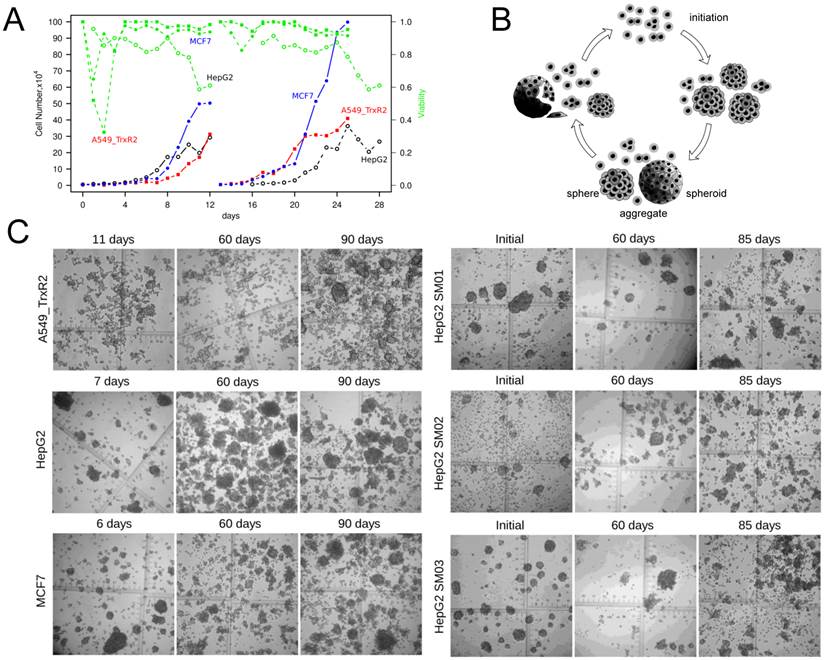
Spheres of 101-150 µm diameters were sufficient to grow independently
Tumor cells shifted to suspension culture following a portion of cell death (Figure 2A). The number of suspension cells increased >20-fold within 10 days. The cells and spheres in suspension could be maintained for >90 days in this continuous, static suspension culture (Figure 2B, 2C). Either because the spheres were resulted from constant cell dividing or because the cell-cell interactions would facilitate cell survival, the spheres could indicate a growth advantage over single cells. Single cells and spheres were selected randomly and traced individually. Randomly selected single cells of HepG2, MCF7 and SM02 could proliferate into colonies with frequencies of 2.6% (2/77), 3.2% (1/31) and 16.9% (14/83), the corresponding frequencies of their 3-16 cells' aggregates were 5.1% (2/39), 4.6% (1/21) and 34.3% (12/35), respectively. There was no significant difference between the growth proportion of single cells and 3-16 cells' aggregates (P>0.05).
The continuous, static suspension culture. (A) Monolayer A549_TR, HepG2 and MCF7 cells shifted to suspension culture by a portion of cell death. (B) Schematic diagram of continuous static suspension culture. Single cells aggregated following seeding in suspension. After initiation (3-7 days depended upon cell lines), cell spheres could be seen. These spheres could be broken by exerting starvation and gentle pipetting. Note: The aggregates include small size of spheres or large multicellular tumor spheroids and we did not distinguish sphere from spheroid in this article. (C) Representative image of spheres (×40) maintained in suspension.
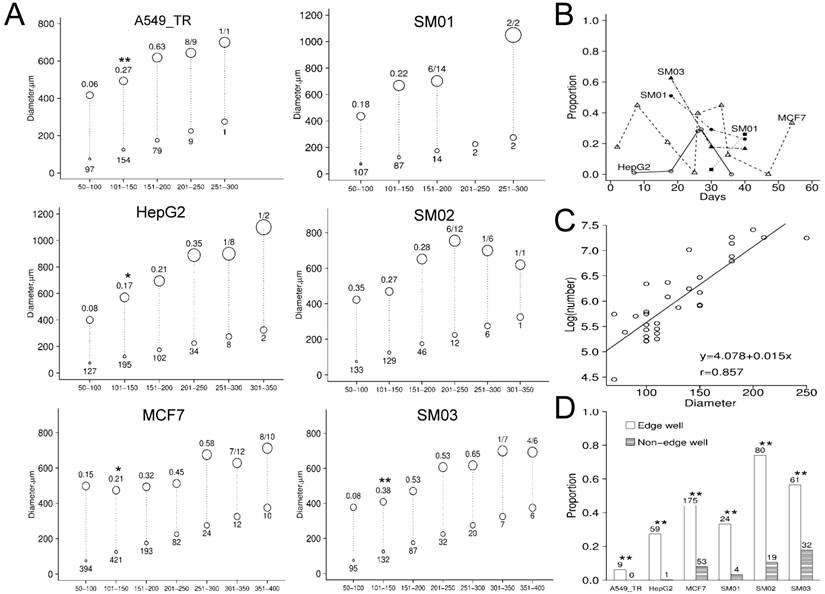
The initial diameters of spheres ranged from 20 to 450 µm and were predominantly in 100-300 µm. The A549, MCF7, HepG2 and SM03 spheres of 101-150 µm diameters indicated elevated proportion of independent growth (Figure 3A). The proportion of growth increased with initial diameters from 50 µm to 250µm, except SM01. Compared with their parental cell (HepG2) the growth proportions were significantly elevated for SM01 (P=0.028, 50-100 µm), SM02 (P<0.001, 50-100 µm) and SM03 (P<0.001, 101-150 µm) spheres. The optimal culture conditions might be cell line-dependent (Figure 3B). For those spheres with a final diameters >1000 µm, presumably resulted from constant cell dividing, their initial diameters were mostly in 100-300 µm. Randomly selected A549_TR spheres were less likely to grow than its tightly packed counterpart (P<0.001). The spheres of 70-250 µm diameters contained 169-2510 cells (Figure 3C). Based upon the averaged diameter, the cell numbers estimated in tracing were >105 cells, >10 folds of those that could initiate tumor. The spheres in the edge wells were more likely to grow than the inner wells in 96-well plate (P<0.001) (Figure 3D).
Characterizing of continuous static suspension culture. (A) The growth proportion of randomly selected spheres that be traced individually in vitro. X-axis indicated the initial diameters categories and the circle size was proportion to the sphere initial diameter. The initial diameter of spheres (bottom circle of the dash line) were mostly <300 µm. Only the final diameters of sphere (averaged diameter displayed as top circle of the dash line) ≥2 folds those of initial were recorded. The number under bottom circle indicated initial spheres number; the number above the top circle indicated the proportion of growth. (B) The growth proportions of individual spheres varied along time. The optimal culture condition could be cell line-dependent. (C) The logarithmic cell number of spheres (30 spheres of different cell lines) was predicted from its diameter (70-250 µm) (P<0.001). (D) The spheres in edge-well of the 96-well plate were more likely to grow. The sphere number was displayed above the bar. Note: *, P<0.05; **, P<0.01
Single sphere can initiate tumor in nude mice
If the sphere could grow independently, it could grow into tumor as well. At first, single sphere transplanting of SM01 (0/18 tumor/site), SM02 (0/16 tumor/site), SM03 (0/18 tumor/site) failed to initiate tumor. However, single spheres in 50% Matrigel could initiate tumor in nude mice (Table 2). The diameters of spheres for transplanting were ~200 µm, containing up to 1186 cells. The TIC frequency was hence estimated. Most spheres, except A549_TR, could initiate tumors after 15-27 days in serum-containing suspension culture; HepG2 and MCF7 spheres retained this capability for 52 days and SM02 spheres for 83 days (Table 2).
Single sphere initiates tumor in nude mice
| cell lines | days in susp | tumor /injection | latency (day) | TIC frequency (95% C.I.) | days in susp | tumor /injection | latency (day) | TIC frequency (95% C.I.) | days in susp | tumor /injection | latency (day) | TIC frequency (95% C.I.)* |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A549_TR | 39 | 0/6 | NA | NA | 60 | 0/3 | NA | NA | 88 | 0/6 | NA | NA |
| HepG2 | 27 | 3/4 | 60-70 | 1/404 (119-1371) | 53 | 2/6 | 104 | 1/1381 (342-5575) | 95 | 0/4 | NA | NA |
| SM01 | 15 | 3/4 | 50-70 | 1/404 (119-1371) | 34 | 0/6 | NA | NA | 83 | 0/6 | NA | NA |
| SM02 | 15 | 2/6 | 50-70 | 1/1381 (342-5575) | 34 | 4/8 | 57-84 | 1/808 (297-2196) | 83 | 4/6 | 55-90 | 1/182 (65-510) |
| SM03 | 15 | 4/6 | 31-70 | 1/510 (182-1428) | 34 | 0/6 | NA | NA | 83 | 0/6 | NA | NA |
| MCF7 | 26 | 9/12 | 21-60 | 1/404 (199-819) | 52 | 5/8 | 35-83 | 1/571 (229-1421) | 94 | 0/4 | NA | NA |
Note: Single sphere with a diameter ~200 µm in 50% Matrigel was used in transplanting. The spheres from same batch were used in repeated transplanting. TIC frequency: tumor-initiating cell frequency estimated via cell number predicted by diameter of sphere; days in susp: days of continuous static culture in suspension before transplanting; *: small spheres containing ~200 cells.
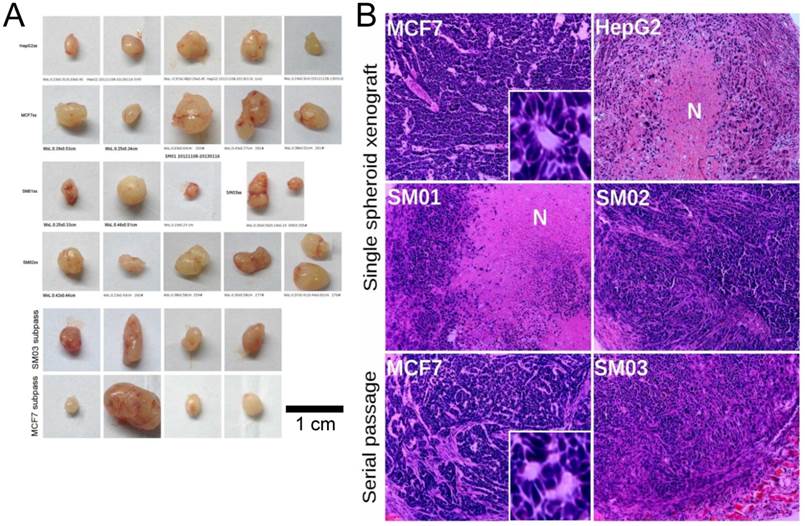
Two MCF7 tumors at single sphere transplanting site were observed (Figure 4A). Necrotic center was observed in HepG2 and SM01 tumors 18 days post transplanting (Figure 4B). A blood vessel was seen near the necrotic center in HepG2 tumor. The secondary transplanted tumors of SM03 and MCF7 (4/4, tumor/site each) appeared within 18 days. The secondary SM03 tumor exhibited similar cell morphology with that of SM02. The secondary passage of MCF7 tumors indicated similar cell morphology and glandular structure with its parental tumors (Figure 4B). The spheres could initiate tumor in nude mice and the structures of tumors were similar to those of their parental cells. If the spheres that could grow constantly and contribute to its elevated tumor-initiating capacity were nucleated from cells of de-differentiation, these cells should express corresponding markers.
Tumor initiated with single spheres. (A) The single cell spheres (of diameter ~200 µm, containing ~1186 cells) in 50% Matrigel could initiate tumor in nude mice (except A549_TR sphere). The secondary transplanted SM03 and MCF7 tumor appeared rapidly (in 18 days). (B) Single spheres could reconstitute structurally similar tumor of its parental cell. N: stands for necrotic core (H&E, ×100).
Cells differentiate in suspension culture
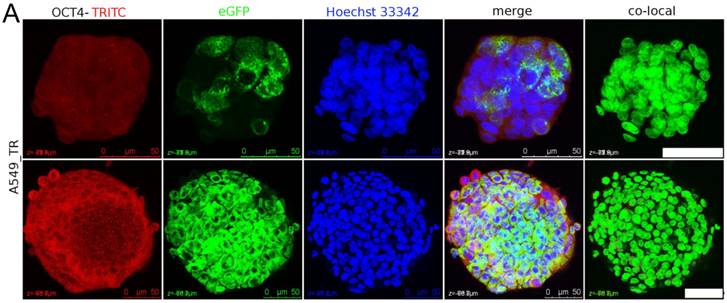
The nuclear staining of OCT4 in sphere was examined. Some A549_TR spheres composed of GFP-positive cells probably originated from single cell. After 26 days in suspension culture the A549_TR sphere displayed sparse OCT4 signal colocalized with nuclei in the inner layer of sphere (Figure 5A). After 40 days in suspension culture, the proportion of A549_TR spheres composed of GFP-positive cells was approximately 7.3% (3/41). Single A549_TR sphere failed to initiate tumor.
The nuclear staining of OCT4 in spheres cells. (A) Sparse OCT4 signal could be detected in nuclei of A549_TR after 26 days (upper panel) and 34 days (lower panel) in continuous static suspension culture. Tightly packed sphere composed of GFP-positive cells was probably originated from single cell (lower panel). The white dots in nuclei in merged channel image were the colocalized nuclear staining of OCT4. Scale bar stands for 50 µm. (B) MCF7, HepG2 and its subpopulation (SM01, SM02 and SM03) (33, 40, 21, 21, 21 days in suspension, respectively) were randomly selected for OCT4 nuclear staining detection. Dispersed OCT4 signal appeared in SM01, SM02 and SM03 spheres. Scale bar stands for 100 µm.
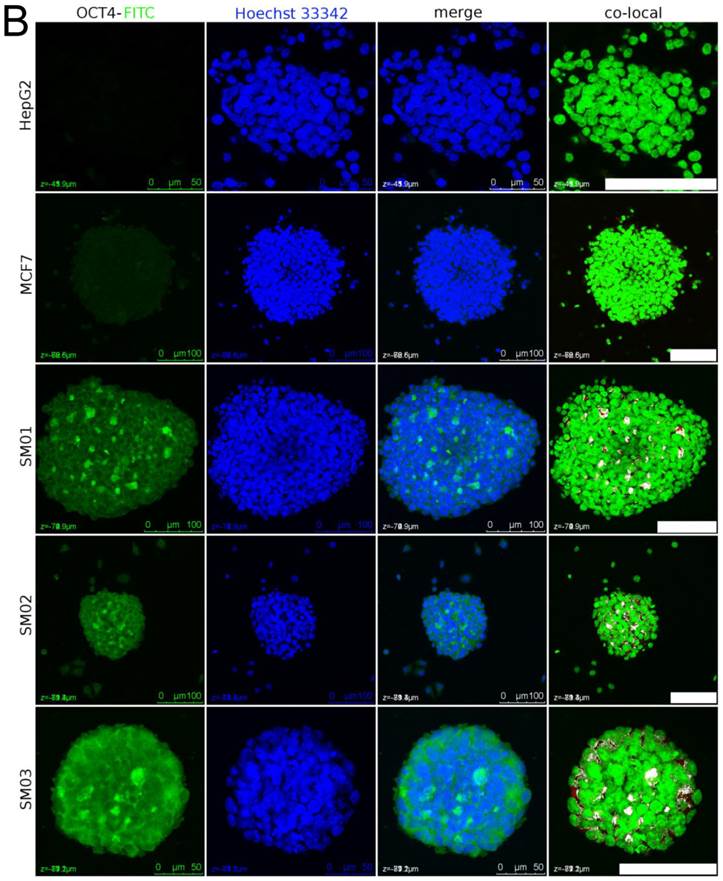
The HepG2 and MCF7 cells' nuclei in spheres did not show colocalized OCT4 signal after 23 and 27 days in suspension, respectively (Figure 5B). Around this period the growth proportions in vitro, ≥2-fold of that initial diameter, of individual MCF7, SM02 and SM03 spheres were 41.7% (40/96), 50% (48/96) and 61.4% (59/96), respectively. In the same period, the tumor-initiating proportions of single sphere were 3/4 (HepG2), 3/4 (SM01), 2/6 (SM02), 4/6 (SM03), 9/12 (MCF7) (Table 2). The estimated TIC frequencies in vivo, enhanced greatly, did not correlate with those of in vitro. It seemed the HepG2 and MCF7 spheres that could initiate tumors were not necessarily those with nuclear staining of OCT4 or the TICs required to initiate tumor were less than detectable; in contrast, constant dividing A549_TR cell itself was insufficient for tumor-initiating.
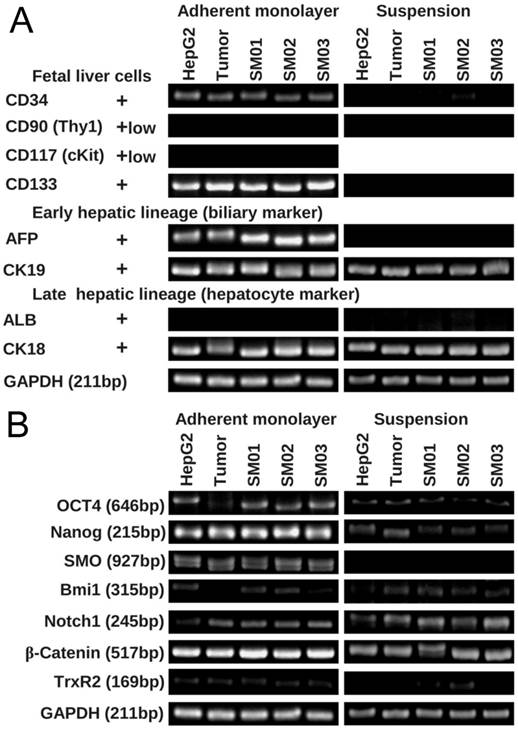
After two weeks static suspension culture, HepG2 cell and its tumor, SM01, SM02 and SM03 reduced in expression of fetal liver cell marker genes (CD34 and CD133), and AFP gene (Figure 6). ALB gene, expressed in differentiated hepatocyte, did not appear in two culture methods, while the CK19 and CK18 seemed unaffected. The expressions of Nanog and SMO genes were also reduced in suspension culture. The expressions of Oct4 and Bmi1 genes increased when HepG2 tumor cell shifted from monolayer to suspension culture.
HepG2 and its sub-populations in suspension were liable to differentiate. (A) The expression of fetal and early hepatic marker genes decreased in HepG2, its tumor and subpopulations (SM01, SM02 and SM03) in two weeks' suspension culture. (B) The expressing of Oct4 and SMO gene decreased in HepG2, its tumor and subpopulations (SM01, SM02 and SM03) in two weeks' suspension culture.
Discussion
Static suspension culture is still used in MTS initiation [18]. The continuous, static suspension culture combined agar coating, suspension cell aggregating and nutrient starvation in this study was simple and efficient. These conditions were aimed to mimic the microenvironment of tumor initiating and/or tumor relapse processes. This method was developed based upon the following evidence and inference. Agar coating was more suitable for transformed cell clones' formation than agarose [19]. Cells could migrate to desirable location within aggregates in suspension [20]. The low glucose could induce cell differentiation and quiescence [21]. Lowering glucose from 16.5 to 0.8 mM in medium, the onset diameter of necrosis in spheroids was reduced from >500 to 150-220 µm [22]. The cells bordering on necrotic center were compromised in metabolism and more resistant to cisplatin [23]. The cells around necrotic core might be hypoxic as well. Hypoxia induced stemness and played multiple roles in cancer progression and cell survival [24-26]. The hypoxia and nutrient starvation triggered autophagy contributed to the liver CD133+ cells' survival [27]. We broke cell spheres with nutrients starvation, and a cell dissociation process which caused 30-40% stem cell loss [28] could be avoided. However, an equivalent number of cell loss occurred during starvation in this process. Whether the cells with more active metabolism were more vulnerable in starvation remain unclear. The serum-containing formula might speed up the cell differentiation [7] as indicated in “mammosphere” [3] and “neurosphere” [7]. Other media and supplements such as Ham's F12 and serum replacement were also worth trying.
Both sphere and colony took advantage of spontaneous cell aggregating. Normal cells would form small spheres but would break apart <72 h [15]. The small colonies formed in soft-agar could not distinguish the progenitors, with limited dividing cycles, from stem-like cells. To reduce non-constant dividing cells, those spheres grown in suspension for >12 days were used. The single cells and spheres were only traced individually for 2-3 weeks; however, the suspension culture period of the same populations exceeded 90 days. In sphere, only the peripheral cells were proliferating [29, 30], this was consistent with our observation that a lag phase existed before the sphere growth. Cells shed from spheres remained round and translucent during observation. The tumor initiating potential of the single cells and spheres were not compared yet. Tightly packed HepG2, MCF7 and A549_TR spheres could be seen in this culture. Both loosely and tightly packed spheres could grow independently, the difference between them was unclear. The question remained whether spheres of different structures were different in tumor initiating potential.
We tested the tumor initiating potential of HepG2 sphere because the HepG2 cell was not a good starting material in TIC's sorting. HepG2 contained approximately 0.7-8% CD133+ cells [31-34], 0.04±0.01% CD90+ cells [35], 4.8% EpCAM+ cells [33], and had no side population cells [36]. However, recent study indicated the HepG2 cells expressed comparable CD13 and calcium channel subunit (α2δ1, identified by 1B50-1 mAb) with Huh7 (53.4 vs 37.1%; 0.5-2.1 vs 0.9-2.2%) [33]. HepG2 cells could not form embryoid body (EB), and grew as small circular mass even located in EB [37]. The combination of known markers could further narrow down the putative liver TIC subpopulation [33, 34], but the underlying mechanisms of cell surface markers were not fully understood yet. In adherent culture, the proportion of purified CD133+ cells decreased from >95 to 58-65% in six weeks, while the proportion of CD133+ cells would rise from <1 to 47-65% in purified CD133- cells (Huh7 harbors 65% CD133+ cells) [32]. This dynamic process would be a cause of the CD133+ cell's content variation in certain cell lines [33]. The diverse stem cell surface markers might only relate to tumor types [4]. The expression of cancer stem cell markers did not correlate with ALDH (aldehyde dehydrogenase) activity and clonogenic capacity; the “colonospheres” could even alter surface marker expression, depending on their parental cell lines [38]. As the marker-based methods were trying to piece together the whole image of TIC, the sphere that could initiate tumor would be helpful in this effort.
The artificial extracellular matrix, Matrigel, was used in transplanting especially in single cell's tumor-initiating [39-41] though the effect of this basement membrane extract (BME) on TIC remained unclear. Matrigel was able to drastically (>5000-fold) enhance tumorigenicity in animal in certain cells [39]. A549 cell in 50% Matrigel (1:1,v/v) was able to produce tumors at 1000 cells level in nude or SCID mice [42]. In this case, single cell and sphere of HepG2 and its three subpopulations could not initiate tumor at 104 cells level without Matrigel while their spheres, containing less cells, initiated tumor in 50% Matrigel. Though differentiation of HepG2 in suspension could partly explain its failure in tumor regeneration at 104 cells level, cell-cell interaction and extracellular matrix were required as well. Based on TIC frequency, Matrigel increased the TIC potential 136 folds in MCF7 and 567 folds in HepG2. The Matrigel (1.6 mg/ml) coated on agar altered the A549_TR sphere growth pattern from 2D into 3D and enhanced the growth rate of tightly packed A549_TR sphere slightly (P=0.062, n=48). EGF, bFGF and other components are usually supplemented in media of sphere culture [38, 43, 44], EGF and bFGF were also reported in Matrigel [40]. The effects of Matrigel on tumor cells and the immune response of host animal should be explained independently. The nude mice, with remaining non-specific immune response, could more closely mimic the tumorigenesis de novo.
Limitations of this study included: tumor-initiating and (de)differentiation in spheres were not studied at single cell's level; spheres for transplanting and for nuclear staining of OCT4 were not at exactly the same time point; the mechanisms of variations along time and among cell lines were unclear; Matrigel's effect in tumor initiation should be explored. We supposed that only those viable cells supported by neighboring cells and isolated from serum could survive repeated starvation and were more likely to re-constitute tumor. It was unknown whether the spheres were originated from clonal expansion and hence indicated a growth advantage that resulted in tumor-initiating. Further data were required to elucidate whether the more tumorigenic cells in sphere or merely the tumor cells by clustering simply into certain structure initiated the tumor. This method is simple and these findings may have potential applications in TIC marker finding and in the study of tumor microenvironment.
Conclusion
Tumor cells could proliferate rapidly and aggregate repeatedly in continuous, static suspension culture; spheres could be maintained in serum-containing media for >90 days even under repeated starvation. Some tumor cells in suspension were slightly more tumorigenic and the spheres with diameter of 101-150 µm were likely to grow independently. The HepG2 and MCF7 spheres of diameter ~200 µm could initiate tumor in nude mice with estimated TIC frequency of 1/1500-1/400. The very cell that initiated tumor and the underlying mechanism of variations along time and among cell lines required further study.
Supplementary Material
Supplementary Table 1 Primers and cycling condition.
Acknowledgements
We are indebted to Bo Xu for the help in cell isolation, to Qiuru Zhang and Xin Chen for routine care for nude mice, to Jian Chen for tumor slice preparation, to Lan Yuan for cofocal microscopy examination. We thank the critical advice on writing from Xiaoqing Zheng. This work is supported by the National Natural Science Foundation (#30472036) of China.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Roesch A, Fukunaga-Kalabis M, Schmidt EC. et al. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell. 2010;141:583-94
2. Perego M, Tortoreto M, Tragni G. et al. Heterogeneous phenotype of human melanoma cells with in vitro and in vivo features of tumor-initiating cells. J Invest Dermatol. 2010;130:1877-86
3. Pece S, Tosoni D, Confalonieri S. et al. Biological and molecular heterogeneity of breast cancers correlates with their cancer stem cell content. Cell. 2010;140:62-73
4. Benayoun L, Gingis-Velitski S, Voloshin T. et al. Tumor-initiating cells of various tumor types exhibit differential angiogenic properties and react differently to antiangiogenic drugs. Stem Cells. 2012;30:1831-41
5. Steiner D, Khaner H, Cohen M. et al. Derivation, propagation and controlled differentiation of human embryonic stem cells in suspension. Nat Biotechnol. 2010;28:361-4
6. Inagaki A, Soeda A, Oka N. et al. Long-term maintenance of brain tumor stem cell properties under at non-adherent and adherent culture conditions. Biochem Biophys Res Commun. 2007;361:586-92
7. Lee J, Kotliarova S, Kotliarov Y. et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell. 2006;9:391-403
8. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-74
9. Freedman VH, Shin SI. Cellular tumorigenicity in nude mice: correlation with cell growth in semi-solid medium. Cell. 1974;3:355-9
10. Fluri DA, Tonge PD, Song H. et al. Derivation, expansion and differentiation of induced pluripotent stem cells in continuous suspension cultures. Nat Methods. 2012;9:509-16
11. Sutherland RM. Cell and environment interactions in tumor microregions: the multicell spheroid model. Science. 1988;240:177-84
12. Sutherland R, Carlsson J, Durand R. et al. spheroids in cancer research. Cancer Res. 1981;41:2980-2984
13. Santini MT, Rainaldi G. Three-dimensional spheroid model in tumor biology. Pathobiology. 1999;67:148-57
14. Hirschhaeuser F, Menne H, Dittfeld C. et al. Multicellular tumor spheroids: an underestimated tool is catching up again. J Biotechnol. 2010;148:3-15
15. Yuhas JM, Li AP, Martinez AO. et al. A simplified method for production and growth of multicellular tumor spheroids. Cancer Res. 1977;37:3639-43
16. Crema A, Ledda M, De Carlo F. et al. Cord blood CD133 cells define an OV6-positive population that can be differentiated in vitro into engraftable bipotent hepatic progenitors. Stem Cells Dev. 2011;20:2009-21
17. Yu SC, Ping YF, Yi L. et al. Isolation and characterization of cancer stem cells from a human glioblastoma cell line U87. Cancer Lett. 2008;265:124-34
18. Zweigerdt R, Olmer R, Singh H. et al. Scalable expansion of human pluripotent stem cells in suspension culture. Nat Protoc. 2011;6:689-700
19. Neugut AI, Weinstein IB. The use of agarose in the determination of anchorage-independent growth. In Vitro. 1979;15:351-5
20. Linser P, Moscona AA. Induction of glutamine synthetase in embryonic neural retina: localization in Muller fibers and dependence on cell interactions. Proc Natl Acad Sci U S A. 1979;76:6476-80
21. Casciari JJ, Sotirchos SV, Sutherland RM. Glucose diffusivity in multicellular tumor spheroids. Cancer Res. 1988;48:3905-9
22. Freyer JP, Sutherland RM. Regulation of growth saturation and development of necrosis in EMT6/Ro multicellular spheroids by the glucose and oxygen supply. Cancer Res. 1986;46:3504-12
23. Frenzel KR, Saller RM, Kummermehr J. et al. Quantitative distinction of cisplatin-sensitive and -resistant mouse fibrosarcoma cells grown in multicell tumor spheroids. Cancer Res. 1995;55:386-91
24. Keith B, Simon MC. Hypoxia-inducible factors, stem cells, and cancer. Cell. 2007;129:465-72
25. Conley SJ, Gheordunescu E, Kakarala P. et al. Antiangiogenic agents increase breast cancer stem cells via the generation of tumor hypoxia. Proc Natl Acad Sci U S A. 2012;109:2784-9
26. Greer SN, Metcalf JL, Wang Y. et al. The updated biology of hypoxia-inducible factor. EMBO J. 2012;31:2448-60
27. Song YJ, Zhang SS, Guo XL. et al. Autophagy contributes to the survival of CD133+ liver cancer stem cells in the hypoxic and nutrient-deprived tumor microenvironment. Cancer Lett. 2013;339:70-81
28. Amit M, Laevsky I, Miropolsky Y. et al. Dynamic suspension culture for scalable expansion of undifferentiated human pluripotent stem cells. Nat Protoc. 2011;6:572-9
29. Yuhas JM, Li AP. Growth fraction as the major determinant of multicellular tumor spheroid growth rates. Cancer Res. 1978;38:1528-32
30. Demicheli R, Foroni R, Ingrosso A. et al. An exponential-Gompertzian description of LoVo cell tumor growth from in vivo and in vitro data. Cancer Res. 1989;49:6543-6
31. Suetsugu A, Nagaki M, Aoki H. et al. Characterization of CD133+ hepatocellular carcinoma cells as cancer stem/progenitor cells. Biochem Biophys Res Commun. 2006;351:820-4
32. Ma S, Chan KW, Hu L. et al. Identification and characterization of tumorigenic liver cancer stem/progenitor cells. Gastroenterology. 2007;132:2542-56
33. Zhao W, Wang L, Han H. et al. 1B50-1, a mAb raised against recurrent tumor cells, targets liver tumor-initiating cells by binding to the calcium channel alpha2delta1 subunit. Cancer Cell. 2013;23:541-56
34. Chen Y, Yu D, Zhang H. et al. CD133(+)EpCAM(+) phenotype possesses more characteristics of tumor initiating cells in hepatocellular carcinoma Huh7 cells. Int J Biol Sci. 2012;8:992-1004
35. Yang ZF, Ho DW, Ng MN. et al. Significance of CD90+ cancer stem cells in human liver cancer. Cancer Cell. 2008;13:153-66
36. Chiba T, Kita K, Zheng YW. et al. Side population purified from hepatocellular carcinoma cells harbors cancer stem cell-like properties. Hepatology. 2006;44:240-51
37. Fong CY, Chak LL, Subramanian A. et al. A three dimensional anchorage independent in vitro system for the prolonged growth of embryoid bodies to study cancer cell behaviour and anticancer agents. Stem Cell Rev. 2009;5:410-9
38. Stuelten CH, Mertins SD, Busch JI. et al. Complex display of putative tumor stem cell markers in the NCI60 tumor cell line panel. Stem Cells. 2010;28:649-60
39. Quintana E, Shackleton M, Sabel MS. et al. Efficient tumour formation by single human melanoma cells. Nature. 2008;456:593-8
40. Benton G, Kleinman HK, George J. et al. Multiple uses of basement membrane-like matrix (BME/Matrigel) in vitro and in vivo with cancer cells. Int J Cancer. 2011;128:1751-7
41. Fridman R, Benton G, Aranoutova I. et al. Increased initiation and growth of tumor cell lines, cancer stem cells and biopsy material in mice using basement membrane matrix protein (Cultrex or Matrigel) co-injection. Nat Protoc. 2012;7:1138-44
42. Bertolini G, Roz L, Perego P. et al. Highly tumorigenic lung cancer CD133+ cells display stem-like features and are spared by cisplatin treatment. Proc Natl Acad Sci U S A. 2009;106:16281-6
43. Svendsen CN, ter Borg MG, Armstrong RJ. et al. A new method for the rapid and long term growth of human neural precursor cells. J Neurosci Methods. 1998;85:141-52
44. Dontu G, Abdallah WM, Foley JM. et al. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 2003;17:1253-70
Author contact
![]() Corresponding author: Huihui Zeng, Professor (zenghhedu.cn), Address: No 38 Xueyuan Road, Beijing 100191, P.R. China. Tel: 86-10-82802878, Fax: 86-10-62015584
Corresponding author: Huihui Zeng, Professor (zenghhedu.cn), Address: No 38 Xueyuan Road, Beijing 100191, P.R. China. Tel: 86-10-82802878, Fax: 86-10-62015584